ZeroFold: Protein-RNA Binding Affinity Predictions from Pre-Structural Embeddings
The accurate prediction of protein-RNA binding affinity remains an unsolved problem in structural biology, limiting opportunities in understanding gene regulation and designing RNA-targeting therapeutics. A central obstacle is the structural flexibil…
Authors: Josef Hanke, Sebastian Pujalte Ojeda, Shengyu Zhang
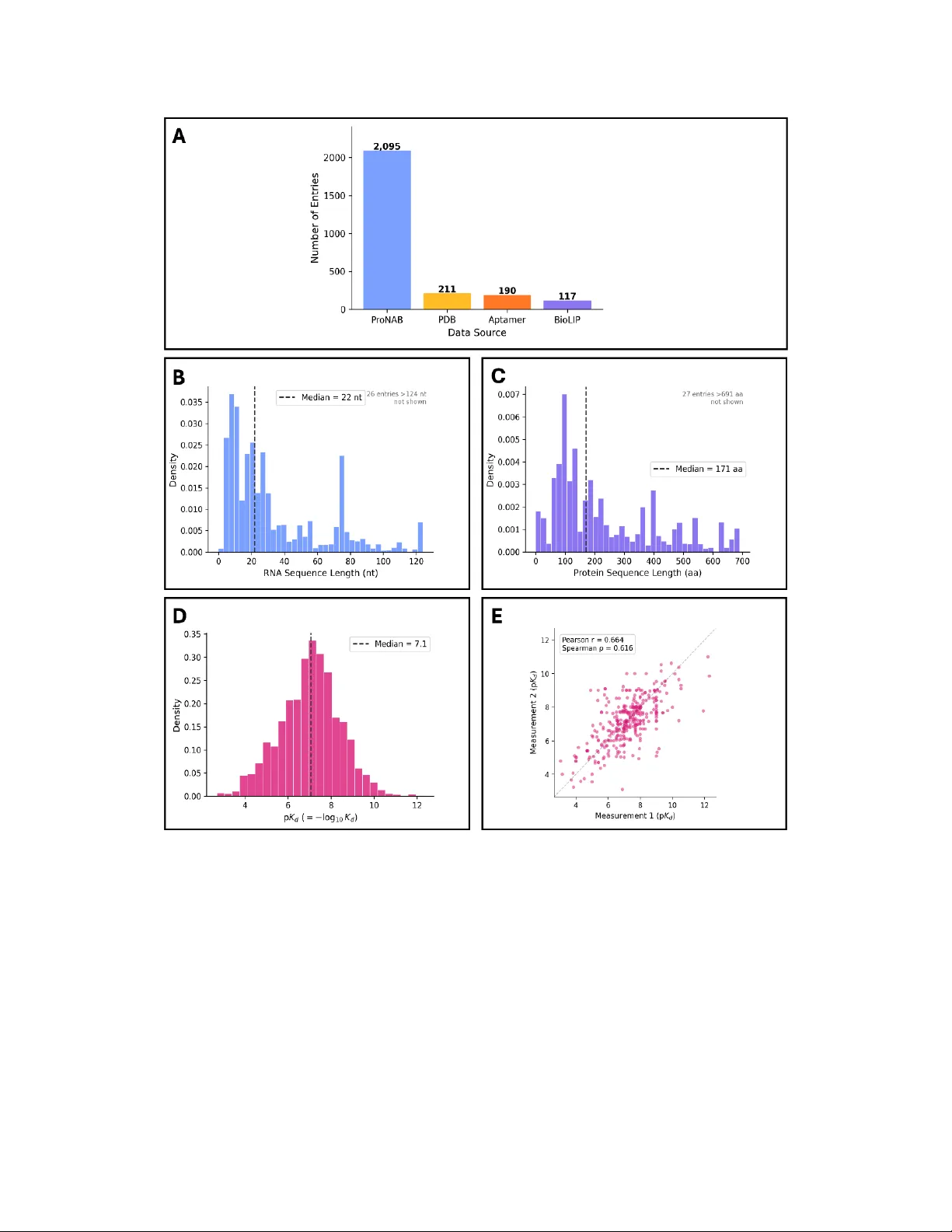
ZeroFold: Protein - RNA Bindi ng Affinity Pre dictions from Pre - Structural Embeddings Josef Hanke 1 , Sebastian Pujalte Ojeda 1 , Shengyu Zhang 1 , Werngard Czechtizky 2 , Leonardo De Maria 2 and Michele Vendruscolo 1,* 1 Centre for Misfolding Di seases, Yusuf Hamied Departmen t of Chemistry, University of Cambridge, Cambridge, UK 2 Medicinal Chemistr y, Research and Ear ly Development, Resp iratory and Immunol ogy, BioPharmaceuticals R&D, Astr aZeneca, Gothenburg, Sweden Abstract The accurate prediction of protein - RNA binding affinity remains an unsolved problem in structural biology, limiting opportunities in understanding gene regulation and designing RNA - targeting therapeutics. A central obstacle is the structural flexibility of RNA, as, unlike prote ins, RNA molecules exist as dynamic conformational ensembles. Thus, committing to a single pr edicted structure discards information relevant to binding. Here , we show t hat this obstacle can be addressed by extracting pre - structural em beddings, which are intermediate representations from a biomolecular foundation model captured before the str ucture decoding step. Pre - structural embeddings implicit ly encode conformational ensemble information without requiring predicted structures. We bu ild ZeroFold , a tra nsformer - based model that combines pre - structural embeddings from Boltz - 2 for both protein and RNA molecules through a cross - modal attention mechanism to predict bi nding affinit y directly from sequen ce. To sup port trainin g and eval uation , we construct PRADB, a cur ated dataset of 2,621 unique protein - RNA pairs with exp erimentally measured affinities drawn from four complementary databases. On a held - out test set constructed with 40% sequence identity thresholds, ZeroFold achieves a Spearman correlation of 0.6 5 , a value approaching the ceiling imposed by experimental measurement noise. Under progressively fairer evaluation conditions that control for training - set overlap, Zer oFold compares favourably with respect to leading structure - based and leading sequence - based predictors, with the performance gap widening as sequence similarity to competitor training data is reduced. These results illustrate how pre - structural embeddings offer a represent ation strategy for flexible biomolecules, opening a route to affi nity predi ction for protein - RNA pairs for which no str uctural data exist. * Correspondence to: mv245@cam.ac.uk Introduction Protein - RNA interactions are fundamental to cellular regul ation, and their misregulation is increasingly recognized as a driver of human disease 1-3 . Long - read sequencing technologies have revealed an unexpectedly diverse repertoire of non - coding RNAs with key regulatory functions across cell types and tissues 4,5 , and the dysregulation of these molecules has been implicated in a wide range of diseases, from defective RNA splicing to aberrant microRNA activity 6,7 . Protein - RNA interactions are central to these processes and are eq ually relevant to the development of peptide - based therapeutics, a clinically validated drug class whose larger interaction surfaces make them well suited to the shallow, extended interfac es t ypical of RNA - binding sites 8,9 . Recent advances in understanding the protein - RNA interaction code, together with computational tools for predicting biomolecular structures and interactions 10 - 12 , have created new opportunities for the rational design of RNA - binding peptides. The greatest difficulty in predicting these interactions arises from the nature of the binding interfaces themselves. Protein - RNA complexes typi cally involve large, often sh allow surface areas that lack the hydrophobic pockets characteristic of traditional ly druggable targets 13 , and the binding is frequently heterogeneous, involving disor dered protein regions. Furthermore, the flexibility of RNA structures often causes them to behave like intrinsically disordered proteins, so that a single three - dimensional conformation cannot adequately represent the bound state 14 . Several computatio nal strategies have been d eveloped to address this p roblem, broadly categorised into structure - based and sequence - based approaches. Structure - based methods extract features at the binding interface, such as contact distances or interaction energies, from experimentally resolved structures and use them to train affinity predicti on models. While these approaches can capture detailed physical interactions, they rely on structural inputs that are available for only a small fraction of known protein - RNA pairs and tend to show limited generalisation to novel sequences. Among these, PRdeltaGPred 15 and PredPRBA 16 both utilise interface structure features in addition to sequence information, and the recent model CoPRA combines protein and RNA language model representations with complex structural information, reporting strong performance on complexes with resolved structures 17 . Sequence - based models avoid this structural dependency but have shown variable performance. PNAB classifies protein - nucleic acid complexes by nucleic acid type and builds stacking heterogeneous ensemble models for each class 18 , PRA - pred uses multi ple linear regression 19 , and the leading sequence - based model DeePNAP encodes sequences directly 20 , although its performance has been shown to vary substantially depending on the test set 17 . A central limitation across all these approaches is the scarcity of high - quality training data, with existing datasets comprising only a few thousand data points. Machine learning has transformed the p rediction of protein structure, protein - protein interactions, and a growing range of biomolecular properties 10,21 - 23 . Yet a ccurately modelling biomolecules th at exist as conformational ensembles, such as RNA and disordered proteins, remains a fundamental challenge 24,25 . Current structure prediction methods output a single conformation, discarding the ensemble informati on that is often cr itical for understanding binding. This limitation is especially acute for protein - RNA interactions , where the flexibil ity of bo th partners means that no single predicted structure can fully capture the determinants of affinity. A key in sight motivating this work is that the intermedi ate representati ons generated by biomolecular foundation models during structure prediction , here termed pre - structural embeddings , encode richer information about binding than the final pr edicted structures themselves. Rather than committing to a single folded conformation, these high - dimensional representations implicitly encode the ensemble of possible conformations and their associ ated binding properties 26,27 , a distinction that is particularly consequential for fl exible molecules such as RNA. Crucially, extracting these embeddings before the structure decoding step avoids the information loss that occurs when a dynamic conformational ensemble is collapsed int o a singl e predicted structure. This principle has recently been val idated for protein - ligand binding affinity prediction, where pre - structural embeddings achieved strong predictive performance 27 , suggesting that it may represent a general strategy for modell ing bi omolecular interactions involving flexible partners. Here we intro duce Zer oFold, a transformer - based model that operationalises thi s principle for protein - RNA binding affinity prediction. ZeroFold extracts pre - structural embeddings from Boltz - 2 28 , a biomolecular foundation model that natively supports protein, RNA, and DNA sequences within a unified trunk a rchitecture. Representati ons are extracted from the fina l trunk layer prior to the structure prediction head, capturing contextually rich per -r esidue and pairwise embeddings that reflect both sequence and evolutionary context ( Figure 1 ). These are processed through separate encoding blocks for each chain, integrated via a cross - modal attention module, and passed to an affinity prediction head. The choice of Boltz - 2 over protein - only foundation models was motivated by its unified treatment of nucleic acid an d protein sequen ces, which makes it well suited to modelling the joint conformational space of protein - RNA complexes, an d by its flexibl e sequence defi nitions, which accommodate non - natural amino acids and modified nucleotide bases relevant to therapeutic design. To address the scarcity of trai ning data, we assembled the Protein - RNA Affinity Database (PRADB), a curated dataset of 4,510 pairs (2,621 unique) of protein and RNA sequences with experimentally measured binding affinities (pK D ), compiled from four complementary sources: ProNAB 29 , BioLiP 2 30 , the UTexas Aptamer Database 31 , and PDBbind+ 32 . We train ZeroFold on PRADB and evaluate it against state - of - the - art structure - based and sequence - based methods. On a test set con structed with strict 40% sequence i dentity thr esholds on both protein and RNA sequences, ZeroFold achieves a Spearman correla tion of 0.6 5 , performing better than CoPRA and DeePNAP even in baseline comparisons. Because the ZeroFold test set contains sequences present in the training datasets of these competing models, this comparison inherently favours those models. To account fo r this, we constructed progressively stricter evaluation subsets that systematically remove sequences with training - set overlap for each competitor. As expected, the performance of CoPRA and DeePNAP decl ines as t his overlap is reduced, whereas the performance of ZeroFold remains stable. These results demonstrate that ZeroFold achieves cutting - edge performance in protein - RNA binding aff inity predic tion, and that th is advantage becomes more pronounced under fairer evaluation conditions. Results ZeroFold training To train and evaluate Ze roFold , we assembled PRADB, a curated dataset of 2,621 unique protein - RNA pairs with experimentally measured b inding affinities (pKD) drawn from four complementary sources: ProNAB 29 , BioLiP2 30 , the UTexas Aptamer Database 31 , and PDBbind+ 32 ( Figure 2A ). ProNAB consti tutes the largest single co ntribution, reflecting i ts broad coverage of RNA - binding protein families, while the remaining three databases provide complementary coverage of structurally characterised complexes and aptamer interactions. The 2 ,621 unique pairs derive from 512 distinct proteins and 1,411 distinct RNA sequences, with protein sequence lengths spanning a wide range that encompasses both compact RNA - binding domains and larger multidomain assemblies ( Fi gure 2C ), and RNA se quence lengths similarly covering a br oad spectrum from short aptamers to longer structured RNAs ( Figure 2B ). The distribution of experimentally measured affinities (pK D ) is approximately unimodal and centred around 7.1, with a standard deviation of 1.35, reflecting a dataset that is neither dominated by very low - affinity nor very high - affinity binders and spans a range well suited to training a regression model ( Figure 2D ). ZeroFold performance PRADB was split into training, validation, and test sets in an approximate 8:1:1 ratio. To en sure fair evaluation and reduce potential data leakage, these splits were constructed such that no protein sequences shared greater than 40% sequence identity with at least 80% coverage across different sets. The same criterion was applied to RNA sequences. The fi nal mode l was trained for 100 epoc hs, wit h each epoch comprising 900 samples from the training set weighted by cluster representation (see Methods). The model performed strongly on the test set, achieving a mean absolute error (MAE) of 1.14, a root m ean squared error (RMSE) of 1.47, a Pearson correlation coefficient ( PCC ) of 0.63, and a Spearman correlation coefficient ( SCC ) of 0.65. Noise and variabi lity in the expe rimental affinity mea surements impose an e ffective upper bound on achievable correlation coefficients of approximately 0. 6 - 0.7 ( Figure 2E ). This limitation reflects the quality and availability of current experimental data rather than a fundamental constraint of the modelling approach. As a result, models reporting substantially higher correlations may reflect overfitting or data leakage from the training set to the test set. The performance of ZeroFold therefore approaches the practical upper l imit imposed by the quality and consistency of the available experimental data, suggesting that further improvements are likely to require larger , higher - quality, and more representative binding affinity datasets. In addition to predictive performance, ZeroFold offers a substantial advantage in computational efficiency over structure - based affinity predi ction pipelines. Both ZeroFold and structure - based approaches require a Boltz - 2 trunk forward pass; however, struc ture - based pipelines additionally incur the cost of structure decoding prior to affinity estimation, whereas ZeroFold passes pre - structural embeddings direct ly to its affinity prediction head, bypassing this step ( Figure 3 ). This reduction in computational overhead enables higher - throughput evaluation of protein - RNA interactions, making ZeroFold particularly well suited to applications such as virtual screening and proteome - wide affinity profiling, where the ability to evaluate lar ge numbers of candidate sequences is critical. Comparison with CoPRA A comparative analysis aga inst existing state - of - the - art predictors was conducted. Most current methods for prot ein - RNA af finity prediction are structu re - based, meaning that predictions are made using experiment ally determined 3D st ructures in which i ntera ction information is already implicitly encoded. This provides a substantial advantage compared with approaches that rely solely on sequence information, particularly because such methods are typically evaluated on complexes with resolved structures rather than on sequence pairs lacking structural data. The leading structure - based protein - RNA aff inity predictor is CoPRA, whose reporte d performance substantially exceeds that of ot her competing methods in the field 17 . To enable a more comparable evaluation on protei n - RNA p airs without expe rimentally resolved structures, predicted complex struct ures were first generated using Boltz - 2 and subsequently used as input for CoPRA. CoPRA reports a PCC of 0.58 and a SCC of 0.59, based on five - fold cross - validation of its PRA310 dataset. Howeve r, the sequence similarity threshold used to separ ate clusters in this benchmark was 70% on the protein sequence, which is substantially more permissive than the 40% ident ity t hreshold on both the RNA and the protein applied when constructing the ZeroFold test set. This more relaxed criterion increases the likelihood that closely related sequences appear across training and evaluat ion folds and, when combined with the use of experimentally resolved structural data, may contribute to inflated repor ted performance metrics. To enabl e a fair comparison between ZeroFold and CoPRA, performance was evaluated on five progressively stricter subsets of the Ze roFold test set ( Table 1 ). In the first setting (A), evaluation was performed on the full Zer oFold test set, regardless of whether any sequences overlapped with the CoPRA PRA310 dat aset. In th e second se tting (B), any protein sequences p resent in both the ZeroFold test set and PRA310 were removed. In the third setting (C), protein sequences with gre ater than 70% sequence identity to sequences in PRA310, the similarity threshold used in the CoPRA be nchmark, were exclud ed. In the fourth setting (D), a stricte r 40% protein sequence identity threshol d was applied. Finally, in the most stringent setting, both protein and RNA sequen ces were required to have less than 40% i dentity to any sequence in the training data, mirroring t he criteria used to construct the Zero Fold test set and therefore providing the most co mparable evaluation to t he repo rted ZeroFold performance. This resulted in too few datapoints to make a meaningful comparison, so it is not repor ted in Table 1 . As expected, the performance of CoPRA decreased, wit h the PCC falling fr om 0.50 in A to 0.22 in D , as the evaluation criteria became more stringent, reflecting the increasing difficulty of predicting affinities without closely related sequences or experimentally resolved structures. In contrast, the performance of ZeroFold remained largely consistent across these subsets, PCC around 0.6. Notably, ZeroFold outperformed CoPRA even in the l east restrictive settings, where identical or highly similar protein sequences were present in PRA310, but not in PRADB, and CoPRA therefore had the grea test potential advantage. Thes e results de monstrate that ZeroFold achieves stronger predictive performance despite operating without experimentally determined structural inputs and under more stringent evaluation condi tions. Comparison with DeePNAP A more direct comparison for ZeroFold is with other sequence - based affinity prediction methods. The current cu tting - edge model of this t ype is DeePNAP 20 , which was trained on the ProNAB dataset, a dataset that also forms part of PRADB. DeePNAP reports a correlation coefficient (R) of 0.92 for K D prediction. However, the C o PRA study reports substantially lower correlation coefficients ( PCC and SCC ) of 0.35 and 0.35, respectively, when evaluating DeePNAP on the PRA201 dataset. This reduction in performance likely reflects di fferences in dataset construction: DeePNAP does not a pply a se quence similarity threshold when genera ting its test set, increas ing the l ikelihood that closely related sequences appear in both training and evaluation data and thereby inflating the reported performance. To enable a fai rer comparison with ZeroFold , the same four test set subsets (A - D) used in the Co PRA analysis were constructed ag ain, but this time using ProNAB as the reference trai ning dataset for sequence similarity filtering. As the filtering criteri a becam e progressively more stringent, removing identical sequences and then increasingly similar p rotein and RNA sequences, the performance of DeePNAP declined sli ghtly, refl ecting the increasing difficulty of predicting affinities for sequences that are les s si milar to those observed during training. However, this decrease was far less pronounced than the performance drop observed for C o PRA under the stricter evaluation settings. DeePNAP was trained on a consider ably larger dataset, which likely contributes to its st ronger generalisation across these subsets and further highlights t he importance of larger training datasets for improving m odel performance. The performance of ZeroFold also decreased on the more stringent subsets, suggesting that these later sub sets are intrinsically more challenging and that the difficulty cannot be explained solely by sequence distance from the training set. Nevertheless, ZeroFold consistently outperformed DeeP NAP across all evaluated metrics. To further characterise model performance across the affinity range, the tes t set was divided into low - , medium - , and high - affinity bands, defined by thresholds at the mean test - set affinity ± one standard deviation. Correlation metr ics were then computed separately within each group ( Table 2 ). The correlation calculated across the full test set was higher than that observed within any individual tercile. This indicates that much of the global correlation arises from between - group variance across the affini ty spectrum, whereas analysing terciles in isolation removes this variance and reveals the weaker within - group ranki ng signal. These results suggest that the model ha s learne d robust coarse - grai ned discrimination: it reliably distinguishes weak binders fro m str ong b inders. However, within - tercile correlation decreases toward the extremes of the affinity range, particularly among high - affinity interactions. In practical terms, this performance profile is well - suited to virt ual screening applications, where t he primary objective is to identify promising candidate binders from a large pool of sequences with unknown affinity. By contrast, the model is likely to be less reliable for fine - grained lead optimisation tasks, with an SCC of only 0.28 in the st ronger binders, where subtle differences in affi nity between already pot ent binders must be resolve d. It is al so important t o note that ZeroFold has a higher SCC than both C o PRA and DeePNAP across all bands. Discussion We have report ed how ZeroFold achiev es stat e - of - the - art performance in protein - RNA binding affinity prediction, with accuracy approaching the practical upper bound imposed by experimental noise and inter - assay variability 29 - 32 . The proximity of model performance to the level of inter - measurement variability suggests that curre nt dat a quali ty may consti tute a major limit ing f actor, and that further gains may require larger and more consistently measured training datasets 29 - 32 , and that further gains will likely require larger and more consistently measured training data rather than architectural improvements alone. A central finding is t hat the advantage of ZeroFold over both structure - based and sequence - based baselines widens as eval uation conditions become fairer. Against CoPRA, it s lead grows as test subsets are progressively filtered to remove sequences with tr ai ning - set overlap, and this holds even though CoPRA has access to experimentally resolved structures 17 . Against DeePNAP, ZeroFold outperforms consiste ntly across all filtered ProNAB su bsets despite operating on a smaller training set 20 . Together, these comparisons indicate that the gains of ZeroFold reflect genuine generalisation rather than favourable dataset compositi on. These resul ts support the view that pre - structural embeddings are a particularly informative representation for modelling interactions involving conformationally heterogeneous partners 26,27 . By extracting intermediate representations from the Bol tz - 2 trunk before the structure decoding step, ZeroFold retai ns ensemble - level information that is discarded when a dynamic system is collapsed into a single predicted conformation 26,28 . This is especially consequential for protein - RNA systems, where RNA f lexibility, context dependence, and b inding - induced rearrangements limit the utility of static structural models 14 . Crucially, the appro ach requires no resolved structure at inference time, making i t directly applicable to the large majority of prot ein - RNA pairs for which structural data do not exist. Several l imitations remain. The available quantitative protein - RNA affinity da ta are modest in scale relative to those underpinning advances in other areas of machine learning for biology, and the integration of measurements from different laboratories and experimental modalities may obscure finer - grained biophysical trends. Prediction accuracy is also likely to vary across protein classes, RNA types, and af finity regimes, and futur e work should characterise these differences systematically. It will additio nally be important to test whether the pre - structural embedding strategy generalises to related tasks such as mutation - effect prediction, binding - site identification, and the modelling of multicomponent ribonucleoprotein assemblies. In summary, ZeroFold demonstrates that pre - structural embeddings offer a general strategy for learning interaction energetics in systems where conformational flexibility is central to function. As the p erformance is approaching the ceiling set by current data quality, progress beyond this point will particularly depend on the generation of larger, more reproducible affinity datasets. Methods ZeroFold comprises four mai n compon ents: t he RNA encoder, the protein encoder, the transformer layers and the affinity prediction hea d. RNA/Protein input representations . Zer oFold employs Boltz -2 28 to extract representations for both protein and nucleic acid chains. These representations were extracted from the final layer of the Boltz - 2 trunk (recycling_steps 3), just before the structure module decoding. The protein and nucleic acid representation s each consist of two components. Firstly, we extracted the single sequence representation {𝑠 ! "#$ }, where 𝑠 ! "#$ % ∈ & 𝑅 & !"# , 𝑐 "#$ = 384 , and 𝑖 & ∈ & {1. . . 𝑁 '(# }&, which is the per - residue (or per - nucleotide) embedding from t he f inal trunk layer. Secondly, we extracted the pair represen tations, {𝑧 !) *$!' } , where 𝑧 !) *$!' ∈ & 𝑅 & $#%& , 𝑐 *$!' = 128 and 𝑖, 𝑗 & ∈ & {1. . . 𝑁 '(# }&, which are t he pairwise embeddings encoding residue – residue (or nucleotide – nucleotide) spatial and evolutionary relationships. RNA - protein encoder . The protein encoder maps Boltz - 2 trunk representations i nto refined single and pai r feature streams. The single representation 𝑠 ! "#$ % and pair r epresentation 𝑧 !) *$!' are each passed through two successive transition layers with residual connections and dropout, preserving the input dimensionality throughout. The RNA encoder follows an identical structure, with the ad dition of a learn ed nuc leic acid type e mbedding (RNA vs DNA, 32 - dimensional) concatenated to the single representation before the fi rst transition layer (which, due to the resulting dimension change from 384 to 416, is applied without a residual connection), allowing the encoder to condition on nucleic acid type. Cross - modal attention module and affin ity head . The cross - modal attent ion module integr ates the encoded protein and RNA representations to model interactions across the protein - RNA interface. The resulting joint representation is then passed to an affinity prediction head to produce a scalar pK D estimate. Datasets ProNAB . The primary dataset used to train ZeroFold was ProNAB, a manually curated database containing over 20,000 experimentally measured binding affi nities for protein - DNA (14,606) and protein - RNA (5,323) complexes 29 . Among the protein - RNA entries, 5,173 contained valid K D measurements, which were co nverted t o pK D values for training. After further fi ltering to retain only valid sequences, specifically com plexes containing unmodified nucleotide bases and natural amino acids, the dataset comprised 3,588 protein - RNA pairs with vali d affinity values. Many protein - RNA sequence pairs appeared multiple times in the dataset with substantially different reported affinity values ( Figure 2E ). These discrepancies likely arise from differences in experimental conditions, including temperature, pH, and assay methodology, which are extensively annotated within the ProNAB database. However, stratifying the data by these experimental variables did not substantially reduce the variability in reported pK D values, suggesting that much of the observed variation reflects measurement noise and experimental uncertainty rat her than systematic differences in the annotated conditions. Because it was not possible to define a consistent set of standard experimental condi tions, and because each measurement still repr esents a valid experiment al observation of the same in teraction under different contexts, all entries were retained to preserve the available experimental evidence and allow the model to learn from the ful l range of reported binding affinities. BioLiP2 . The next largest source of affinity data was BioLiP2 30 . This database contains biologically relevant protein - ligand interactions derived from the Protein Data Bank (PDB), annotated with binding affinity measurements collected from external databases and manual literature curation. BioLiP2 contains 425 protein - RNA complexes with reported b inding affinity data. UTexas aptamer database . The aptamer database cu rated by the University of Texas compiles binding aff inity measurements reported in the literature between 1990 and 2022 31 . Within this resource, 258 protein - RNA complexes were associated with l abelled binding affi nity values. PDBbind+ . The final dataset used was PDBbi nd+ (v2020), which was conside red the most reliable source of a ffinity data due to i ts high level of curation and the availability of experimentally determined 3D structural information alongside binding affinity annotation s 32 . This dataset contains 239 protein - RNA complexes with repor ted binding affiniti es. PRADB . The union of the four previously described databases was taken to produce a redundant set of 4,510 protein - RNA pairs. For pairs associated with multiple affinity measurements, the value repor ted in PDBbind+ was pref erentially selected due to its higher level of curation; if no PDBbind+ measuremen t was available, the median affinity across the remaining sources was used. After resolving these redundancies, the final Protein - RNA Affinity DataBase (PRADB) comprised 2,621 unique protein - RNA se quence pair s. The distributions of protein sequence lengths, RNA sequence lengths, and affinity valu es are shown in Figure 2B -D . Clustering. To preven t dat a le akage across trai n, v alidation, and test splits , al l pr otein and nuclei c acid sequences were clustered independently using MMseqs2 easy - cluster 33 , with a 40% sequence identity threshol d and 80% coverage requirement. This yielde d 347 pr otein clusters and 1,151 nucl eic aci d cl usters, with each data point assigned both a protein cluster label and a nucleic acid cluster l abel. Sp lit assign ment was then p erformed at the cluster level, wh ere no cluster representative could appear i n more than one split, ensuring that no sequence in the evaluation sets shares more than 40% identi ty with any traini ng sequence. Following an initial split assignment, the spl its were audited using bidirectional MMseqs2 easy - search to detect leakage cases that greedy clustering can miss: two sequences above the identity threshold can occupy different clusters if each is more similar to a different representative. This search re vealed 23 entries in the validation and test set s with sequence identities exceeding 40% t o tr aining counterparts; these were removed, yielding final split sizes of 2,104 training, 210 validation, and 299 test data points. Sample weighti ng . To counteract the over - representation of highly sampled sequence families in the training set, we follow a similar protocol to AlphaFold - Multimer, where each data point was assigned a sample weight inversely proportional to the size of its protein and nucleic acid clusters: 𝑤 !) = + |- . % | · + |/ . ' | where 𝑃 𝐶 ! is the protein cluster 𝑖 and 𝑃 𝐶 ) is the RNA cluster 𝑗 . Weights were normalised so that they sum to unity across the training set. This scheme down - wei ghts comple xes from densely sampled families (such as ribosomal proteins paired with rRNA) and up - weights unique or rare sequence pairs, encouraging the model to learn from the full diversity of the dataset rather than fitting the dominant clusters. Funding. This work was supported by AstraZeneca through a PhD s tudentship. Part of this work was performed usi ng resources prov ided by the Cambridge Service for Data Driven Discovery (CSD3) operated by the University of Cambridge Research Computing Service ( www.csd3.cam.ac.uk ). References 1 Van Nostrand, E. L. et al. A large - scale bi nding and functional map of human RNA - binding proteins. Nature 583 , 711 - 719 (2020). 2 Corley, M., Burns, M. C. & Yeo, G. W. How RNA - binding proteins interact with RNA: Molecules and mechani sms. Molecular ce ll 78 , 9 - 29 (2020). 3 Gebauer, F., Schwa rzl, T., Valcárc el, J. & Hentze, M. W. RNA - binding proteins in human genetic disease. Nature Reviews Geneti cs 22 , 185 - 198 (2021). 4 Glinos, D. A. et al. Trans criptome variation in human tissues revealed by long - read sequencing. Nature 608 , 353 - 359 (2022). 5 Liu, S. et al. Nap - seq reveals multiple classes of structured noncoding rnas with regulatory functions. Nat. Commun. 15 , 2425 (2024). 6 Tao, Y., Zhang, Q., Wang, H., Ya ng, X. & Mu, H. Alternative s plicing and related RNA binding proteins in human health and disease. Signal Transduct. Target. Ther. 9 , 26 (2024). 7 Vaghf, A., Khansarinejad , B., Ghaznavi - Rad, E. & Mondanizadeh, M. The role of micrornas in dise ases and related si gnaling pathways. Mol. Biol. Rep . 49 , 6789 - 6801 (2022). 8 D’Aloisio, V., Dognini , P., Hutcheon, G. A. & Coxon, C. R. Peptherdia: Database a nd structural composition analysis of approved peptide therapeuti cs and diagnostics. Drug Discov. Today 26 , 1409 - 1419 (2021). 9 Walker, M. J. & Varani, G. in Methods in enzymology Vol. 623 339 - 372 (Elsevier, 2019). 10 Abramson, J. et al. Accurate struct ure prediction of biomol ecular interactions wi th AlphaFold 3. Nature 630 , 493 - 500 (2024). 11 Roca - Martínez, J., Dh ondge, H., Sattl er, M. & Vranken, W. F. Deciphering the rrm - RNA recognition code: A computational analysis. PLoS Comput. Biol. 19 , e1010859 (2023). 12 Roca - Martínez, J., Ka ng, H. - S., Sattler, M. & Vrank en, W. Analysis of the in ter - domain orientation of tandem rrm domains with diverse linkers: Connecti ng experimental with AlphaFold2 predicted mod els. NAR Genom. Bioinform. 6 , lqae002 (2024). 13 Seufert, L., Benzing, T., Ignarski, M. & Müller, R. - U. RNA - binding pr oteins and their role in kidney disease. Nat. Rev. Nephrol. 18 , 153 - 170 (2022). 14 Ganser, L. R., K elly, M. L., Her schlag, D. & Al - Hashimi, H. M. The roles of structu ral dynamics in the cellular functions of rnas. Nat. Re v. Mol. Cell Biol. 20 , 474 - 489 (2019). 15 Hong, X. et al. An updated dataset and a structure - based prediction model for protein – RNA binding affinity. Prot eins 91 , 1245 - 1253 (2023). 16 Deng, L., Yang, W. & Liu, H. Predprba: Prediction of protein - RNA binding affinity usi ng gradient boosted regression trees. Front. Genet. 10 , 637 (2019). 17 Han, R. et al. Copra: Bridging c ross - domain pretrained sequence models with complex structures for protein - rna binding affinity prediction. Proceedings of the AAAI Conf erence on Artificial Intelligence 39 , 246 - 254 (2025). 18 Yang, W. & Deng, L. in 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 58 - 63 (IEEE). 19 Harini, K., Sekijima, M. & Gromiha, M. M. Pra - pred: Structure - based prediction of protein - RNA binding affinity. Int. J. Biol. Macromol. 259 , 129490 (2024). 20 Pandey, U. et al. Deepnap: A deep l earning method to predic t protein – nucleic acid binding affinity from their sequences. J. Chem. Inf. Model. 64 , 1806 - 1815 (2024). 21 Evans, R. et al. Protein complex p rediction with AlphaFol d - multimer. biorxiv , 2021.2010. 2004.463034 (2021). 22 Jumper, J. et al. Highly accurate prot ein structure predicti on with AlphaFold. nature 596 , 583 - 589 (2021). 23 Watson, J. L. et al. De novo design of prot ein structure and funct ion with rfdiffusion. Nature 620 , 1089 - 1100 (2023). 24 Akdel, M. et al. A structural bi ology community assessment o f AlphaFold2 applicati ons. Nat. Struct. Mol. Biol. 29 , 1056 - 1067 (2022). 25 Ghafouri, H. et al. Toward a unified framework for de termining conformational ensembles of disordered proteins. Nat. Methods , 1 - 15 (2026). 26 Brotzakis, Z. F., Zhang, S. , Murtada, M. H. & Vendruscolo, M. AlphaFold prediction of structural ensembles of disordered proteins. Nat. Commun. 16 , 1632 (2025). 27 Zhang, S. et al. Sequence - based virtual screening using transf ormers. Nat. Commun. 16 , 6925 (2025). 28 Passaro, S. et al. Boltz - 2: Towards accurate and efficient binding affinity prediction. BioRxiv (2025). 29 Harini, K., Srivastava, A., Kulandaisamy, A. & Gromiha, M. M. Pronab: Database for binding affinities of protein – nucleic acid complexes and their mutants. Nucleic Acids Res. 50 , D1528 - D1534 (2022). 30 Zhang, C., Zhang, X., Freddo lino, L. & Zhang, Y. Biol ip2: An updated structure database for biologically relevant ligand – protein interactions. Nucleic Acids Res. 52 , D404 - D412 (2024). 31 Askari, A. et al. Utexas aptamer da tabase: The collection and long - term preservation of aptamer sequence information. Nucleic Acids Re s. 52 , D351 - D359 (2024). 32 Liu, Z. et al. Forging the basis for developing protein – ligand interaction scoring functions. Acc. Chem. Res. 50 , 302 - 309 (2017). 33 Kallenborn, F. et al. Gpu - accelerated homology search with MMseqs2. Nat. Methods 22 , 2024 - 2027 (2025). Figure 1. Architecture of ZeroFold . ZeroFold predicts protein - RNA bi nding affinities from sequences by operating on pre - structural embeddings extracted from the trunk of Boltz - 2 prior to its structure decoding step. These e mbeddings encode rich confo rmational ensemble information for both the protein and RNA chains, and they are used to construct a heterogeneous graph in which protein residue s and RNA nucleotides for m nodes and pairwise rel ationships form edges. Separate encoding blocks refin e the single and pair repr esentations for eac h cha in be fore a cross - modal attent ion module integrates info rmation acros s the protein - RNA interface. The resulting joint representation is passed to an affinity prediction head to produce a scalar binding affinity estimate. Components enclosed in dotted - line boxes h ave fixed parameters and are not updated during training; those enclosed in solid - line boxes are trainable. T he Boltz - 2 tr unk is fixed throughout, with ZeroFold trained on the downstream encoding, attention, and prediction components. Figure 2 . Composition and characteristics of PRADB. (A) Breakdown of the 2,621 unique protein - RNA pairs in PRADB by source database, i llustrating the dominance o f ProNAB and the complementary coverage provided by BioLiP2, the UTexas Aptamer Database, and PDBbind+. (B) Distribu tion of RNA sequence lengths across 1,411 uniqu e RNA sequences, refle cting t he broad range of RNA classes represented in the dat aset. (C) Distribution of protein sequence lengths across 512 unique proteins, highlighting the diversity o f RNA - binding prot ein families included. (D) Distributi on of experimentally measured binding affinities (pK D ) across all 2,621 protein - RNA pairs, showing the range and central te ndency of affinity val ues used for model training and evaluation. (E) Agreement between indep endent replicate af finity measurements for the 353 protein - RNA pairs with two or more experi mental values, illustrating t he degree o f experimental noise that sets a practical upper bound on achievabl e model performance. Figure 3 . Computational efficiency of ZeroFold compared with Boltz -2. Inference time per protein - RNA pai r for ZeroFol d and a structure - based pipeline in which Boltz - 2 is used to generate predicted complex structures, which are then used as input for affinity estimation. Both pipeli nes require a Boltz - 2 trunk f orward pass; the structure - based pipeline addit ionally performs structure decoding prior to affinity estimation, whereas Z eroFold passes pre - structural embeddings directly to its affinity prediction head, bypass ing this step. Bars represent mean inference time per sample; error bars indicate standard deviation across independent runs. The reduction in runtime achieved by ZeroFold r eflects the elimination of the structure decoding step, enabling hi gher - throughput evaluation of protein - RNA pairs compared wi th structure - based approaches. Table 1. ZeroFold performanc e vs CoPRA and Dee P NAP. (A - D) Comparison of ZeroFold performance on the full test set (A) and progressi vely stricter subsets (B - D) against CoPRA and Dee P NAP. These subsets impose increasin gly stringent constraints on s equence overlap between the subsets and the training datasets of CoPRA (PRA310) and Dee P NAP (ProNAB). Table 2. Performance of models on s tratified t est set. Correlation of models on the test set stratified by aff inity band. Band boundaries are defined as the test - set mean ±1 standard deviation (mean = 7.10, SD = 1.35; low < 5.75, me dium 5.75 - 8.45, high ≥ 8.45).
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment