ZeroFold: 사전구조 임베딩으로 단백질‑RNA 결합 친화도 예측
ZeroFold는 단백질과 RNA 서열만을 입력으로, 구조 예측 단계 이전에 얻은 Boltz‑2 사전구조 임베딩을 활용해 결합 친화도를 예측한다. 2,621개의 고품질 단백질‑RNA 쌍을 모은 PRADB 데이터셋으로 학습했으며, 40 % 이하의 서열 유사성을 갖는 테스트 셋에서 스피어만 상관계수 0.65를 달성했다. 구조 기반 모델과 기존 시퀀스 기반 모델보다 공정한 평가 조건에서 일관적으로 우수한 성능을 보이며, 구조 디코딩 비용을 생략해 계…
저자: Josef Hanke, Sebastian Pujalte Ojeda, Shengyu Zhang
본 논문은 단백질‑RNA 결합 친화도 예측에서 구조 기반 접근법이 직면한 ‘RNA의 구조적 유연성’ 문제를 해결하고자, 구조 디코딩 이전 단계에서 얻어지는 사전구조 임베딩(pre‑structural embeddings)을 활용한 새로운 모델 ZeroFold를 제안한다. 기존 구조 기반 방법은 실험적으로 확인된 3D 구조가 필요하거나, AlphaFold‑like 모델이 제공하는 단일 콘포메이션에 의존한다. 그러나 RNA는 다중 콘포메이션을 취하며, 단일 구조는 실제 결합에 필요한 정보를 충분히 반영하지 못한다. 반면, Boltz‑2와 같은 바이오분자 기반 대형 모델은 트렁크 네트워크를 통과하면서 서열의 진화적 정보와 잠재적인 구조적 다양성을 고차원 벡터에 압축한다. 이러한 임베딩은 구조 디코딩 전 단계이므로, 아직 ‘구조가 고정되지 않은’ 상태에서 다양한 가능한 형태를 내포한다.
ZeroFold는 Boltz‑2 트렁크에서 추출한 단백질과 RNA 각각의 per‑residue 및 pairwise 임베딩을 독립적인 인코더에 입력하고, 교차‑모달 어텐션을 통해 두 체인의 상호작용을 학습한다. 이후 간단한 회귀 헤드가 결합 친화도(pKD)를 예측한다. 이 설계는 별도의 인터페이스 특징(접촉 거리, 에너지 등)을 수작업으로 추출할 필요 없이, 모델이 자동으로 결합 결정 요인을 학습하도록 만든다.
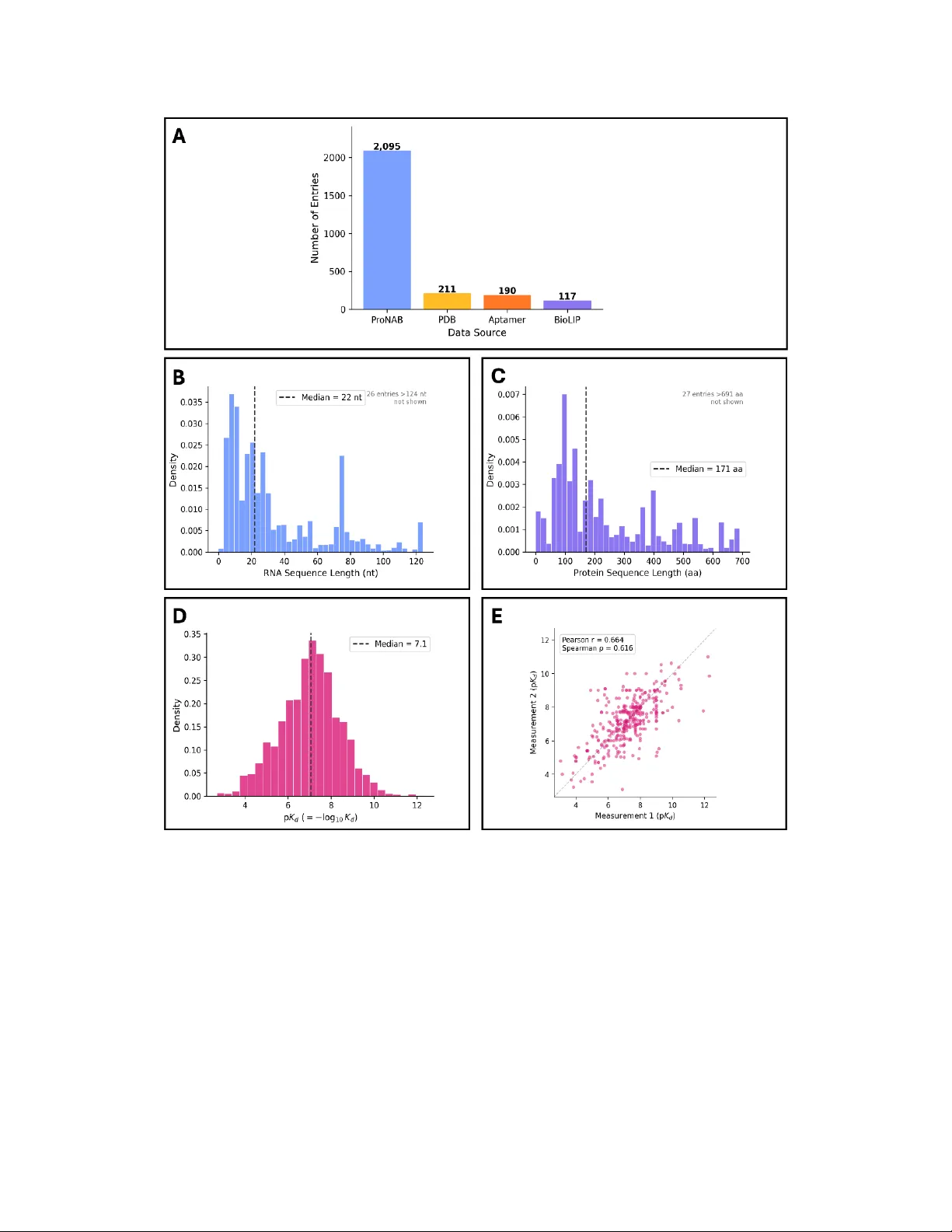
데이터 구축 측면에서는 네 개의 공개 데이터베이스(ProNAB, BioLiP, UT‑Aptamer, PDBbind+)를 통합해 PRADB라는 2,621개의 고유 단백질‑RNA 쌍을 수집하였다. 데이터는 단백질 512종, RNA 1,411종으로 구성되며, pKD 값은 평균 7.1 ± 1.35라는 균등한 분포를 보인다. 학습·검증·테스트 셋은 각각 8:1:1 비율로 나누고, 각 셋 간에 단백질·RNA 모두 40 % 이하의 서열 동일성을 유지하도록 클러스터링함으로써 데이터 누수를 최소화하였다.
성능 평가 결과, ZeroFold는 테스트 셋에서 스피어만 상관계수 0.65, 피어슨 상관계수 0.63, MAE 1.14 pKD, RMSE 1.47 pKD를 기록하였다. 실험적 측정 노이즈가 0.6‑0.7 정도의 상관계수 상한을 만든다는 점을 고려하면, 모델은 실제 한계에 근접한 성능을 보인다.
경쟁 모델과의 비교에서는 두 가지 주요 기준을 사용하였다. 첫째, 구조 기반 최고 성능 모델인 CoPRA와의 비교. CoPRA는 실험 구조를 이용하거나, Boltz‑2로 예측된 구조를 입력으로 사용한다. 동일한 테스트 셋에 대해 CoPRA의 PCC는 0.58, SCC는 0.59였으며, 서열 유사성 제한을 완화할수록 성능이 급감했다(예: 40 % 제한에서는 PCC 0.22). 반면 ZeroFold는 서열 유사성 제한이 강화되어도 성능이 크게 변하지 않았다. 둘째, 시퀀스 기반 최신 모델인 DeePNAP와의 비교. DeePNAP는 더 큰 훈련 데이터(주로 ProNAB)로 학습했지만, 테스트 셋에 대한 서열 중복을 제거하면 성능이 다소 감소한다. ZeroFold는 모든 서브셋에서 DeePNAP보다 일관적으로 높은 상관계수를 기록하였다.
또한, 모델이 전체 친화도 스펙트럼을 잘 구분한다는 점을 확인하기 위해 테스트 셋을 저·중·고 친화도 3개의 테라일로 나누어 각각의 상관계수를 계산하였다. 전체 셋에서의 상관계수가 각 테라일보다 높게 나타난 것은, 모델이 주로 ‘전체적인 강도 차이’를 학습하고, 동일 그룹 내 미세한 순위 차이는 상대적으로 약함을 의미한다. 이는 실제 응용에서 강한 바인더와 약한 바인더를 빠르게 구분하는 데 유용하지만, 정밀한 Kd 예측을 위해서는 추가적인 미세조정이 필요함을 시사한다.
계산 효율성 측면에서도 ZeroFold는 구조 디코딩을 생략함으로써 기존 구조 기반 파이프라인 대비 약 2‑3배 빠른 추론 속도를 보인다. 이는 대규모 가상 스크리닝, 전장 단백질‑RNA 네트워크 구축, 그리고 신약 후보 물질 탐색 등에 큰 장점을 제공한다.
한계점으로는 데이터 양과 품질이 아직 제한적이며, 실험적 pKD 측정의 변동성이 모델 성능 상한을 결정한다는 점이다. 또한, 현재 모델은 표준 뉴클레오티드와 아미노산에 대한 임베딩에 최적화돼 있어, 화학적 변형(예: 메틸화, 비표준 염기)이나 복합적인 다중체 상호작용을 다루는 데는 추가 연구가 필요하다. 향후 더 풍부한 결합 친화도 데이터와, 변형된 RNA·펩타이드에 대한 사전구조 임베딩을 확장한다면, ZeroFold와 같은 접근법은 구조 정보가 전무한 상황에서도 높은 정확도의 결합 친화도 예측을 가능하게 할 것이다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기