Fair splits flip the leaderboard: CHANRG reveals limited generalization in RNA secondary-structure prediction
Accurate prediction of RNA secondary structure underpins transcriptome annotation, mechanistic analysis of non-coding RNAs, and RNA therapeutic design. Recent gains from deep learning and RNA foundation models are difficult to interpret because curre…
Authors: Zhiyuan Chen, Zhenfeng Deng, Pan Deng
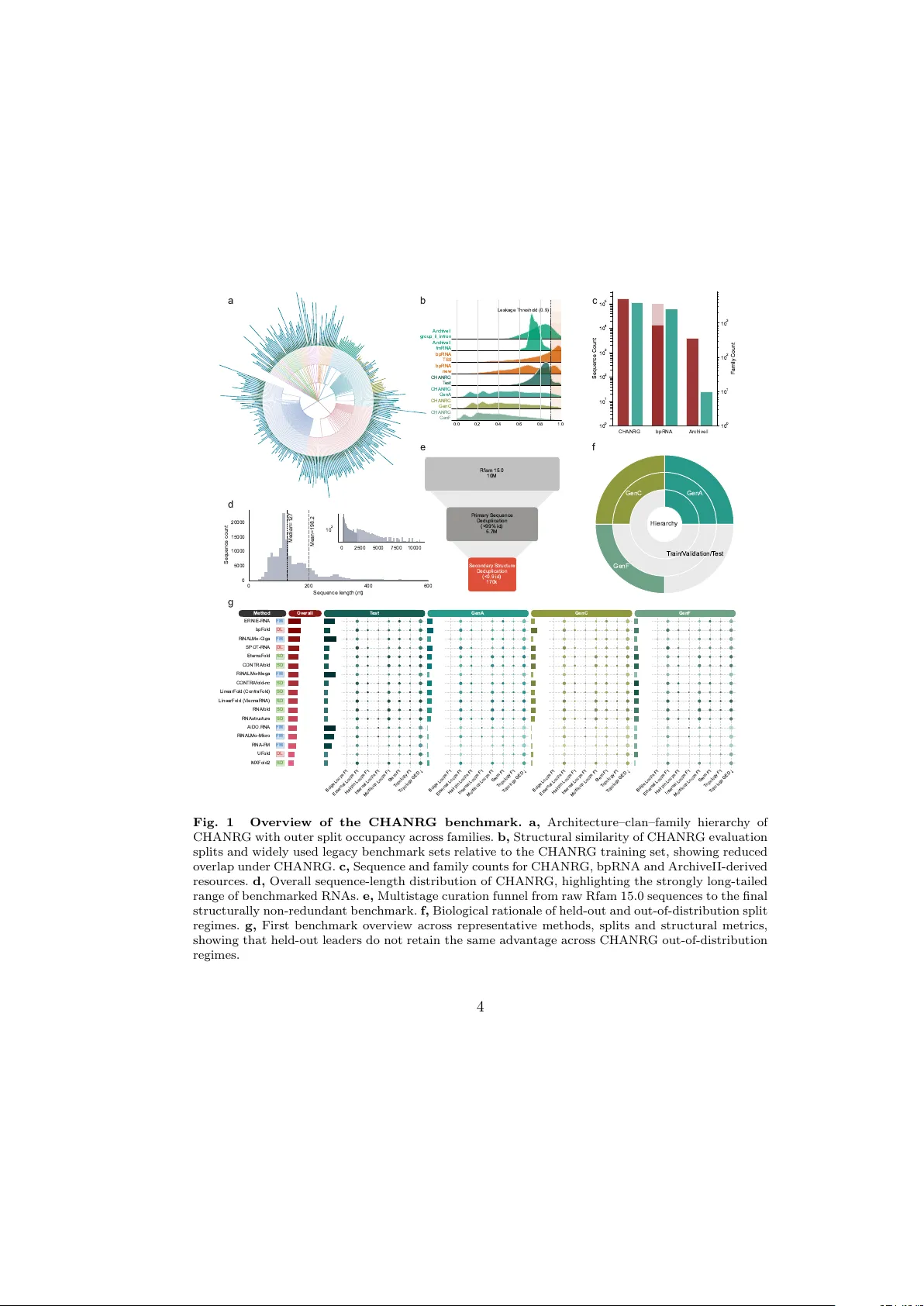
F air splits flip the leaderb oard: CHANR G rev eals limited generalization in RNA secondary-structure prediction Zhiyuan Chen 1,2,3 , Zhenfeng Deng 4,5,6 , P an Deng 7 , Y ue Liao 8 , Xiu Su 9 , P eng Y e 2,10* , Xih ui Liu 1* 1* Departmen t of Electrical and Computer Engineering, The Universit y of Hong Kong, P ok F u Lam, Hong Kong Island, Hong Kong SAR, China. 2 AI4Science Center, Shanghai Artificial Intelligence Laboratory , 129 Longw en Road, Xuh ui, 200232, Shanghai, China. 3 Departmen t of P athology , Stanford Medicine, 265 Campus Driv e, Stanford, 94305, Califorina, US. 4 Departmen t of Molecular Medicine, The Hospital for Sick Children (Sic kKids), Peter Gilgan Centre for Research and Learning, 686 Bay Street, T oronto, On tario, M5G 0A4, Canada. 5 Departmen t of Molecular Genetics, Universit y of T oron to, 1 King’s College Circle, T oron to, M5S 1A8, Ontario, Canada. 6 V ector Institute for Artificial Intelligence, 661 Univ ersity Av enue, T oronto, M5G 1M1, Ontario, Canada. 7 Zhongguancun Academy , 17 Second Ring Road, Daniufang, Haidian, 100094, Beijing, China. 8 Sc ho ol of Computing, National Universit y of Singap ore, 13 Computing Driv e, 117417, Singap ore. 9 Big Data Institute, Cen tral South Univ ersity , 932 Lushan South Road, Changsha, 410083, Hunan, China. 10 Departmen t of Information Engineering, Chinese Univ ersity of Hong Kong, Sha Tin, New T erritories, Hong Kong SAR, China. *Corresp onding author(s). E-mail(s): yepeng@p jlab.org.cn ; xih uiliu@eee.hku.hk; ; Con tributing authors: this@zyc.ai ; zfev an.deng@mail.utoronto.ca ; dengpan@bza.edu.cn ; liaoyue.ai@gmail.com ; xiusu1994@csu.edu.cn ; 1 Abstract Accurate prediction of RNA secondary structure underpins transcriptome anno- tation, mec hanistic analysis of non-co ding RNAs, and RNA therap eutic design. Recen t gains from deep learning and RNA foundation mo dels are difficult to in terpret b ecause current benchmarks ma y ov erestimate generalization across RNA families. W e present the Comprehensive Hierarchical Annotation of Non- co ding RNA Groups (CHANRG), a benchmark of 170,083 structurally non- redundan t RNAs curated from more than 10 million sequences in Rfam 15.0 using structure-aw are deduplication and arc hitecture-aw are split design. Across 29 predictors, foundation-mo del methods achiev ed the highest held-out accu- racy but lost most of that adv antage out of distribution, whereas structured deco ders and direct neural predictors remained markedly more robust. This gap p ersisted after controlling for sequence length and reflected b oth loss of structural cov erage and incorrect higher-order wiring. T ogether, CHANRG and a padding-free, symmetry-a ware ev aluation stack pro vide a stricter and batch- in v ariant framew ork for developing RNA structure predictors with demonstrable out-of-distribution robustness. Keyw ords: RNA secondary structure prediction, b enc hmark, out-of-distribution generalization, non-coding RNA, structure-aw are deduplication, top ology-a ware ev aluation, conv olutional neural netw ork 1 In tro duction The secondary structure of RNA, defined b y its pattern of in tramolecular base pairs, is a central determinant of RNA folding and function and underlies the div erse catalytic and regulatory roles of RNA molecules in biology [ 1 – 5 ]. By shaping three-dimensional conformations and conformational dynamics, it also informs the design of RNA-based therap eutics and RNA-guided molecular to ols [ 6 – 9 ]. Exp erimen tal assays pro vide v alu- able structural evidence, but they remain condition-dep endent and incomplete across transcripts, biological states, and structural resolutions [ 10 – 13 ]. Computational pre- diction therefore complements exp erimen ts, enables transcriptome-scale annotation, and guides design in RNA therap eutics and synthetic biology [ 14 , 15 ]. Curren t RNA secondary-structure predictors can b e group ed op erationally in to three classes. Structured deco ders (SD) produce the final structure under ther- mo dynamic, statistical, or hybrid structured optimization, as represented by Eter- naF old [ 16 ], CONTRAfold [ 17 ], RNAfold [ 18 ], and RNAstructure [ 19 ]. Direct neu- ral predictors (DL), for instance bpF old [ 20 ], SPOT-RNA [ 21 ] and UF old [ 22 ], learn con tact maps from sequence without a pretrained RNA language mo del. F oundation-mo del (FM) predictors couple pretrained RNA encoders to learned struc- ture heads [ 23 – 26 ]. Although prior work has reported impro ved generalization for RNA language mo dels [ 25 ], these results were obtained on b enchmark settings that differ from the structure-aw are, genome-aw are, and hierarchical out-of-distribution regimes considered here, and ma y not fully predict transfer under stricter ev aluation condi- tions. Although all three classes can achiev e strong held-out p erformance, it remains 2 unclear whether recent gains reflect transferable structure learning or impro v ed fitting to p ermissiv e b enchmark settings [ 20 , 27 , 28 ]. As curren t predictors can already achiev e strong held-out p erformance on famil- iar datasets, the more pressing question is whether they generalize across families, structural regimes, and reference genomes that were not represen ted during mo del dev elopment. Ev aluation practice has not kept pace with mo del developmen t [ 29 , 30 ]. First, man y widely used bpRNA-derived b enc hmark datasets were constructed from older source collections relative to recen t Rfam releases [ 31 , 32 ]. Second, these datasets are t ypically deduplicated primarily b y sequence iden tity , so structurally similar RNAs can remain on b oth sides of the ev aluation b oundary ev en when primary-sequence similarit y is modest [ 27 , 33 ]. Finally , pair-lev el scores can mask higher-order struc- tural errors, including incorrect junction wiring and top ological mismatc hes [ 34 , 35 ]. These limitations motiv ate a b enchmark that selects ev aluation examples with gen- uine structural nov elty relative to training, controls leak age through shared sequence or reference-genome con text, and ev aluates predictions hierarc hically from base-pair reco very to higher-order top ology . The b enchmark design in this work builds on a long lineage of curated RNA resources and annotation standards, including succes- siv e Rfam releases and curation up dates [ 32 , 36 ]. It is also informed b y established RNA reference databases and analysis to ols, including CR W, SRPDB, tmRDB, and V ARNA [ 37 – 40 ]. Recent comparativ e studies emphasize that robust b enc hmarking should test transfer b eyond within-family in terp olation and in to new-family or lo w- similarit y settings [ 27 – 29 ]. They also show that pairwise o verlap alone do es not fully capture structural fidelity [ 34 , 35 ]. V ariable-length contact-map prediction in tro duces a second, less visible limitation. Dense padded tensors w aste substantial memory and computation because contact maps scale quadratically with sequence length. When batches are padded to the longest RNA they con tain, dense conv olution can also make predictions depend on batc h composition rather than on sequence conten t alone, as quan tified later in this w ork (Fig. 5 c). This batc h-context dep endence undermines repro ducibilit y and mak es b enc hmark-scale ev aluation unnecessarily expensive (Fig. 5 c). A rigorous b enc h- mark for RNA secondary-structure prediction therefore requires b oth stronger dataset design and a compute path that resp ects v ariable-length inputs (Fig. 1 b,f; Fig. 5 a–c). Here w e in tro duce the Comprehensiv e Hierarc hical Annotation of Non-co ding RNA Groups (CHANR G), a b enc hmark curated from Rfam 15.0 with structure-a ware dedu- plication based on bpRNA-CosMoS [ 32 , 33 ]. The architecture-a ware split design is biologically motiv ated by hierarchical RNA structure classification sc hemes defined in RNArc hitecture [ 41 ]. CHANRG includes held-out in-distribution V alidation and T est splits together with three biologically distinct out-of-distribution regimes that prob e transfer to a held-out architectural regime, clans absent from training, and genome- sparse families under limited within-family source div ersity . W e b enchmark 29 pre- dictors spanning structured deco ders, direct neural predictors, and foundation-mo del predictors under standardized preprocessing and scoring. All foundation-mo del base- lines were instan tiated through the MultiMolecule framework [ 42 ]. T o supp ort faithful v ariable-length ev aluation, we also provide a padding-free, symmetry-a ware reference 3 implemen tation that remov es padded p ositions from the computational graph and a voids redundan t computation on symmetric con tact maps. Using CHANR G, w e show that foundation-mo del predictors achiev e the highest held-out accuracy but lose most of that adv antage out of distribution, whereas structured and direct neural predictors remain markedly more robust (Fig. 2 a–c). T ogether, CHANR G and the accompany- ing reference implementation pro vide a communit y resource for rigorous ev aluation and a practical framework for dev eloping RNA secondary-structure predictors that generalize b ey ond familiar training families (Fig. 1 – 5 ). ArchiveII group_ii_intron Leakage Threshold (0.9) ArchiveII tmRNA bpRNA TS0 bpRNA new CHANRG T est CHANRG GenA CHANRG GenC 0.0 0.2 0.4 0.6 0.8 1.0 CHANRG GenF CHANRG bpRNA ArchiveII 1 0 0 1 0 1 1 0 2 1 0 3 1 0 4 1 0 5 Sequence Count 1 0 0 1 0 1 1 0 2 1 0 3 Family Count 0 200 400 600 Sequence length (nt) 0 5000 10000 15000 20000 Sequence count Median=127 Mean=198.2 0 2500 5000 7500 10000 1 0 2 Rfam 15.0 10M Primary Sequence Deduplication (<99% id) 5.7M Secondary Structure Deduplication (<0.9 id) 170k GenA GenC GenF T rain/V alidation/T est Hierarchy Method Overall T est GenA GenC GenF MXFold2 UFold RNA-FM RiNALMo-Micro AIDO.RNA RNAstructure RNAfold LinearFold (V iennaRNA) LinearFold (ContraFold) CONTRAfold-nc RiNALMo-Mega CONTRAfold EternaFold SPOT -RNA RiNALMo-Giga bpFold ERNIE-RNA SD DL FM FM FM SD SD SD SD SD FM SD SD DL FM DL FM Bulge Loops F1 External Loops F1 Hairpin Loops F1 Internal Loops F1 Multiloop Loops F1 Stem F1 T opology F1 T opology GED Bulge Loops F1 External Loops F1 Hairpin Loops F1 Internal Loops F1 Multiloop Loops F1 Stem F1 T opology F1 T opology GED Bulge Loops F1 External Loops F1 Hairpin Loops F1 Internal Loops F1 Multiloop Loops F1 Stem F1 T opology F1 T opology GED Bulge Loops F1 External Loops F1 Hairpin Loops F1 Internal Loops F1 Multiloop Loops F1 Stem F1 T opology F1 T opology GED a b c d e f g Fig. 1 Ov erview of the CHANRG b enc hmark. a, Arc hitecture–clan–family hierarch y of CHANRG with outer split occupancy across families. b, Structural similarity of CHANR G ev aluation splits and widely used legacy benchmark sets relativ e to the CHANRG training set, sho wing reduced ov erlap under CHANRG. c, Sequence and family counts for CHANR G, bpRNA and Arc hiveII-derived resources. d, Overall sequence-length distribution of CHANRG, highlighting the strongly long-tailed range of benchmark ed RNAs. e, Multistage curation funnel from ra w Rfam 15.0 sequences to the final structurally non-redundant benchmark. f, Biological rationale of held-out and out-of-distribution split regimes. g, First b enchmark overview across represen tative methods, splits and structural metrics, showing that held-out leaders do not retain the same adv antage across CHANR G out-of-distribution regimes. 4 2 Results 2.1 CHANRG: a structure-a w are, leak age-controlled b enc hmark for RNA secondary-structure prediction W e constructed CHANRG to address a cen tral limitation of existing RNA secondary- structure b enc hmarks, as sequence-only deduplication is insufficien t b ecause RNAs with mo dest primary-sequence similarit y can still share highly similar secondary- structure top ology , and inferred structural annotations ma y not alw ays b e supported b y evolutionary evidence [ 27 , 33 , 43 ]. As a result, structurally redundant examples can p ersist across ev aluation b oundaries and inflate apparent generalization. CHANRG therefore explicitly remov es b oth sequence and structural redundancy while com bin- ing held-out in-distribution splits with biologically distinct out-of-distribution regimes. Ev aluation uses held-out in-distribution V alidation and T est splits together with three out-of-distribution regimes that prob e transfer to a held-out architectural regime (GenA), clans absen t from training (GenC), and genome-sparse families under lim- ited within-family source div ersity (GenF). Performance is assessed using a multiscale metric ladder spanning base-pair F 1 for local contact re co v ery , stem F 1 for helix-lev el reco very , top ology F 1 for higher-order structural organization, and top ology GED, a lo wer-is-better structural edit distance [ 44 ]. An ov erview of b enc hmark construction, split design, dataset properties, metric hierarch y , and the first b enc hmark summary app ears in Fig. 1 . Starting from Rfam release 15.0, we applied a m ulti-stage curation pip eline to 10,025,911 sequences drawn from 4,178 source families [ 32 ]. After integrit y screening, 10,025,740 sequences remained, follow ed by 5,670,054 after sequence-lev el deduplica- tion and 170,083 after structure-a ware deduplication. This pip eline comprised in tegrity screening, high-stringency sequence-level deduplication, and structure-aw are dedupli- cation based on bpRNA-CosMoS similarit y scores [ 33 ]. Th us, ev en after stringen t sequence-lev el filtering, structure-aw are pruning remov ed an additional 33-fold of residual redundancy , indicating that man y non-iden tical RNAs still shared highly sim- ilar secondary structures. Figure 1 e summarizes this curation funnel, whereas Fig. 1 c situates CHANR G relativ e to widely used legacy resources in b oth sequence coun t and family count. T o test whether structural leak age remained after curation, w e compared structural-similarit y distributions of CHANRG ev aluation sets against the training set with those of commonly used legacy benchmark sets, including bpRNA-deriv ed and Arc hiveII family-fold settings [ 21 , 27 , 31 , 45 ]. These distributions sho w ed that CHANR G ev aluation splits are less structurally coupled to training than these legacy resources (Fig. 1 b). This distinction is imp ortan t b ecause sequence-level fil tering alone can still lea ve highly similar folds on b oth sides of an ev aluation b oundary [ 27 , 33 ]. Structure-a ware curation therefore c hanges not only the size of the b enchmark, but also the effective no velt y of the examples used to assess generalization. Split design combines the hierarc hical organization of non-co ding RNAs with a reference-genome-a ware rule that separates developmen t from ev aluation within fami- lies [ 32 , 36 ]. Figure 1 a visualizes the architecture–clan–family hierarch y of CHANRG, and Fig. 1 f summarizes the biological rationale of the held-out and out-of-distribution 5 splits. GenA contains sequences annotated as “complex unclassified” and therefore prob es transfer to a held-out architectural regime. GenC contains sequences from clans absent from training and therefore prob es broader evolutionary distance b eyond the training clan hierarc hy . F or the remaining families, V alidation and T est are con- structed so that no tw o sequences from the same reference genome appear together within a family . F amilies that cannot provide sufficient genome diversit y for this split are assigned to GenF, yielding a distinct family-level stress test under sparse phyloge- netic cov erage. Sequences not assigned to V alidation, T est, or one of the three OOD regimes are retained for training. The final dataset comprises 123,223 T rain sequences, 14,070 V alidation sequences, 14,070 T est sequences, 12,499 GenA sequences, 4,424 GenC sequences, and 1,797 GenF sequences. Sequence lengths are strongly long-tailed, with split-sp ecific medians of 128 n t in T est, 211 nt in GenA, 93 nt in GenC, and 89 n t in GenF (Fig. 1 d). Pseudo- knot prev alence is low ov erall but heterogeneous across ev aluation regimes, reaching 2.8% in T est and 2.7% in GenA, compared with 0.2% in GenC and 0.3% in GenF. Unlik e legacy benchmarks built primarily around sequence-level curation, CHANR G com bines structural deduplication with biologically distinct OOD splits [ 27 , 31 , 33 ]. T ogether, this scale, structural diversit y , and split design create a stringen t test b ed for RNA secondary-structure generalization, and the first benchmark ov erview in Fig. 1 g previews how these design choices reshape model comparisons. 2.2 Standard held-out leaderb oards ov erestimate generalization W e next b enc hmark ed a canonical 17-mo del cohort comprising 8 structured dec oders, 3 direct neural predictors, and 6 foundation-mo del predictors. Structured deco ders pro- duce the final structure under explicit folding constraints or structured optimization, direct neural predictors infer structure directly from sequence without a pretrained RNA language mo del, and foundation-model predictors combine a pretrained RNA enco der with a learned structure head. T o a void ov erweigh ting multiple structure heads attac hed to the same pretrained bac kb one, class-level summaries use one U-Net head p er foundation-mo del backbone or scale [ 46 ], whereas full results for all 29 ev aluated mo dels are rep orted in Extended Data. T o summarize out-of-distribution b eha vior, w e define OOD mean as the unw eighted mean across GenA, GenC, and GenF. Held-out leaderb oards and out-of-distribution ev aluation fav or different predictor classes. F oundation-mo del predictors achiev ed the highest mean T est base-pair F 1 , reac hing 0 . 6731 across the canonical cohort, whereas direct neural predictors reac hed 0 . 3495 and structured decoders reached 0 . 3015 (Fig. 2 c). Ho wev er, foundation-model p erformance fell to 0 . 1796 on OOD mean , corresp onding to an absolute loss of 0 . 4935 and a retention of only 26 . 7% relativ e to T est. By con trast, direct neural predictors retained 82 . 5% of their T est p erformance, decreasing from 0 . 3495 to 0 . 2883, whereas structured deco ders retained 92 . 3%, decreasing only from 0 . 3015 to 0 . 2784. Con- v entional held-out ev aluation therefore substan tially ov erestimates the robustness of foundation-mo del predictors (Fig. 2 a–c). This observ ation contrasts with prior reports of improv ed generalization in RNA language mo dels [ 25 ], suggesting that b enc hmark design critically affects the measured transfer b ehavior. 6 0.0 0.2 0.4 0.6 0.8 1.0 Base-pair F (Test) 0.0 0.2 0.4 0.6 0.8 1.0 Base-pair F (OOD mean) 100% 75% 50% 25% MXFold2 EternaFold SPOT-RNA bpFold RiNALMo-Giga ERNIE-RNA Class FM SD DL Test GenA GenC GenF OOD mean Retention EternaFold [SD] CONTRAfold [SD] CONTRAfold-nc [SD] LinearFold (ViennaRNA) [SD] RNAfold [SD] LinearFold (ContraFold) [SD] RNAstructure [SD] MXFold2 [SD] bpFold [DL] SPOT-RNA [DL] UFold [DL] RiNALMo-Giga [FM] AIDO.RNA [FM] RiNALMo-Mega [FM] ERNIE-RNA [FM] RiNALMo-Micro [FM] RNA-FM [FM] 0.32 0.32 0.31 0.29 0.31 0.96 0.32 0.31 0.31 0.29 0.30 0.96 0.31 0.31 0.31 0.30 0.30 0.99 0.30 0.28 0.30 0.28 0.29 0.96 0.30 0.28 0.30 0.28 0.29 0.96 0.30 0.30 0.29 0.29 0.29 0.99 0.30 0.28 0.30 0.27 0.28 0.96 0.27 0.17 0.16 0.13 0.16 0.58 0.41 0.39 0.41 0.28 0.36 0.89 0.38 0.35 0.34 0.25 0.31 0.83 0.27 0.11 0.20 0.27 0.19 0.72 0.76 0.25 0.17 0.23 0.21 0.28 0.73 0.06 0.11 0.19 0.12 0.17 0.71 0.15 0.17 0.19 0.17 0.24 0.71 0.33 0.28 0.23 0.28 0.40 0.62 0.10 0.12 0.22 0.15 0.23 0.51 0.10 0.12 0.22 0.15 0.28 0.0 0.2 0.4 0.6 0.8 1.0 Base-pair F / Retention SD DL FM 0.0 0.2 0.4 0.6 0.8 1.0 Base-pair F Test OOD mean MXFold2 EternaFold SPOT-RNA bpFold RiNALMo-Giga ERNIE-RNA 0.0 0.2 0.4 0.6 0.8 1.0 Base-pair F (per sequence) Split Test GenA GenC GenF a b c d Fig. 2 Standard held-out leaderb oards ov erestimate generalization. a, T est versus OOD mean base-pair F 1 for the canonical 17-model cohort. Eac h point is one mo del, colored by pre- dictor class (FM, SD, or DL). Dashed guide lines indicate equal p erformance and 75%, 50%, and 25% reten tion relative to T est. b, Heatmap of base-pair F 1 across T est, GenA, GenC, and GenF, together with OOD mean and retention (OOD mean /T est), for the canonical cohort. Held-out lead- ers lose their adv antage across CHANRG OOD regimes. c, Class-level base-pair F 1 on T est and OOD mean . Poin ts denote individual models and error bars denote 95% b ootstrap confidence inter- v als o ver per-mo del means. d, Per-sequence base-pair F 1 distributions across T est, GenA, GenC, and GenF for six representativ e methods: MXF old2 and EternaF old (SD), SPOT-RNA and BPfold (DL), and RiNALMo-Giga and ERNIE-RNA (FM). OOD mean denotes the unw eighted mean across GenA, GenC, and GenF. Mo del-lev el comparisons make this in version visually explicit. In the canonical 17- mo del cohort, foundation-mo del predictors cluster tow ard high T est accuracy but weak OOD mean p erformance, whereas structured deco ders and direct neural predictors lie closer to the reten tion diagonal (Fig. 2 a). The corresponding heatmap sho ws that held- out leaders lose their adv antage across GenA, GenC, and GenF rather than on only one OOD split (Fig. 2 b). Among foundation-model predictors, RiNALMo-giga-U-Net [ 25 ] ac hieved the highest T est base-pair F 1 of 0 . 7579, but its p erformance fell to 0 . 2509 on GenA, 0 . 1651 on GenC, and 0 . 2260 on GenF, for an ov erall OOD mean of 0 . 2140. Within the main-text foundation-mo del cohort, ERNIE-RNA-U-Net [ 26 ] provided the strongest out-of-distribution performance, reaching OOD mean = 0 . 2807. Among struc- tured deco ders, EternaF old [ 16 ] ac hieved the strongest aggregate OOD p erformance, with a T est base-pair F 1 of 0 . 3189 and an OOD mean of 0 . 3064, whereas RNAfold [ 18 ] remained comparably stable at 0 . 3013 on T est and 0 . 2886 on OOD mean . Among direct neural predictors, BPfold [ 20 ] ac hieved the strongest o verall OOD performance, with a 7 T est score of 0 . 4065 and an OOD mean of 0 . 3608. The highest-scoring model on the con- v entional T est split therefore did not b elong to the class that generalized b est across arc hitectural, clan-level, and family-level shift. Bo otstrap resampling ov er p er-model means yielded the same qualitativ e in v ersion, indicating that the class-level contrast was not driven by a single mo del v ariant. On T est, the 95% confidence interv al for the foundation-mo del class [0 . 6004 , 0 . 7300] did not ov erlap the structured-deco der interv al [0 . 2910 , 0 . 3106] (Fig. 2 c). On OOD mean , this relationship rev ersed, with the foundation-mo del in terv al [0 . 1417 , 0 . 2247] lying en tirely below the structured-decoder in terv al [0 . 2418 , 0 . 3004]. The same inv ersion held at higher structural abstraction. F or top ology F 1 , which measures reco v ery of ste ms, lo ops, and their connections, foundation-mo del predictors dominated on T est [0 . 3517 , 0 . 4870] but not on OOD mean , where their in terv al [0 . 0550 , 0 . 0935] fell b elo w the structured-decoder interv al [0 . 0982 , 0 . 1208] (Fig. 2 d). F or top ology GED, a lo wer-is-better structural edit distance on the lo op–helix graph, foundation-model pre- dictors were b est on T est [0 . 3212 , 0 . 4598] but worst on OOD mean , where their interv al [0 . 6873 , 0 . 7598] la y w ell ab ov e the structured-deco der interv al [0 . 5111 , 0 . 5462]. Th us, the apparent sup eriority of foundation-model predictors under held-out ev aluation in verts systematically under CHANRG’s out-of-distribution regimes. Held-out leaderb oard rank is also a po or proxy for out-of-distribution robustness within the foundation-mo del class. Among foundation-mo del predictors, the Spearman correlation b etw een T est rank and OOD mean rank was weak ( ρ = 0 . 200, P = 0 . 704). By con trast, structured deco ders preserved their ranking muc h more consisten tly across held-out and OOD ev aluation ( ρ = 0 . 905, P = 2 . 0 × 10 − 3 ). Because the direct-neural class contains only three methods, w e do not o verin terpret within-class rank stability for that group. These results indicate that leaderb oard p osition on standard held-out sets is a p o or pro xy for cross-regime robustness among foundation-mo del predictors, whereas structured deco ders remain substantially more stable (Fig. 2 a–c). 2.3 The generalization gap is not explained b y sequence length A natural concern is that the OOD deficit simply reflects differences in sequence length across splits. That explanation is incomplete. GenA is longer than T est, with median lengths of 211 and 128 nt, resp ectiv ely , but GenC and GenF are both shorter than T est, with median lengths of 93 and 89 nt (Fig. 3 a). If length were the dominan t driver of OOD failure, performance should reco ver on the shorter OOD regimes, yet large deficits p ersist there. Split comp osition therefore suggests that length contributes to difficult y , but cannot b y itself explain the ov erall generalization pattern. W e next repeated the benchmark after restricting all splits to RNAs b et ween 50 and 200 nt. The main class-level result p ersisted under this control (Fig. 3 b). Within the canonical cohort, foundation-mo del predictors achiev ed a mean T est base-pair F 1 of 0 . 7016 but only 0 . 2074 on OOD mean under length matching. By contrast, struc- tured deco ders achiev ed 0 . 3212 on T est and 0 . 3156 on OOD mean , whereas direct neural predictors achiev ed 0 . 3870 on T est and 0 . 3299 on OOD mean . Thus, con trol- ling the ev aluated length range does not rescue foundation-model performance out of distribution. 8 1 0 2 1 0 3 1 0 4 Sequence length (nt) Test GenA GenC GenF Test GenA GenC GenF OOD mean Retention EternaFold [SD] CONTRAfold [SD] CONTRAfold-nc [SD] RNAfold [SD] LinearFold (ViennaRNA) [SD] LinearFold (ContraFold) [SD] RNAstructure [SD] MXFold2 [SD] bpFold [DL] SPOT-RNA [DL] UFold [DL] RiNALMo-Giga [FM] AIDO.RNA [FM] RiNALMo-Mega [FM] ERNIE-RNA [FM] RiNALMo-Micro [FM] RNA-FM [FM] 0.33 0.46 0.29 0.29 0.35 1.00 0.33 0.46 0.29 0.28 0.34 1.00 0.32 0.45 0.29 0.30 0.34 1.00 0.32 0.42 0.28 0.28 0.33 1.00 0.32 0.42 0.28 0.28 0.33 1.00 0.32 0.43 0.27 0.29 0.33 1.00 0.32 0.42 0.28 0.27 0.32 1.00 0.31 0.27 0.15 0.14 0.18 0.60 0.45 0.57 0.40 0.28 0.41 0.93 0.41 0.53 0.32 0.25 0.37 0.89 0.30 0.17 0.19 0.27 0.21 0.69 0.78 0.36 0.15 0.23 0.25 0.31 0.75 0.10 0.10 0.20 0.13 0.18 0.73 0.21 0.17 0.20 0.19 0.26 0.73 0.52 0.27 0.23 0.34 0.47 0.64 0.18 0.11 0.23 0.17 0.27 0.57 0.15 0.12 0.23 0.16 0.29 0.0 0.2 0.4 0.6 0.8 1.0 Base-pair F / Retention SD DL FM 0.0 0.2 0.4 0.6 0.8 1.0 Base-pair F (50 200 nt) Test OOD mean Test GenA GenC GenF 0.00 0.05 0.10 0.15 Base-pair F (matched full) MXFold2 EternaFold SPOT-RNA bpFold RiNALMo-Giga ERNIE-RNA MXFold2 EternaFold SPOT-RNA bpFold RiNALMo-Giga ERNIE-RNA micro mega giga 0.0 0.2 0.4 0.6 0.8 1.0 Base-pair F bpFold EternaFold RNAfold Split Test OOD mean micro mega giga 0.0 0.2 0.4 0.6 0.8 1.0 Topology F bpFold EternaFold RNAfold a b c d e Fig. 3 The CHANR G generalization gap is not explained b y sequence length or model scale. a, Split-sp ecific sequence-length distributions (log-scale x-axis) show that GenA is longer than T est, whereas GenC and GenF are shorter, ruling out a simple “OOD = longer RNAs” explanation. b, Base-pair F 1 heatmap for the canonical 17-model cohort after restricting all splits to RNAs b etw een 50 and 200 nt, with OOD mean and retention columns. c, Class-lev el base-pair F 1 on the length-matched subset, showing that the held-out/OOD inv ersion p ersists after con trolling the ev aluated length range. Poin ts denote individual mo dels and error bars denote 95% bo otstrap confidence interv als ov er p er- model means. d, Change in base-pair F 1 after length matc hing ( F matched 1 − F full 1 ) for six representativ e methods. Length control improv es sev eral structured and direct neural baselines on GenA but does not rescue foundation-model transfer on GenC or GenF. e, Scaling within the RiNALMo-U-Net family improv es T est performance muc h more than OOD p erformance for b oth base-pair F 1 and top ology F 1 . Horizon tal reference lines indicate BPfold, EternaF old, and RNAfold on the corresponding metric. OOD mean denotes the un weigh ted mean across GenA, GenC, and GenF. Mo del-lev el comparisons yielded the same conclusion. F or RiNALMo-giga-U-Net, base-pair F 1 increased from 0 . 7579 to 0 . 7832 on T est and from 0 . 2509 to 0 . 3565 on GenA after length matching, but remained v ery lo w on GenC at 0 . 1525 and on GenF at 0 . 2298. By con trast, several structured and direct neural baselines impro ved substan tially on GenA under the same control, including BPfold from 0 . 3876 to 0 . 5687, RNAfold from 0 . 2802 to 0 . 4193, and EternaF old from 0 . 3171 to 0 . 4608 (Fig. 3 c). Length matching therefore reveals an informativ e class asymmetry: long sequences partly penalize some structured and direct neural metho ds on GenA, whereas foundation-mo del failure p ersists on the shorter GenC and GenF regimes. These results argue against a simple geometric explanation for the held-out versus OOD inv ersion. The decisiv e observ ation is GenC: although its sequences are shorter than those in T est, foundation-mo del predictors remain muc h w orse there than on held- out ev aluation. The generalization deficit revealed by CHANRG is therefore b etter explained b y structural and evolutionary shift than b y sequence length alone. W e next 9 ask ed whether the same benchmark result might instead reflect insufficient foundation- mo del scale. 2.4 Scaling foundation mo dels improv es held-out accuracy more than out-of-distribution robustness W e next examined whether increasing foundation-mo del capacity is sufficient to close the generalization gap revealed by CHANRG. T o v ary scale while holding the bac kb one family and prediction head fixed, w e compared RiNALMo-micro-U-Net, RiNALMo- mega-U-Net, and RiNALMo-giga-U-Net (Fig. 3 d). This comparison isolates scale from head c hoice and therefore provides a direct test of whether larger foundation mo dels solv e the b enc hmark’s central OOD problem. Scaling substantially improv ed held-out base-pair accuracy , with T est F 1 increasing from 0 . 6222 in RiNALMo-micro-U-Net to 0 . 7122 in RiNALMo-mega-U-Net and 0 . 7579 in RiNALMo-giga-U-Net. The corresp onding gains on OOD mean w ere m uch smaller, increasing only from 0 . 1460 to 0 . 1697 and 0 . 2140, respectively . F rom micro to giga, scale therefore improv ed T est base-pair F 1 b y 0 . 1357, but improv ed OOD mean b y only 0 . 0680. The same asymmetry was visible across the individual OOD regimes: scaling impro ved GenA substantially , but pro duced little improv ement on GenC and only mo dest impro vemen t on GenF. T op ology-a ware ev aluation show ed the same pattern more strongly . T est top ology F 1 increased from 0 . 3602 in RiNALMo-micro-U-Net to 0 . 4566 in RiNALMo-mega-U- Net and 0 . 5304 in RiNALMo-giga-U-Net. Ov er the same range, OOD mean top ology F 1 increased only from 0 . 0582 to 0 . 0665 and 0 . 0911. T op ology GED likewise impro ved more on T est than out of distribution, decreasing from 0 . 4617 to 0 . 3608 and 0 . 2722 on T est, but only from 0 . 7594 to 0 . 7248 and 0 . 6570 on OOD mean . Thus, increasing foundation-mo del scale improv es held-out structural reconstruction muc h more than transferable higher-order organization. Ev en the largest RiNALMo-U-Net mo del remained less robust out of distribution than several lo wer-scoring baselines. RiNALMo-giga-U-Net ac hieved the highest T est base-pair F 1 in the benchmark at 0 . 7579, y et its OOD mean of 0 . 2140 remained well b elo w BPfold (0 . 3608), EternaF old (0 . 3064), and RNAfold (0 . 2886). The same ranking held at higher structural abstraction, where RiNALMo-giga-U-Net reached OOD mean top ology F 1 = 0 . 0911, compared with 0 . 1552 for BPfold, 0 . 1227 for EternaF old, and 0 . 1207 for RNAfold, and had a worse OOD mean top ology GED (0 . 6570) than all three baselines. Increasing capacit y therefore does not resolve the central b enc hmark result. Within the curren t foundation-mo del pip eline, increasing scale impro ves held-out fit- ting muc h more than out-of-distribution robustness. Because neither sequence length nor mo del scale is sufficient to explain the observ ed OOD gap, we next examined how these failures app ear across the structural abstraction ladder. 10 SD DL FM 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 Stem F Test OOD mean SD DL FM 0.0 0.1 0.2 0.3 0.4 0.5 Topology F SD DL FM 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 Topology GED better 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 Precision 0.0 0.2 0.4 0.6 0.8 1.0 Recall F =0.3 F =0.5 F =0.7 F =0.9 SD DL FM Test OOD mean Stem F Hairpin Internal Multiloop External Topology F 0.0 0.2 0.4 0.6 0.8 1.0 1.2 Retention (OOD mean / Test) Class SD DL FM Stem F Hairpin Internal Multiloop External Topology F MXFold2 EternaFold SPOT-RNA bpFold RiNALMo-Giga ERNIE-RNA 0.58 0.68 0.69 0.48 0.51 0.52 0.96 0.93 0.93 1.27 0.88 0.94 0.85 0.88 0.88 0.62 0.94 0.78 0.82 0.91 0.86 0.72 0.97 0.85 0.31 0.45 0.21 0.04 0.18 0.17 0.45 0.49 0.31 0.10 0.42 0.27 0.0 0.2 0.4 0.6 0.8 1.0 1.2 Retention (OOD / Test) GenA · 109 nt LMFL01000010.1_72055-71947 Target bp=0.64 stem=0.00 topo=0.05 GED=0.42 RiNALMo-Giga bp=0.72 stem=0.29 topo=0.19 GED=0.57 RNAfold bp=0.82 stem=0.33 topo=0.23 GED=0.40 bpFold GenC · 98 nt AE017226.1_854285-854382 bp=0.81 stem=1.00 topo=0.54 GED=0.30 bp=0.97 stem=1.00 topo=0.79 GED=0.02 bp=0.97 stem=1.00 topo=0.79 GED=0.02 Target TP FP FN A U G C a b c d e Fig. 4 Hierarc hical ev aluation reveals cov erage failure and helix miswiring. a, Class-level stem F 1 , topology F 1 , and topology GED on T est and OOD mean . P oints denote individual mo dels and error bars denote 95% bo otstrap confidence interv als o ver p er-mo del means. Lo wer topology GED indicates b etter agreement. b, Precision–recall shift from T est to OOD mean for structured deco ders (SD), direct neural predictors (DL), and foundation-mo del predictors (FM). Gray contours indicate iso- F 1 v alues. c, Retention relativ e to T est for stem F 1 , hairpin-loop F 1 , in ternal-lo op F 1 , m ultilo op F 1 , external-loop F 1 , and topology F 1 , stratified by metho d class. Poin ts denote individual models. d, Motif-specific reten tion (OOD mean /T est) for six representativ e metho ds: MXF old2 and EternaF old (SD), SPOT-RNA and BPfold (DL), and RiNALMo-Giga and ERNIE-RNA (FM). e, Two pre- specified case studies lo calizing OOD errors to global loop–helix organization. Left, Case 1 (GenA, 109 nt; RF01527; LMFL01000010.1 72055-71947 ) shows moderate contact reco very but incorrect global architecture for RiNALMo-Giga. Right, Case 2 (GenC, 98 nt; RF03162; AE017226.1 854285-854382 ) shows perfect helix recov ery but failed multiloop wiring. T arget pairs, true p ositives (TP), false posi- tives (FP), and false negatives (FN) are sho wn for each prediction. OOD mean denotes the un weigh ted mean across GenA, GenC, and GenF. 11 2.5 Hierarchical ev aluation rev eals cov erage failure and helix miswiring Base-pair F 1 established the existence of a generalization gap, but it understated its structural depth. T o lo calize this gap, we mo ved from pair-lev el accuracy to a mul- tiscale set of structural diagnostics spanning stem F 1 , topology F 1 , topology GED, precision and recall, and motif-level p erformance (Fig. 4 a). Here, stem F 1 asks whether the correct helical segmen ts are recov ered, top ology F 1 asks whether stems, lo ops, and their connections are assembled into the correct higher-order organization, and top ol- ogy GED measures ho w far the predicted lo op–helix graph lies from the reference, with lo wer v alues indicating b etter agreement. A mo del can therefore achiev e high stem F 1 while still failing top ologically if it reco vers the right helices but connects them incor- rectly . These diagnostics rev ealed t wo distinct failure modes among foundation-mo del predictors, namely cov erage failure, in which true interactions are omitted, and wiring failure, in which reco vered helices are assembled in to incorrect global architectures. The strongest foundation-mo del predictor illustrates the scale of this collapse. F or RiNALMo-giga-U-Net, pair exact matc h decreased from 0 . 0904 on T est to 0 . 0009 on OOD mean . Ov er the same comparison, stem F 1 decreased from 0 . 6947 to 0 . 2157, top ology F 1 decreased from 0 . 5304 to 0 . 0910, and topology GED w orsened from 0 . 2722 to 0 . 6570. At the class lev el, foundation-model predictors dropp ed from mean top ology F 1 = 0 . 4240 on T est to 0 . 0729 on OOD mean , while top ology GED increased from 0 . 3869 to 0 . 7245 (Fig. 4 b). By contrast, structured deco ders c hanged muc h less, from mean top ology F 1 = 0 . 1269 on T est to 0 . 1125 on OOD mean , and from mean topology GED = 0 . 5401 to 0 . 5270, whereas direct neural predictors c hanged from 0 . 1431 to 0 . 1136 in top ology F 1 and from 0 . 5803 to 0 . 5756 in topology GED. Th us, the OOD deficit of foundation-model predictors is not only a pair-reco very problem, but a larger collapse in higher-order structural organization. The gap b et ween stem F 1 and topology F 1 further localizes where this collapse o ccurs. Across foundation-mo del predictors, mean stem F 1 fell from 0 . 6087 on T est to 0 . 1890 on OOD mean , corresp onding to 31 . 1% retention. Over the same comparison, top ology F 1 fell from 0 . 4240 to 0 . 0729, corresp onding to only 17 . 2% retention. By con trast, structured decoders retained 89 . 8% of stem F 1 and 88 . 6% of top ology F 1 , while direct neural predictors retained 81 . 4% and 79 . 4%, resp ectiv ely . F oundation- mo del predictors therefore preserve some helical signal out of distribution, but fail m uch more severely when those helices must b e assembled in to the correct global wiring diagram. Decomp osing base-pair F 1 in to precision and recall revealed the first failure mo de, co verage failure. Across foundation-mo del predictors, mean precision and recall shifted from 0 . 7968 and 0 . 6108 on T est to 0 . 3477 and 0 . 1403 on OOD mean , moving the class to ward a markedly high-precision, low-recall regime (Fig. 4 c). The corresp onding precision-to-recall ratio increased from 1 . 30 to 2 . 48, indicating that OOD predictions b ecame increasingly conserv ative and omitted man y true in teractions. By con trast, structured deco ders remained comparatively balanced, with mean precision and recall c hanging only from 0 . 3121 and 0 . 3149 on T est to 0 . 2840 and 0 . 2893 on OOD mean . Direct neural predictors also remained far more balanced than foundation-model pre- dictors, shifting from 0 . 3660 and 0 . 3501 on T est to 0 . 3019 and 0 . 2877 on OOD mean . 12 Co verage failure is therefore a prominent characteristic of foundation-model predictors rather than a generic feature of all predictor classes. Motif-lev el reten tion quantified the second failure mo de, wiring failure, at scale. Relativ e to T est, foundation-mo del predictors retained 31 . 1% of stem F 1 , 31 . 3% of hairpin-lo op F 1 , 10 . 1% of internal-loop F 1 , 23 . 5% of multiloop F 1 , 48 . 8% of external- lo op F 1 , and 17 . 2% of top ology F 1 on OOD mean (Fig. 4 d). The weak est reten tion therefore o ccurred precisely in junction-dep enden t and higher-order motifs, especially in ternal loops and multiloops. By contrast, structured decoders retained 89 . 8% of stem F 1 , 131 . 3% of hairpin-lo op F 1 , 89 . 8% of internal-loop F 1 , 95 . 2% of multiloop F 1 , 92 . 8% of external-lo op F 1 , and 88 . 6% of top ology F 1 . The apparent gain in hairpin- lo op reten tion for structured deco ders likely reflects relative enrichmen t of simpler hairpin-dominated structures in the OOD regimes rather than a literal increase in ra w predictive capacit y . Overall, these motif-level results show that the OOD gap in foundation-mo del predictors is concentrated in structural con texts that dep end on correct higher-order assembly rather than lo cal helix detection alone. Tw o pre-sp ecified case studies lo calized these errors to global lo op–helix organiza- tion (Fig. 4 e). In a 109-nt GenA RNA from RF01527 ( LMFL01000010.1 72055-71947 ), RiNALMo-giga-U-Net achiev ed base-pair F 1 = 0 . 638 but top ology F 1 = 0 . 045, stem F 1 = 0 . 000, and topology GED = 0 . 417. On the same RNA, RNAfold ac hieved base-pair F 1 = 0 . 719, topology F 1 = 0 . 185, and stem F 1 = 0 . 286, whereas BPfold ac hieved base-pair F 1 = 0 . 821, top ology F 1 = 0 . 227, and stem F 1 = 0 . 333. Th us, the foundation-mo del recov ered many individual contacts, but assembled them in to a fundamen tally differen t architecture. In a 98-nt GenC RNA from RF03162 ( AE017226.1 854285-854382 ), RiNALMo-giga-U-Net correctly recov ered all helices (stem F 1 = 1 . 000) and still ac hieved base-pair F 1 = 0 . 808, but failed completely on the m ultilo op junction (m ultilo op F 1 = 0 . 000), yielding top ology F 1 = 0 . 542 and top ology GED = 0 . 302. By contrast, RNAfold and BPfold b oth ac hieved base-pair F 1 = 0 . 969, stem F 1 = 1 . 000, top ology F 1 = 0 . 792, and top ology GED = 0 . 020, with substan tial m ultilo op recov ery in both cases. These examples sho w that man y OOD errors do not arise from complete structural ignorance. Instead, foundation-model predictors often reco ver lo cal helices while failing to place them in to the correct global arrangement. T ogether, these analyses sho w that OOD failure is a coupled loss of structural co verage and structural wiring fidelit y . 2.6 Padding-free, symmetry-a w are computation enables faithful CHANR G-scale ev aluation CHANR G spans RNAs from 19 to 10,799 nt. Because con tact maps scale quadratically with sequence length, this broad range pro duces extreme v ariation in contact-map area and makes dense padding especially costly in heterogeneous batc hes. Under dense tensor execution, eac h sequence is padded to the longest RNA in the batc h, so the w asted contact-map area for sequence i is L 2 max − L 2 i . Padding therefore consumes a substan tial fraction of the compute budget and creates a setting in whic h predictions for the same RNA can dep end on whic h longer RNAs happ en to share its batc h (Fig. 5 a). 13 Conventional (Padded tensor) Ours (NestedTensor) Input y = W x + b Output L=5 L=5 L=3 L=3 conv conv take triu ( y + y ) / 2 valid pad contaminated y i j y j i ½ wasted valid (clean) contaminated padding y j i p r e - s y m . discarded 2 4 8 16 32 64 Batch size 0 20 40 60 80 100 Contact-map padding ratio (%) 2 4 8 16 32 64 Batch size 1.6 1.8 2.0 2.2 2.4 2.6 2.8 3.0 Flipped candidate-pair fraction (%) Test GenA GenC GenF Latency (ms) Memory (GB) 0 20 40 60 80 100 Latency (ms) 3.3× Padded tensor NestedTensor 0 1 2 3 4 Memory (GB) 6.7× a b c d Fig. 5 P adding-free, symmetry-a w are computation enables faithful CHANRG-scale ev aluation. a, Comparison of con ven tional padded-tensor execution and the NestedT ensor reference implementation. In con ven tional dense execution, shorter RNAs are embedded in a larger L max × L max contact map, so con volution mixes v alid and padded regions and symmetry enforcement by taking the upper triangle discards half of the dense output. In the NestedT ensor path, padded p ositions are excluded from the computation graph and symmetry is enforced at the output lev el b y av eraging the predicted con tact map with i ts transp ose, o = ( y + y ⊤ ) / 2. b, Con tact-map padding ratio across real ev aluation-set batc h con texts from T est, GenA, GenC, and GenF, stratified b y batch size. c, Flipp ed candidate-pair fraction under dense padded inference across batch sizes for T est, GenA, GenC, and GenF, showing that batch-con text dep endence persists even at batc h size 2. d, Controlled systems benchmark on synthetic batches spanning representativ e sequence lengths. Relative to dense padded tensors, the NestedT ensor implementation reduces inference latency by 3.3-fold and allo cated GPU memory b y 6.7-fold. Panels b and c use real ev aluation-set contexts, whereas panel d uses synthetic batches to isolate systems cost. W e first quan tified the padding burden across CHANRG ev aluation. Across 185,472 sequence–batc h contexts derived from 32,416 ev aluable sequences in T est, GenA, GenC, and GenF, the mean padding ratio was 0.380, 40.1% of con texts con tained more than 50% padding, and 14.5% con tained more than 75% padding (Fig. 5 b). Mean padding increased from 0.152 at batch size 2 to 0.534 at batch size 32, and on the T est split reached 0.647 at batch size 32. Dense ev aluation therefore sp ends a large fraction of its compute budget on padded contact-map area under realistic CHANR G batc hing. W e next asked whether dense padding changes mo del predictions and whether this effect can b e mitigated simply by reducing batc h size. Using an ERNIE-RNA mo del finetuned for RNA secondary structure prediction [ 26 ], we re-ev aluated the same ev aluation sequences across batch sizes 2, 4, 8, 16, 32, and 64 under dense padded inference. Across batch sizes, batc h context flipp ed 1.90–2.13% of candidate pair decisions on av erage at a threshold of 0.5, despite only small perturbations in the 14 con tinuous outputs, with mean logit MAE of 0.00382–0.00404 and mean probabilit y MAE of 8 . 85 × 10 − 4 –9 . 40 × 10 − 4 . This effect was already presen t at batc h size 2 and c hanged little as batch size increased, sho wing that reducing dense batch size do es not eliminate the artifact. Av eraged across batch sizes, the flipped fraction was 1.83% on T est, 2.24% on GenA, 1.73% on GenC, and 2.89% on GenF (Fig. 5 c). The largest relativ e disturbance occurred in GenF, indicating that the artifact is not restricted to the longest split. Because the n umber of candidate pairs scales with sequence length, this corresp onded to 72.0–79.3 flipp ed upp er-triangular entries p er sequence on av er- age. Dense padding therefore breaks batc h-context in v ariance for v ariable-length RNA structure prediction even in small batches. T o remov e this confound, we implemen ted a mask-aw are NestedT ensor execu- tion path that excludes padded p ositions from the computational graph rather than masking them after dense computation. W e coupled this representation with a symmetry-aw are triangular conv olution that ev aluates only the nonredundant half of the con tact map while preserving symmetric output by construction (Fig. 5 d). P adding burden and batc h-context inv ariance were quantified on ev aluation-set batc h con texts from T est, GenA, GenC, and GenF, whereas memory and latency w ere bench- mark ed separately on synthetic batches spanning represen tative sequence lengths. This design eliminates the mechanism underlying the batch-con text effect by preven t- ing padded p ositions from en tering the computation, while sim ultaneously reducing w asted memory and arithmetic on padded and symmetric regions. The computational gains were substan tial even for the same U-Net arc hitecture. In the con trolled systems b enc hmark, the padding-free implementation reduced the effectiv e w orkload from 60.7 to 10.2 billion FLOPs, reduced allo cated GPU memory from 4.61 to 0.69 GB, and reduced forward latency from 109.4 to 33.3 ms (Fig. 5 e). This corresponds to a 3.3-fold inference sp eedup together with a 6.7-fold reduction in allocated memory . During training, the same change reduced total step time from 229.5 to 115.0 ms and reduced p eak training memory from 6.33 to 1.06 GB. Padding- free computation therefore impro ves repro ducibilit y , mak es CHANR G-scale ev aluation practical, and remov es a hidden source of batc h-context dep endence from v ariable- length prediction (Fig. 5 c,d). 3 Discussion CHANR G changes what counts as progress in RNA secondary-structure prediction. Across a structure-aw are, leak age-controlled benchmark, the mo dels with the highest held-out base-pair accuracy did not generalize best to biologically distinct out-of- distribution regimes (Fig. 2 a–c). This div ergence indicates that con ven tional held-out leaderb oards can substantially ov erestimate robustness when structural redundancy and related-source leak age are not adequately controlled [ 27 , 33 ]. By combining up dated source co v erage, structure-aw are deduplication, genome-a ware split construc- tion, and hierarchical ev aluation, CHANR G makes this gap directly measurable and pro vides a stronger basis for future metho d comparison. These findings contrast with prior rep orts of strong generalization in RNA language models [ 25 ], underscoring the imp ortance of b enc hmark design. 15 The b enc hmark also reveals that high-capacity represen tation learning is not yet matc hed b y equally transferable structural bias. F oundation-model predictors achiev ed the strongest p erformance on the held-out T est split (Fig. 2 a,c), consistent with strong within-distribution sequence-to-structure fitting. How ever, their sharp decline on GenA, GenC, and GenF, together with the larger deterioration in top ology F 1 and top ology GED than in stem F 1 , indicates that curren t foundation-mo del pip elines do not yet reco ver higher-order structure robustly outside familiar families (Fig. 2 b,c; Fig. 4 a,d). By contrast, structured deco ders and the strongest direct neural baselines retained substantially more p erformance across OOD regimes in CHANRG (Fig. 2 a,c; Fig. 4 a,d), suggesting that explicit structural constrain ts and task-aligned inductive bias remain imp ortant for transfer. The scaling analysis supp orts the same interpreta- tion, b ecause increasing foundation-mo del capacity improv ed held-out accuracy m uch more than out-of-distribution robustness (Fig. 3 e). CHANR G is therefore both a b enc hmark and a practical ev aluation framew ork. Its split design distinguishes held-out in terp olation from transfer to arc hitectural, clan-lev el, and genome-sparse family regimes (Fig. 1 f ), and its metric ladder sepa- rates lo cal con tact reco very from helix recov ery , topology recov ery , and higher-order structural damage (Fig. 1 g; Fig. 4 a). These distinctions mak e it possible to c ho ose predictors according to the in tended use case rather than according to a single held-out leaderb oard. F or targets drawn from familiar and w ell-represented families, foundation-mo del predictors curren tly provide the highest held-out lo cal structural accuracy (Fig. 2 a). F or structurally nov el or family-distant targets, ho wev er, structured deco ders and the strongest direct neural predictors pro vide more reliable topology and more stable b enc hmark p erformance across OOD regimes (Fig. 2 a,c; Fig. 4 a,d). The accompan ying padding-free, symmetry-aw are reference implementation further makes CHANR G-scale ev aluation batch-in v ariant, efficien t, and practical for future mo del dev elopment and comparison (Fig. 5 a–d). CHANR G is designed as a stringen t stress test of structural generalization, not as an exhaustiv e taxonomy of RNA nov elty . Its OOD splits are op erationally defined and capture sp ecific forms of arc hitectural and evolutionary shift, and structure-a ware deduplication is only as complete as the structural representation used to define it. In particular, the current framew ork pro vides the strongest con trol ov er non-crossing secondary-structure top ology , whereas pseudoknot-inv olved in teractions remain dif- ficult to reco ver in current comparativ e ev aluations [ 29 ]. The b enc hmark likewise ev aluates transfer through curated annotation and structural nov elty rather than through direct downstream biological utility . F uture progress will therefore require not only larger or more expressive mo dels, but also stronger transferable structural priors, ric her b enc hmark tasks, and tighter links b et ween b enc hmark robustness and exp erimen tally v alidated RNA function. 16 References [1] Tinoco, I. Jr, Bustamante, C.: How RNA folds. Journal of Molecular Biology 293 (2), 271–281 (1999) h ttps://doi.org/10.1006/jmbi.1999.3001 [2] Mortimer, S.A., Kidw ell, M.A., Doudna, J.A.: Insigh ts in to RNA structure and function from genome-wide studies. Nature Reviews Genetics 15 (7), 469–479 (2014) https://doi.org/10.1038/nrg3681 [3] Kruger, K., Grabowski, P .J., Zaug, A.J., Sands, J., Gottsc hling, D.E., Cec h, T.R.: Self-splicing RNA: auto excision and auto cyclization of the rib osomal RNA inter- v ening sequence of tetrah ymena. Cell 31 (1), 147–157 (1982) h ttps://doi.org/10. 1016/0092- 8674(82)90414- 7 [4] Doudna, J.A., Cech, T.R.: The chemical repertoire of natural rib ozymes. Nature 418 (6894), 222–228 (2002) h ttps://doi.org/10.1038/418222a [5] Isaacs, F.J., Dwyer, D.J., Collins, J.J.: RNA synthetic biology . Nature Biotech- nology 24 (5), 545–554 (2006) https://doi.org/10.1038/n bt1208 [6] Dethoff, E.A., Ch ugh, J., Musto e, A.M., Al-Hashimi, H.M.: F unctional complexity and regulation through RNA dynamics. Nature 482 ( 7385), 322–330 (2012) https: //doi.org/10.1038/nature10885 [7] Sullenger, B.A., Nair, S.: F rom the RNA world to the clinic. Science 352 (6292), 1417–1420 (2016) https://doi.org/10.1126/science.aad8709 [8] P ardi, N., Krammer, F.: mRNA v accines for infectious diseases - adv ances, chal- lenges and opp ortunities. Nature Reviews Drug Disco v ery 23 (11), 838–861 (2024) h ttps://doi.org/10.1038/s41573- 024- 01042- y [9] Jinek, M., East, A., Cheng, A., Lin, S., Ma, E., Doudna, J.: RNA-programmed genome editing in human cells. Elife 2 , 00471 (2013) https://doi.org/10.7554/ eLife.00471 [10] Ding, Y., T ang, Y., Kw ok, C.K., Zhang, Y., Bevilacqua, P .C., Assmann, S.M.: In viv o genome-wide profiling of RNA secondary structure rev eals no vel reg- ulatory features. Nature 505 (7485), 696–700 (2014) https://doi.org/10.1038/ nature12756 [11] Rouskin, S., Zubradt, M., W ashietl, S., Kellis, M., W eissman, J.S.: Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505 (7485), 701–705 (2014) https://doi.org/10.1038/nature12894 [12] Spitale, R.C., Flynn, R.A., Zhang, Q.C., Crisalli, P ., Lee, B., Jung, J.-W., Kuc helmeister, H.Y., Batista, P .J., T orre, E.A., Ko ol, E.T., Chang, H.Y.: Struc- tural imprints in viv o decode RNA regulatory mec hanisms. Nature 519 (7544), 17 486–490 (2015) https://doi.org/10.1038/nature14263 [13] Bevilacqua, P .C., Ritc hey , L.E., Su, Z., Assmann, S.M.: Genome-wide analysis of RNA secondary structure. Annual Review of Genetics 50 (1), 235–266 (2016) h ttps://doi.org/10.1146/annurev- genet- 120215- 035034 [14] Sato, K., Hamada, M.: Recent trends in RNA informatics: a review of mac hine learning and deep learning for RNA secondary structure prediction and RNA drug disco very . Briefings in Bioinformatics 24 (4) (2023) h ttps://doi.org/10.1093/ bib/bbad186 [15] W ang, X., Gu, R., Chen, Z., Li, Y., Ji, X., Ke, G., W en, H.: Uni-rna: Univ ersal pre-trained mo dels rev olutionize rna researc h. bioRxiv (2023) h ttps://doi.org/10.1101/2023.07.11.548588 h ttps://www.biorxiv.org/conten t/early/2023/07/12/2023.07.11.548588.full.p df [16] W a yment-Steele, H.K., Kladw ang, W., Strom, A.I., Lee, J., T reuille, A., Bec k a, A., Eterna Participan ts, Das, R.: RNA secondary structure pack ages ev aluated and impro ved b y high-throughput experiments. Nature Metho ds 19 (10), 1234–1242 (2022) https://doi.org/10.1038/s41592- 022- 01605- 0 [17] Do, C.B., W o ods, D.A., Batzoglou, S.: CONTRAfold: RNA secondary struc- ture prediction without ph ysics-based models. Bioinformatics 22 (14), 90–8 (2006) h ttps://doi.org/10.1093/bioinformatics/btl246 [18] Lorenz, R., Bernhart, S.H., H¨ oner Zu Siederdissen, C., T afer, H., Flamm, C., Stadler, P .F., Hofac ker, I.L.: ViennaRNA pack age 2.0. Algorithms for Molecular Biology 6 (1), 26 (2011) https://doi.org/10.1186/1748- 7188- 6- 26 [19] Reuter, J.S., Mathews, D.H.: RNAstructure: softw are for RNA secondary struc- ture prediction and analysis. BMC Bioinformatics 11 (1), 129 (2010) https://doi. org/10.1186/1471- 2105- 11- 129 [20] Zh u, H., T ang, F., Quan, Q., Chen, K., Xiong, P ., Zhou, S.K.: Deep generalizable prediction of RNA secondary structure via base pair motif energy . Nature Com- m unications 16 (1), 5856 (2025) https://doi.org/10.1038/s41467- 025- 60048- 1 [21] Singh, J., Hanson, J., Paliw al, K., Zhou, Y.: RNA secondary structure predic- tion using an ensemble of tw o-dimensional deep neural netw orks and transfer learning. Nature Communications 10 (1), 5407 (2019) https://doi.org/10.1038/ s41467- 019- 13395- 9 [22] F u, L., Cao, Y., W u, J., Peng, Q., Nie, Q., Xie, X.: UF old: fast and accurate RNA secondary structure prediction with deep learning. Nucleic Acids Research 50 (3), 14 (2022) https://doi.org/10.1093/nar/gk ab1074 [23] Chen, J., Hu, Z., Sun, S., T an, Q., W ang, Y., Y u, Q., Zong, L., Hong, L., Xiao, J., 18 Shen, T., King, I., Li, Y.: In terpretable RNA foundation model from unannotated data for highly accurate RNA structure and function predictions (2022) h ttps: //doi.org/10.48550/arXiv.2204.00300 2204.00300 [q-bio.QM] [24] Zou, S., T ao, T., Mahbub, S., Ellington, C.N., Algayres, R.J., Li, D., Zhuang, Y., W ang, H., Song, L., Xing, E.P .: A large-scale foundation model for RNA func- tion and structure prediction (2024). https://doi.org/10.1101/2024.11.28.625345 . https://doi.org/10.1101/2024.11.28.625345 [25] P eni ´ c, R.J., Vla ˇ si ´ c, T., Hub er, R.G., W an, Y., ˇ Siki ´ c, M.: RiNALMo: general- purp ose RNA language mo dels can generalize well on structure prediction tasks. Nature Comm unications 16 (1), 5671 (2025) https://doi.org/10.1038/ s41467- 025- 60872- 5 [26] Yin, W., Zhang, Z., Zhang, S., He, L., Zhang, R., Jiang, R., Liu, G., W ang, J., Zhang, X., Qin, T., Xie, Z.: ERNIE-RNA: an RNA language model with structure-enhanced represen tations. Nature Comm unications 16 (1), 10076 (2025) h ttps://doi.org/10.1038/s41467- 025- 64972- 0 [27] Qiu, X.: Sequence similarit y gov erns generalizabilit y of de nov o deep learning mo dels for RNA secondary structure prediction. PLoS Computational Biology 19 (4), 1011047 (2023) h ttps://doi.org/10.1371/journal.p cbi.1011047 [28] Szikszai, M., Wise, M., Datta, A., W ard, M., Mathews, D.H.: Deep learning mo dels for RNA secondary structure prediction (probably) do not general- ize across families. Bioinformatics 38 (16), 3892–3899 (2022) h ttps://doi.org/10. 1093/bioinformatics/btac415 [29] Just yna, M., Antczak, M., Szac hniuk, M.: Machine learning for RNA 2D structure prediction b enc hmarked on exp erimental data. Briefings in Bioinformatics 24 (3) (2023) https://doi.org/10.1093/bib/bbad153 [30] Sc hneider, B., Sw eeney , B.A., Bateman, A., Cern y , J., Zok, T., Szac hniuk, M.: When will RNA get its AlphaF old momen t? Nucleic Acids Research 51 (18), 9522– 9532 (2023) https://doi.org/10.1093/nar/gk ad726 [31] Danaee, P ., Rouches, M., Wiley , M., Deng, D., Huang, L., Hendrix, D.: bpRNA: large-scale automated annotation and analysis of RNA secondary structure. Nucleic Acids Research 46 (11), 5381–5394 (2018) https://doi.org/10.1093/nar/ gky285 [32] On tiv eros-Palacios, N., Co ok e, E., Na wro cki, E.P ., T rieb el, S., Marz, M., Riv as, E., Griffiths-Jones, S., Petro v, A.I., Bateman, A., Sw eeney , B.: Rfam 15: RNA families database in 2025. Nucleic Acids Research 53 (D1), 258–267 (2025) https: //doi.org/10.1093/nar/gk ae1023 [33] Lasher, B., Hendrix, D.A.: bpRNA-CosMoS: a robust and efficien t RNA structural 19 comparison metho d using k-mer based cosine similarity . Bioinformatics 41 (4) (2025) https://doi.org/10.1093/bioinformatics/btaf108 [34] Zhao, Y., W ang, J., Zeng, C., Xiao, Y.: Ev aluation of RNA secondary structure prediction for b oth base-pairing and top ology . Biophysics Rep orts 4 (3), 123–132 (2018) https://doi.org/10.1007/s41048- 018- 0058- y [35] Mathews, D.H.: Ho w to benchmark RNA secondary structure prediction accuracy . Metho ds 162-163 , 60–67 (2019) https://doi.org/10.1016/j.ymeth.2019.04.003 [36] Kalv ari, I., Nawrocki, E.P ., On tiveros-P alacios, N., Argasinsk a, J., Lamkiewicz, K., Marz, M., Griffiths-Jones, S., T offano-Nio c he, C., Gautheret, D., W einberg, Z., Riv as, E., Eddy , S.R., Finn, R.D., Bateman, A., Petro v, A.I.: Rfam 14: expanded co verage of metagenomic, viral and microRNA families. Nucleic Acids Research 49 (D1), 192–200 (2021) h ttps://doi.org/10.1093/nar/gk aa1047 [37] Cannone, J.J., Subramanian, S., Sc hnare, M.N., Collett, J.R., D’Souza, L.M., Du, Y., F eng, B., Lin, N., Madabusi, L.V., M¨ uller, K.M., P ande, N., Shang, Z., Y u, N., Gutell, R.R.: The comparativ e RNA w eb (CR W) site: an online database of comparative sequence and structure information for rib osomal, intron, and other RNAs. BMC Bioinformatics 3 (1), 2 (2002) h ttps://doi.org/10.1186/ 1471- 2105- 3- 2 [38] Rosen blad, M.A., Goro dkin, J., Knudsen, B., Zwieb, C., Sam uelsson, T.: SRPDB: Signal recognition particle database. Nucleic Acids Research 31 (1), 363–364 (2003) https://doi.org/10.1093/nar/gkg107 [39] Zwieb, C., Goro dkin, J., Kn udsen, B., Burks, J., W o wer, J.: tmRDB (tmRNA database). Nucleic Acids Research 31 (1), 446–447 (2003) https://doi.org/10. 1093/nar/gkg019 [40] Dart y , K., Denise, A., Pon ty , Y.: V ARNA: Interactiv e drawing and editing of the RNA secondary structure. Bioinformatics 25 (15), 1974–1975 (2009) https: //doi.org/10.1093/bioinformatics/btp250 [41] Boccaletto, P ., Magn us, M., Almeida, C., Zyla, A., Astha, A., Pluta, R., Baginski, B., Janko wsk a, E., Dunin-Hork awicz, S., Wirec ki, T.K., Boniec ki, M.J., Stefaniak, F., Bujnic ki, J.M.: RNArc hitecture: a database and a classification system of RNA families, with a fo cus on structural information. Nucleic Acids Research 46 (D1), 202–205 (2018) https://doi.org/10.1093/nar/gkx966 [42] Chen, Z., Zhu, S.Y.: MultiMolecule. h ttps://doi.org/10.5281/zeno do.12638419 . h ttps://doi.org/10.5281/zeno do.12638419 [43] Riv as, E., Clemen ts, J., Eddy , S.R.: A statistical test for conserv ed RNA structure sho ws lack of evidence for structure in lncRNAs. Nature Metho ds 14 (1), 45–48 (2017) https://doi.org/10.1038/nmeth.4066 20 [44] Sanfeliu, A., F u, K.-S.: A distance measure b etw een attributed relational graphs for pattern recognition. IEEE T ransactions on Systems, Man, and Cybernetics SMC-13 (3), 353–362 (1983) h ttps://doi.org/10.1109/TSMC.1983.6313167 [45] Sloma, M.F., Mathews, D.H.: Exact calculation of lo op formation probabilit y iden tifies folding motifs in RNA secondary structures. RNA 22 (12), 1808–1818 (2016) https://doi.org/10.1261/rna.053694.115 [46] Ronneberger, O., Fischer, P ., Bro x, T.: U-net: Conv olutional net works for biomed- ical image segmentation. In: Nav ab, N., Hornegger, J., W ells, W.M., F rangi, A.F. (eds.) Medical Image Computing and Computer-Assisted In terven tion – MICCAI 2015, pp. 234–241. Springer, Cham (2015). https://doi.org/10.1007/ 978- 3- 319- 24574- 4 28 21
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment