Conditioning Protein Generation via Hopfield Pattern Multiplicity
Protein sequence generation via stochastic attention produces plausible family members from small alignments without training, but treats all stored sequences equally and cannot direct generation toward a functional subset of interest. We show that a…
Authors: Jeffrey D. Varner
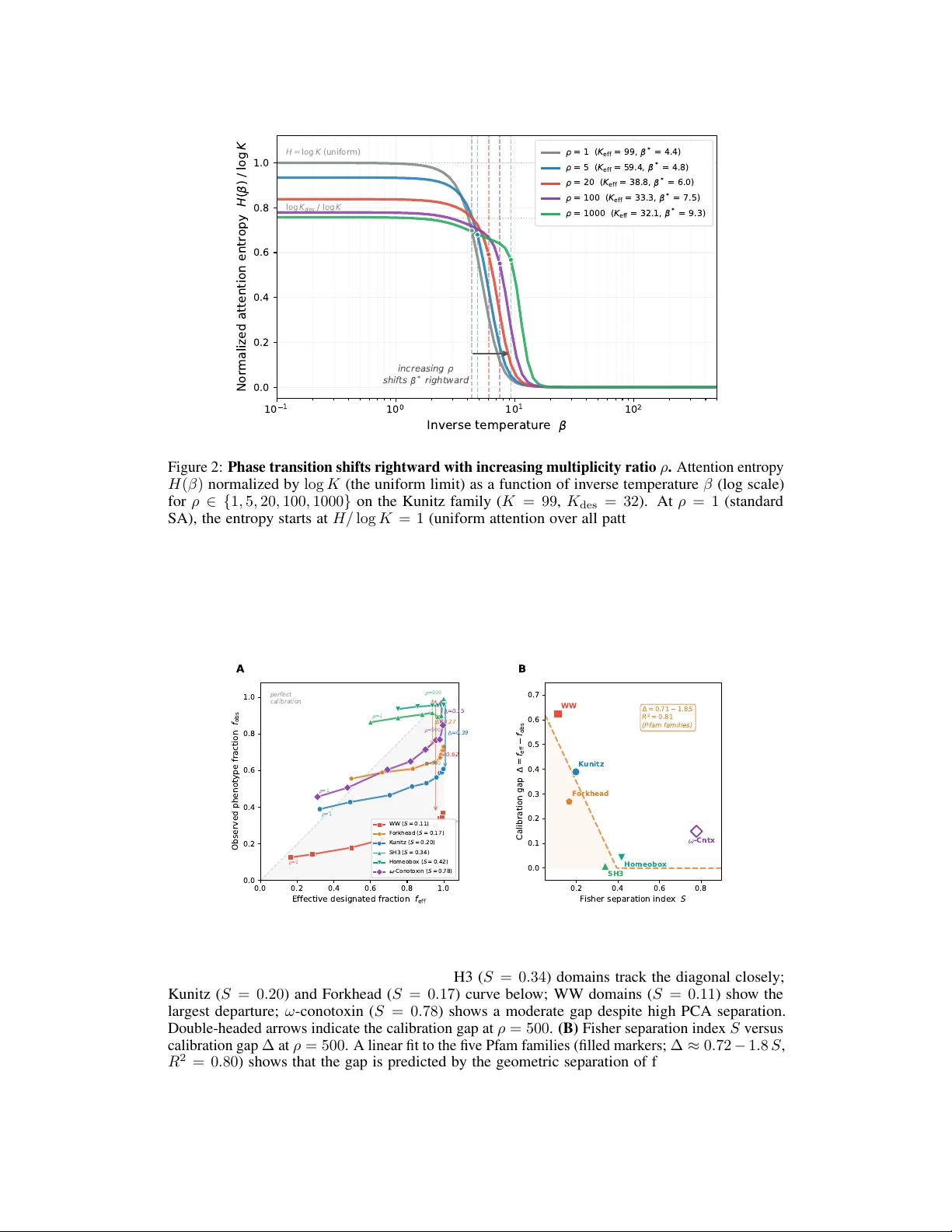
Conditioning Pr otein Generation via Hopfield Patter n Multiplicity Jeffr ey D. V arner R.F . Smith School of Chemical and Biomolecular Engineering Cornell Univ ersity , Ithaca, NY 14850 jdv27@cornell.edu Abstract Protein sequence generation via stochastic attention produces plausible family members from small alignments without training, b ut treats all stored sequences equally and cannot direct generation tow ard a functional subset of interest. W e show that a single scalar parameter , added as a bias to the sampler’ s attention logits, continuously shifts generation from the full family to w ard a user-specified subset, with no retraining and no change to the model architecture. A practitioner supplies a small set of sequences (for e xample, hits from a binding screen) and a multiplicity ratio that controls ho w strongly generation f av ors them. The method is agnostic to what the subset represents: binding, stability , specificity , or any other property . W e find that the conditioning is exact at the le v el of the sampler’ s internal representation, but that the decoded sequence phenotype can fall short because the dimensionality reduction used to encode sequences does not alw ays preserve the residue-le v el v ariation that defines the functional split. W e term this discrepancy the calibration gap and sho w that it is predicted by a simple geometric measure of how well the encoding separates the functional subset from the rest of the family . Experiments on fiv e Pfam families (Kunitz, SH3, WW , Homeobox, and Forkhead domains) confirm the monotonic relationship between separation and gap across a fourfold range of geometries. Applied to omeg a-conotoxin peptides targeting a calcium channel inv olved in pain signaling, curated seeding from 23 characterized binders produces over a thousand candidates that preserve the primary pharmacophore and all experimentally identified binding determinants. These results sho w that stochastic attention enables practitioners to expand a handful of experimentally characterized sequences into div erse candidate libraries without retraining a generativ e model. 1 Introduction Generating novel protein sequences that fold correctly and perform a desired function is a central goal of computational protein engineering. Se v eral classes of methods now address this problem. Deep generativ e models, including variational autoencoders [ 1 ], autoregressi ve language models [ 2 ], and diffusion models [ 3 ], learn sequence distributions from lar ge databases and can produce div erse candidates, but require substantial training data and GPU resources. Structure-conditioned inv erse folding methods [ 4 ] design sequences for a giv en backbone but require a target structure as input. Protein language models [ 5 ] trained on e v olutionary databases can score or sample sequences, but conditioning them on a specific functional property typically requires fine-tuning on labeled data. Profile HMMs [ 6 ] capture family-le vel statistics from alignments b ut, as we show belo w , produce sequences with low structural confidence and poor compositional fidelity . For the scarce-data regime (families with tens to hundreds of members, typical of Pfam seed alignments), most learned models lack sufficient training signal. Preprint. Stochastic attention (SA) was de veloped for this regime [ 7 ]. SA treats the ener gy function of a modern Hopfield network, constructed directly from a small f amily alignment, as a Boltzmann density and samples from it using Lange vin dynamics. The method requires no training, runs on a laptop, and produces sequences that preserv e family-le vel amino acid composition, per -position conserv ation, and predicted three-dimensional fold. A ke y limitation, ho wev er , is that SA treats all sequences in the alignment equally . Ev ery family member contrib utes the same weight to the ener gy landscape, so the sampler explores the full f amily distribution without preference for an y functional subset. In practice, experimentalists often want something more specific: not just “a plausible Kunitz domain, ” but a Kunitz domain that inhibits trypsin ; not just “a plausible SH3 domain, ” but one that binds a particular proline-rich motif. Family alignments define a shared fold (all 99 Kunitz sequences share the same disulfide frame work and double-loop architecture), b ut the members dif fer in which target they bind, and that specificity is encoded in a small number of positions (e.g., the P1 contact residue) rather than in the global fold. The alignment captures structure; the practitioner wants to condition on function. The question is: how can we condition the generative pr ocess on a functional subset without r etraining? A seemingly simple answer is to curate the input, restricting the alignment to only those sequences that a practitioner has identified (through e xperimental screens, literature annotation, or functional assays) as belonging to the functional subset of interest. SA then generates new sequences in their image. W e show that this strategy works: ev en three designated input sequences suffice for complete phenotype transfer in the Kunitz domain. But hard curation discards all remaining family members, losing the fold-lev el constraints they pro vide. A more principled approach would retain the full family while continuously upweighting the designated subset. SA w orks by encoding each sequence in the alignment as a column in a matrix (the memory matrix), then running a physics-inspired sampler that generates new sequences by taking noisy steps through the landscape defined by these stored sequences. At each step, the sampler computes an attention distrib ution o ver the stored sequences and mov es tow ard a weighted av erage of them. The ke y observation we e xploit is that adding a bias term to these attention scores deepens the energy wells around specific sequences, causing the sampler to spend more time near them. Concretely , we assign a multiplicity weight r k > 0 to each stored sequence and add log r k to its attention score before the softmax normalization. A single parameter , the multiplicity ratio ρ = r designated /r background , controls the strength of this bias: ρ = 1 recov ers standard SA (no preference), while increasing ρ shifts generation to ward the designated subset. No sequences are duplicated, no parameters are trained, and no architectural changes are needed. The method is agnostic to what the designated subset represents (binding, thermostability , organism of origin); it simply biases the sampler tow ard the sequences the user points to. In this study , we developed the theory of multiplicity-weighted SA and tested it on fiv e Pfam protein families (K unitz, SH3, WW , Homeobox, and Forkhead domains) spanning a range of family sizes ( K = 55 – 420 ) and functional-subset geometries. W e sho wed that the multiplicity weights enter the sampler as a simple logit bias, preserving the analytic score function and O ( dK ) cost of the original method. The ener gy-lev el conditioning is exact: the softmax attention on designated patterns matches the tar get fraction to within 0 . 3% at all multiplicity ratios tested. Howe v er , we found that the decoded sequence phenotype can fall short of this target, because the PCA encoding that compresses sequences into the sampler’ s state space does not always preserve the residue-level variation that defines the functional split. W e termed this discrepancy the calibration gap and showed that it is predicted by a simple geometric quantity , the Fisher separation inde x S , which measures ho w well PCA separates designated from background sequences. Across five families spanning a fourfold range of S , the gap decreased monotonically with increasing separation ( R 2 = 0 . 80 ). This pro vides a practitioner’ s decision rule: compute S before generating any sequences; if S is high, multiplicity weighting transfers the phenotype directly; if S is low , use hard curation instead. 2 Results W e e v aluated SA on six protein families spanning a range of sizes, functions, and PCA-space geometries (T able 1 ). Five Pf am seed families (K unitz domains (PF00014, K = 99 ), SH3 domains (PF00018, K = 55 ), WW domains (PF00397, K = 420 ), Homeobox domains (PF00046, K = 136 ), and Forkhead domains (PF00250, K = 246 )) were used to benchmark both hard curation and multiplicity-weighted conditioning, with designated subsets defined by kno wn functional-site residues. A sixth family , the ω -conotoxin O-superfamily ( K = 74 , curated from SwissProt), was used 2 to test hard curation on a therapeutically relevant peptide system targeting the Cav2.2 calcium channel. For each f amily , we split sequences into designated and background subsets based on an experimentally v alidated phenotype marker , built conditioned memory matrices, and generated sequences via Langevin dynamics at the entropy-inflection in verse temperature β ∗ . The Fisher separation index S , a measure of ho w well PCA separates designated from background sequences, varied from 0.11 (WW , heavily interlea ved) to 0.42 (Homeobox, well-separated), providing a natural axis along which to test the limits of multiplicity-weighted conditioning. Hard curation and the small-set amplifier . W e restricted the memory matrix to a user-designated functional subset of K unitz domains (PF00014; K = 99 sequences, L = 53 aligned positions) and tested whether subset-specific phenotype transfers to generated sequences (Fig. 1 ). Kunitz domains are serine protease inhibitors whose binding specificity depends on the P1 residue at the primary contact site with the target protease: L ys or Arg at P1 confers strong trypsin inhibition, while other residues (Gly , Gln, Asn) do not [ 8 ]. W e designated the 32 sequences with K/R at P1 as the functional subset ( K des = 32 ) and the remaining 67 as background ( K bg = 67 ), built separate memory matrices from each group, found β ∗ for each via entropy inflection, and generated 930 sequences per condition (30 chains × 5,000 steps, thinned after burn-in). W e observed that 100% of designated- subset-conditioned sequences had K/R at P1; 0% of background-conditioned sequences did; and full-family generation produced 41% K/R, close to the natural proportion ( 32 / 99 = 32% ), showing that the designated phenotype was fully inherited while the fold-le vel composition w as approximately preserved. Sequence-lev el inspection showed that the top strong-binder-conditioned sequences preserved all highly conserved positions ( 11 / 11 ), all six cysteines, and carried K/R at P1, while introducing 19-26 substitutions concentrated at v ariable sites; per-position Shannon entrop y tracked the stored MSA closely ( r = 0 . 988 for full-family , r = 0 . 932 for strong-conditioned), with the lar gest deviation at P1 where conditioning collapsed di v ersity to K/R (Fig. 5 ). T o determine the minimum input size required for reliable transfer , we subsampled K des ∈ { 3 , 5 , 8 , 10 , 15 , 20 , 25 , 32 } from the Kunitz K/R subset (3 replicates each), b uilt curated memory matrices, and generated sequences. W e found that P1 phenotype fidelity was 100% at every sample size, including K des = 3 , but that small input sets reduced diversity: pairwise sequence diversity increased from 0.36 ( K des = 3 ) to 0.50 ( K des = 32 ), and KL diver gence decreased from 0.047 to 0.009 ov er the same range, which is consistent with fewer stored patterns constraining the PCA subspace and reducing the volume accessible to the Langevin chain (Fig. 1 ). These results support the “small-set amplifier” framing: a practitioner with as fe w as 5–10 experimentally characterized sequences (for instance, hits from a phage display screen) can generate hundreds of candidates that maintain the input phenotype while introducing sequence div ersity (pairwise di versity ≥ 0 . 45 for K des ≥ 10 ). Multiplicity-weighted conditioning and the calibration gap. W e tested multiplicity-weighted generation on the Kunitz family ( K = 99 , K des = 32 strong trypsin binders, K bg = 67 background), sweeping ρ from 1 (standard SA, no bias) to 1,000 (Fig. 2 , T able 2 ). For each ρ , we computed the multiplicity vector r , found β ∗ ( r ) via entropy inflection on the weighted attention distrib ution, and generated 930 sequences (30 chains). T wo quantities were tracked at each ρ : the mean softmax attention weight on designated patterns ( ¯ a des , measured in continuous PCA space during sampling) and the fraction of decoded sequences with K/R at P1 ( f obs , measured after in verse PCA and argmax decoding). The attention weight tracked f eff with < 0 . 3% deviation at all ρ values, consistent with Proposition 1 : the energy-lev el conditioning worked as intended. Howe v er , the decoded phenotype fraction told a dif ferent story . P1 K/R fraction increased from 0.41 ( ρ = 1 ) to only 0.63 ( ρ = 500 ), far short of the f eff = 0 . 996 that the attention weights achiev ed. This discrepancy between what the energy deli vers in PCA space and what the decoder recovers in sequence space is the calibration gap. Sequence di v ersity decreased modestly from 0.56 to 0.51 ov er this range and KL div ergence increased from 0.007 to 0.017. The phase transition β ∗ shifted from 4.4 at ρ = 1 ( K eff = 99 ) to 9.3 at ρ = 1 , 000 ( K eff = 32 . 1 ), and the entropy curves at different ρ values formed a family of sigmoidal profiles with systematically rightward-shifted inflection points, consistent with β ∗ ( r ) depending on K eff ( r ) rather than the ra w pattern count K (Fig. 2 ). T o understand why f obs lagged f eff , we measured all three layers of the signal chain (Eq. 7 ) independently across a fine ρ sweep ( ρ ∈ [1 , 1000] , 17 values, 5 replicates each), recording at each step: (i) the total softmax attention weight on designated patterns ( ¯ a des ), (ii) the continuous K/R probability at P1 in the PCA-reconstructed one-hot vector ( f soft ), and (iii) the hard-decoded residue identity ( f obs ). W e found that the attention gap was ∆ attn ≈ 0 across all ρ values ( < 0 . 3% ), indicating that the energy-le vel conditioning was e xact; the PCA gap was the dominant component, with f soft effecti v ely flat at ∼ 0 . 27 regardless of ρ , varying by 3 less than 0.04 across a thousandfold range of multiplicity ratios; and the ar gmax gap w as neg ativ e ( ≈ − 0 . 25 ), indicating that discrete decoding partially reco vered signal lost in the PCA reconstruction. These observations suggest that the PCA encoding mapped designated and background sequences to ov erlapping regions at the P1 position: ev en when 99.8% of attention weight was on the designated patterns, the continuous one-hot reconstruction could not resolve K vs. G at P1 because this variation was not well-captured by the top d = 80 principal components. The Fisher separation inde x for the Kunitz designated/background split w as S = 0 . 20 , consistent with moderate but incomplete geometric separation. Cross-family validation and the separation index. W e repeated the multiplicity analysis on four additional Pfam families (SH3 domains (PF00018; K = 55 , split by conserved T rp at the binding groov e), WW domains (PF00397; K = 420 , split by specificity-loop residue), Homeobox domains (PF00046; K = 136 , split by Gln at position 50 in the recognition helix), and Forkhead domains (PF00250; K = 246 , split by His/Asn at the H3 recognition helix)), yielding five families with distinct PCA-space geometries (T able 1 , T able 2 ). The calibration gap decreased monotonically with increasing separation index: WW domains ( S = 0 . 11 , functional subsets heavily interleav ed in PCA space) exhibited the largest gap ( ∆ = 0 . 64 ); Forkhead domains ( S = 0 . 17 ) a moderate gap ( ∆ = 0 . 27 ); Kunitz domains ( S = 0 . 20 ) a similar gap ( ∆ = 0 . 39 ); and both SH3 ( S = 0 . 34 , ∆ = 0 . 01 ) and Homeobox ( S = 0 . 42 , ∆ = 0 . 04 ) domains sho wed gaps near zero (Fig. 3 ). A linear fit across all fi ve Pfam f amilies yielded ∆ ≈ 0 . 72 − 1 . 8 S with R 2 = 0 . 80 ; the relationship is monotonic, with families abov e S > 0 . 3 achieving near -complete phenotype transfer via multiplicity weighting alone and families belo w S < 0 . 2 showing gaps of 0 . 27 – 0 . 64 . Extending the multiplicity sweep to ω -conotoxin ( S = 0 . 78 , pharmacologically defined binder split) yielded ∆ = 0 . 15 , consistent with the monotonic trend. The attention weights tracked f eff closely in all six families, indicating that the multiplicity-weighted energy operates as e xpected reg ardless of the downstream calibration gap, while hard curation achiev ed 100% phenotype transfer in all families reg ardless of separation index, as expected since curated memory matrices are b uilt from designated-subset-only PCA, which by construction captures subset-specific variation in its leading components. T o test whether increasing β beyond β ∗ could push more phenotype signal through the PCA bottleneck, we swept β /β ∗ from 0.5 to 3.0 at three fix ed ρ values ( ρ = 10 , 50 , 200 ) on the Kunitz domain and observ ed a consistent tradeof f: higher β increased P1 K/R fraction at the cost of reduced div ersity . At ρ = 200 and β = 3 β ∗ , P1 K/R reached 0.71 (versus 0.61 at β ∗ ) while di versity dropped from 0.53 to 0.46, suggesting that the practitioner can tune ( ρ, β /β ∗ ) jointly to navig ate the fidelity–div ersity tradeof f. Application to ω -conotoxin Ca v2.2 binders. W e applied SA to the ω -conotoxin O-superfamily (disulfide-rich peptides from predatory cone snails that block voltage-gated N-type calcium channels (Cav2.2) in sensory neurons [ 9 ]) to test whether curated memory conditioning generalizes be yond classical protein domains to therapeutically relev ant peptide families (T able 5 , Fig. 4 ). Cav2.2 mediates pain signaling in dorsal root ganglion (DRG) neurons, and channel block attenuates synaptic transmission between peripheral nociceptors and the spinal cord. This f amily has direct therapeutic rele vance– ω -conotoxin MVIIA (ziconotide) is an FD A-approv ed non-opioid analgesic for intractable pain [ 10 ]–and is moti v ated by prior computational modeling of A TP-gated calcium dynamics in DRG neurons that identified Cav2.2 as a central node in the pain signaling netw ork [ 11 ]. The primary Cav2.2 pharmacophore is T yr13 (MVIIA numbering): the hydroxyl group contacts the channel pore directly and its substitution abolishes binding, while basic residues (L ys, Ar g) distributed across the peptide contrib ute electrostatic complementarity with the negati v ely charged channel entrance. W e compiled 74 SwissProt ω -conotoxin sequences, computed a multiple sequence alignment with MAFFT [ 12 ], retained 26 positions after gap filtering, designated 23 sequences as strong Cav2.2 binders (confirmed or predicted N-type channel block ers, including MVIIA, GVIA, CVID, CVIE, CVIF , and close homologs) and 2 sequences as weak or non-N-type-selecti v e (MVIIC and MVIID, which preferentially block P/Q-type channels with IC 50 ≥ 200 nM against Cav2.2), and ran two independent SA experiments (50 chains × 5 000 steps, α = 0 . 01 ), generating 1 550 sequences per condition: one seeded from the full family ( K = 74 , β ∗ = 3 . 23 ) and one from the strong binders only ( K = 23 , β ∗ = 2 . 72 ). Pharmacophore inheritance was high under strong-binder seeding and attenuated under full-family seeding: in the full-family input only 33.8% of the 74 sequences carried T yr at the pharmacophore position (col. 13), and full-family-seeded generation raised this modestly to 46.9%, whereas 82.6% of the 23 strong binders carried T yr13 and strong- binder-seeded SA raised this to 98.3% (T able 5 ). The basic-residue (L ys + Arg) fraction was also preserved: strong-binder generation produced 20.1% basic residues versus 19.7% in the input, while 4 full-family generation produced only 12.0%, matching the lower K/R content of the diverse full- family input. Per-position amino acid frequency heatmaps sho wed the conditioning ef fect directly (Fig. 4 ): the strong-binder input and strong-binder-generated heatmaps were similar throughout, with T yr at position 13, conserved Cys frame w ork at positions 14–16, and v ariable-loop div ersity at positions 9–12 all retained; the residual heatmaps (generated − input) sho wed small de viations across all positions, suggesting that SA reproduced the input frequenc y statistics rather than collapsing to a consensus sequence. Sequence-le vel inspection of the fiv e highest-confidence strong-seeded sequences (ranked by ESMFold pLDDT) showed that all fi ve preserved e very highly conserved position ( 6 / 6 ), all six cysteines, and carried T yr at the pharmacophore position, while introducing 5-11 substitutions at v ariable sites; the per -position entropy correlation between stored and strong- seeded sequences was r = 0 . 745 , lo wer than the full-seeded correlation ( r = 0 . 997 ), reflecting the reduced di versity at co-v arying loop positions that contribute to Cav2.2 binding specificity (Fig. 6 ). W e further validated the generated sequences against published structure–acti vity relationship (SAR) data by measuring agreement with experimentally identified binding determinants at 12 positions (T able 6 ): SA strong-seeded sequences preserved the primary pharmacophore (T yr13: 98.3%), all six disulfide-forming cysteines ( ≥ 97 . 4% ), and enriched basic residues at loop 2 positions critical for channel interaction (L ys2: 97.5% K/R; Arg10: 82.3% K/R) relati v e to both the 23-sequence input and full-family generation, indicating that curated memory conditioning transferred both global pharmacophore statistics and the specific residue-lev el binding determinants identified by prior mutagenesis work [ 13 – 15 ]. Structural v alidation showed that the generated conotoxin sequences adopted the expected inhibitor cystine knot fold, with the disulfide-constrained loop architecture retained across all three top SA strong-seeded variants (Fig. 7 ). T ogether , these results indicate that curated memory conditioning transferred Ca v2.2 pharmacophore statistics from a set of 23 characterized sequences to a library of 1 550 candidates, with no retraining and without requiring the Cav2.2 tar get structure for sequence generation. AlphaFold2 multimer complex pr ediction. W e tested whether SA-generated sequences produce complex-le vel predictions comparable to natural ω -conotoxins using ColabF old AlphaFold2-multimer with the Cav2.2 pore-domain target (T able 7 ). SA strong-conditioned ( n = 10 ; pTM 0 . 221 ± 0 . 007 , iPTM 0 . 105 ± 0 . 008 , interface-pLDDT 37 . 2 ± 5 . 4 ) and SA full-conditioned ( n = 10 ; pTM 0 . 219 ± 0 . 008 , iPTM 0 . 104 ± 0 . 007 , interface-pLDDT 38 . 2 ± 4 . 0 ) sequences were statistically indistinguishable from natural controls ( n = 5 ; pTM 0 . 228 ± 0 . 004 , iPTM 0 . 108 ± 0 . 007 , interface- pLDDT 40 . 7 ± 3 . 5 ) on all four metrics (exact permutation tests; all p > 0 . 05 ). SA-generated sequences thus match natural ω -conotoxins under multimer prediction, indicating that the generation process does not de grade predicted binding geometry . The absolute scores are modest across all groups, reflecting the kno wn dif ficulty of predicting small-peptide–channel comple xes rather than a deficiency of the generated sequences; experimental binding assays remain necessary to assess functional activity . Structural and linguistic validation. W e predicted folds for 50 sequences from each source (stored, SA full-family , SA strong-conditioned, SA weak-conditioned, and HMM baseline) using ESMFold and computed TM-scores against the experimental Kunitz domain structure (1BPI, chain A) to verify that SA-generated sequences encode plausible three-dimensional structures (T able 3 ). W e found that SA-generated sequences were structurally comparable to natural sequences: mean pLDDT was 90 . 4 ± 1 . 2 (SA full) and 90 . 7 ± 1 . 1 (SA strong) versus 89 . 3 ± 3 . 2 for stored sequences, with 100% of SA sequences exceeding the pLDDT > 70 confidence threshold; mean TM-scores were 0 . 84 ± 0 . 01 (SA full) and 0 . 83 ± 0 . 01 (SA strong) versus 0 . 83 ± 0 . 03 for stored sequences, all well abov e the TM > 0 . 5 same-fold threshold (Fig. 7 ); whereas HMM-emitted sequences achieved only 60 . 4 ± 13 . 1 mean pLDDT (20% abov e threshold) and 0 . 60 ± 0 . 10 mean TM-score, which suggests that profile HMM emission does not reliably produce foldable sequences (T able 3 ). AlphaFold2-ptm predictions via ColabFold [ 16 , 17 ] were consistent with these results (T able 4 ): K unitz TM-scores were 0 . 83 – 0 . 85 (pLDDT 93 – 95 ) and ω -conotoxin TM-scores were 0 . 35 – 0 . 47 (pLDDT 68 – 79 ) across stored and SA-generated sequences, consistent with the ESMF old values and the shorter peptide length of conotoxins. W e further assessed sequence plausibility using ESM2-650M pseudo-perplexity computed via masked marginal scoring over all positions and found that SA-generated sequences achiev ed pseudo-perplexity comparable to natural sequences (SA full: 4 . 78 ± 0 . 64 ; SA strong: 4 . 72 ± 0 . 60 ; stored: 4 . 97 ± 0 . 98 ), while HMM-emitted sequences were approximately threefold worse ( 16 . 1 ± 4 . 2 )–the slightly lo wer perplexity of conditioned SA sequences relati ve to stored sequences is consistent with SA ’ s tendency to generate near the center of the family distribution, away from outlier 5 sequences present in natural alignments (T able 3 ). W e also compared SA against HMMER3 [ 6 ] profile HMM emission (150 sequences, hmmemit –seed 42 ) and bootstrap resampling from stored patterns and found that SA full-family generation preserved the natural P1 K/R proportion ( 0 . 41 ± 0 . 01 ) and achiev ed the lo west KL di ver gence ( 0 . 0069 ± 0 . 0001 ), while HMM emission produced sequences with lower P1 K/R fraction ( 0 . 15 ± 0 . 03 ) and higher compositional di ver gence ( 0 . 030 ± 0 . 011 ); bootstrap resampling preserved composition (KL ≈ 0 . 001 ) but produced no no vel sequences; and SA strong-binder conditioning achiev ed complete P1 K/R fraction ( 1 . 0 ± 0 . 0 )–a capability not av ailable to unconditional baselines (T able 3 ). These results indicate that SA generated structurally v alid, language-model-plausible sequences that exceeded classical baselines on both fidelity and functional conditioning. 3 Discussion W e ha v e sho wn that pattern multiplicity in the modern Hopfield energy provides a principled, training- free mechanism for conditioning protein sequence generation on functional subsets. The theoretical framew ork is exact: multiplicity weights tilt the Boltzmann distribution through a simple logit bias, and the resulting Lange vin dynamics sample from a kno wn distrib ution with no approximation. A single scalar ρ continuously interpolates between unconditioned generation and hard subset curation, and the practitioner can compute the required ρ for any tar get ef fecti ve designated fraction directly from the family composition (Eq. 4 ). On the Kunitz domain, curated memory achieved 100% phenotype transfer with as few as three designated input sequences; multiplicity-weighted generation on the full family increased phenotype fidelity from the natural proportion (32%) to 63% at ρ = 500 ; and the cross-family validation on fiv e Pfam families sho wed a quantitati ve relationship between PCA-space geometry and conditioning effecti v eness. The central finding is the calibration gap and its decomposition. The gap is not a f ailure of the multiplicity-weighted energy (the attention weights track f eff with < 0 . 3% deviation across all families and ρ values tested) b ut a well-characterized property of the PCA encoding that compresses 20 L -dimensional sequence space into d principal components. When the designated and background subsets ov erlap in PCA space (low S ), the energy-le vel tilt does not propagate to the decoded residue identity; when they separate (high S ), it does. The cross-family v alidation across fi ve Pf am families produced a monotonic negati ve relationship between S and ∆ ( R 2 = 0 . 80 for a linear fit), indicating that the separation index is a useful a priori predictor of conditioning effecti veness. This yields a practitioner’ s decision rule: compute S (a cheap operation requiring only PCA and pairwise cosine similarities, no generation); if S > 0 . 3 , multiplicity weighting preserves fold-le v el constraints from the full family while achie ving high phenotype transfer (SH3 and Homeobox domains, ∆ < 0 . 05 ); if S < 0 . 2 , hard curation is necessary because the PCA bottleneck attenuates the signal (WW and Forkhead domains, ∆ = 0 . 27 – 0 . 64 ); the intermediate re gime ( 0 . 2 < S < 0 . 3 , K unitz domains) admits a tradeoff controlled by ( ρ, β /β ∗ ) . Hard curation achiev es complete phenotype transfer with as few as three designated input sequences. Many e xperimental campaigns (phage display , yeast surface display , functional selections, or e v en literature curation) produce exactly this kind of small, functionally characterized set. The practitioner brings external knowledge (“these sequences bind my tar get, ” “these v ariants are thermostable, ” “these hits came from my screen”) and SA conv erts this small set into a lar ge, diverse candidate library while preserving the input phenotype. This “small-set amplifier” capability complements high-throughput screening: the screen identifies a fe w hits, SA expands them computationally , and the expanded library can be fed back into a second round of selection or directly into functional assays. The method itself is agnostic to the meaning of the designation; it biases the sampler to w ard the designated patterns regardless of whether the underlying phenotype is binding, stability , expression, or any other property . The scaling study shows that e v en K des = 3 suffices for phenotype transfer; div ersity improv es with more input sequences b ut saturates near K des = 20 . The ω -conotoxin experiment extends the small-set amplifier result to a therapeutically relev ant peptide family and illustrates a practically important design choice: whether to seed SA from the full family or from a curated functional subset. Full-family seeding is appropriate when the goal is to generate novel family members with broad structural div ersity; curated seeding is necessary when the goal is to preserve a specific pharmacophore. In the ω -conotoxin case, the O-superfamily is structurally coherent (all members share the C–C–CC–C–C disulfide frame work) b ut functionally heterogeneous (members block N-type, P/Q-type, or sodium channels with varying selecti vity). 6 Full-family seeding dilutes the Cav2.2-specific T yr pharmacophore because the PCA encoding reflects the full structural div ersity; curated seeding from the 23 N-type blockers concentrates the PCA subspace on Ca v2.2-relev ant variation, allowing SA to regenerate it with 98% fidelity . This generalizes the separation-index interpretation: ev en when S is moderate, hard curation can achie ve high pharmacophore transfer by restricting the memory to the functional subset of interest. The cost (lo wer sequence di versity and reduced no velty , 0.46 vs. 0.60) reflects the narrower PCA basis, consistent with the Kunitz scaling study . The AlphaFold2-multimer analysis sho ws that SA-generated ω -conotoxins are indistinguishable from natural sequences under complex prediction: all four metrics (pTM, iPTM, confidence, interf ace pLDDT) ov erlap between generated and control groups with no significant dif ferences. This is consistent with the structural and pharmacophore validation: generation preserves the properties that determine predicted binding geometry . The absolute scores are modest across all groups, a kno wn limitation of multimer prediction for small-peptide–channel complexes; e xperimental binding assays are needed to determine whether the preserved pharmacophore translates to functional Cav2.2 block. Sev eral limitations should be ackno wledged. The cross-family regression spans fi ve Pfam families (plus ω -conotoxin) with R 2 = 0 . 80 for a linear fit; while the monotonic trend is clear, additional families would help characterize the functional form of the S – ∆ relationship, which may be nonlinear (the gap appears to decay steeply below S ≈ 0 . 3 and flatten near zero above it). Our functional splits are defined by single mark er positions (P1 in K unitz, T rp in SH3, specificity-loop residue in WW , Gln at position 50 in Homeobox, H/N at the recognition helix in Forkhead); multi-position phenotypes, where specificity depends on coordinated residue identities at several positions, may exhibit different calibration behavior . W e hav e not e xperimentally validated the binding activity of generated sequences; the phenotype fidelity reported here is a necessary but not sufficient con- dition for functional binding. For the ω -conotoxin family , we validated SA-generated sequences against published SAR data from alanine-scanning and site-directed mutagenesis studies [ 13 – 15 ] and found that strong-seeded generation preserved all experimentally identified binding determinants (T able 6 ); for the Kunitz domain, deep mutational scanning data for the BPTI–trypsin interaction are no w a v ailable [ 18 ] and could provide a quantitativ e binding-landscape comparison in future work. Computational binding affinity predictions (e.g., via AlphaFold-Multimer or FoldX) could provide an additional intermediate v alidation tier between sequence-lev el markers and experimental assays. Finally , we tested interface-weighted PCA encoding as a strategy to close the calibration gap, but found it neutral at lo w weights and destructi ve at high weights; the PCA bottleneck reflects the geometric entanglement of functional and structural v ariation in the family , not the absence of interface-position components. Looking forward, the multiplicity framew ork opens sev eral directions. The binary split (desig- nated/background) generalizes naturally to multi-class conditioning by assigning dif ferent multiplic- ities to different functional categories. The PCA bottleneck that limits conditioning ef fectiv eness might be reduced by alternativ e sequence encodings. Learned representations from protein language models, for instance, may better separate functional subsets than linear PCA. The β le ver suggests that adaptiv e temperature schedules, increasing β during sampling to sharpen the multiplicity bias, could improv e phenotype transfer without permanently sacrificing di versity . These e xtensions remain within the analytic, training-free framework that makes stochastic attention practical for the scarce-data regime. 4 Materials and Methods 4.1 Fr om Hopfield Energy to Multiplicity-W eighted Score Function Let X = [ m 1 , . . . , m K ] ∈ R d × K be a memory matrix whose columns are unit-norm, PCA-encoded protein sequences from a f amily alignment of K members, each represented in d dimensions. The modern Hopfield energy [ 19 ] at state ξ ∈ R d is E ( ξ ) = 1 2 ∥ ξ ∥ 2 − 1 β log P k exp( β m ⊤ k ξ ) , where β > 0 is an inv erse temperature. The score function ∇ ξ log p β ( ξ ) = X softmax( β X ⊤ ξ ) − ξ is exact and computed by a single attention operation; applying the unadjusted Langevin algorithm (ULA) with step size α ∈ (0 , 1) yields the stochastic attention update [ 7 ]: ξ t +1 = (1 − α ) ξ t + α X softmax β X ⊤ ξ t + p 2 α/β ϵ t , ϵ t ∼ N ( 0 , I d ) . (1) 7 W e extend this frame work to condition generation on a functional subset by assigning a multiplicity weight r k > 0 to each stored pattern. The multiplicity-weighted Hopfield energy is E r ( ξ ) = 1 2 ∥ ξ ∥ 2 − 1 β log K X k =1 r k exp β m ⊤ k ξ , (2) where r = ( r 1 , . . . , r K ) ⊤ . When r k = 1 for all k , Eq. ( 2 ) reduces to the standard energy . Patterns with higher multiplicity deepen the energy wells in their vicinity; this is equi v alent to storing r k copies of pattern m k without the O ( d · P k r k ) memory cost. Proposition 1 (Score function) . The score function of p r ( ξ ) ∝ exp( − β E r ( ξ )) is ∇ ξ log p r ( ξ ) = X softmax( β X ⊤ ξ + log r ) − ξ , wher e log r = (log r 1 , . . . , log r K ) ⊤ is the elementwise log- multiplicity . The proof follo ws from the identity r k exp( a k ) = exp( a k + log r k ) : the weights are absorbed into the softmax logits. The resulting Langevin update is ξ t +1 = (1 − α ) ξ t + α X softmax β X ⊤ ξ t + log r + p 2 α/β ϵ t . (3) The only change from Eq. ( 1 ) is the addition of log r k to each logit before the softmax. The memory matrix X is unchanged, the noise schedule is unchanged, and the sampler retains its O ( dK ) per-step cost. 4.2 Multiplicity Ratio and Phase T ransition For a binary functional split (designated subset vs. background), we parameterize the multiplicity vector by a single scalar ρ = r designated /r background , setting r k = ρ for patterns in the user- designated subset and r k = 1 otherwise. The ef fecti v e designated fraction (the share of the softmax probability mass attrib utable to designated patterns when all patterns ha v e equal similarity to ξ ) is f eff ( ρ ) = K des ρ/ ( K des ρ + K bg ) , where K des and K bg = K − K des are the number of designated and background patterns. At ρ = 1 , f eff = K des /K ; as ρ → ∞ , f eff → 1 . Inv erting gives the multiplicity ratio needed for a target fraction f : ρ ( f ) = f · K bg K des (1 − f ) . (4) The standard SA frame w ork exhibits a phase transition at critical in verse temperature β ∗ that depends on the PCA dimension d and the number of stored patterns. Under multiplicity weighting, the relev ant quantity is the ef fecti ve number of patterns K eff ( r ) = ( P k r k ) 2 / P k r 2 k , which equals K when all r k are equal and con ver ges to K des as ρ → ∞ . Remark 2 (Phase transition: analytic baseline and multiplicity shift) . The companion paper [ 7 ] establishes, via a concentration-of-measure ar gument on the softmax attention entropy , that for the unweighted SA sampler ( ρ = 1 ) the phase transition satisfies β ∗ ≈ 1 . 57 + 0 . 28 √ d, (5) with R 2 = 0 . 97 across eight Pfam f amilies spanning d ∈ [18 , 186] , and is nearly independent of K for K ≥ 30 . At ρ = 1 , Eq. ( 5 ) therefore giv es the analytic value directly from the alignment. For ρ > 1 , the logit biases log r k pre-concentrate the attention distrib ution at β = 0 : the weighted entropy at zero temperature is H r (0) = log K eff ( r ) < log K , a lo wer starting entropy than the unweighted case. Reaching the retrie v al-phase inflection from this pre-concentrated starting point requires a higher in verse temperature, so β ∗ ( ρ ) ≥ β ∗ (1) . F or intermediate ρ , β ∗ ( ρ ) is located as the inflection of the weighted attention entropy H r ( β ) via a one-dimensional sweep, a negligible computation relativ e to sequence generation. Empirically , β ∗ increases monotonically from 4.4 ( ρ = 1 , K eff = 99 ) to 9.3 ( ρ = 1000 , K eff = 32 . 1 ) in the Kunitz domain, consistent with Remark 2 : more in v erse temperature is required as the logit biases concentrate probability mass on fewer ef fecti v e patterns. 8 4.3 The Calibration Gap The signal chain from multiplicity weights to decoded sequence phenotype passes through se veral transformations: ρ exact − − − → f eff exact − − − → ¯ a des lossy − − − → f soft nonlinear − − − − − → f obs , (6) where ¯ a des is the mean attention weight on designated patterns during sampling, f soft is the continuous-valued residue probability at the marker position, and f obs is the fraction of gener- ated sequences displaying the designated phenotype after ar gmax decoding. W e define the calibration gap ∆ = f eff − f obs and decompose it into three independently measurable components: ∆ = ( f eff − ¯ a des ) | {z } ∆ attn ≈ 0 + (¯ a des − f soft ) | {z } ∆ PCA + ( f soft − f obs ) | {z } ∆ argmax . (7) T o predict the PCA gap across families, we define the Fisher separation index S = ( ¯ c within − ¯ c between ) / [ 1 2 ( σ within + σ between )] , where ¯ c and σ denote the mean and standard de viation of pairwise cosine similarities within and between functional groups in PCA space. For each e xperiment, we followed the stochastic attention pipeline of [ 7 ] without modification to the core sampler . Seed alignments were do wnloaded from InterPro/Pfam in Stockholm format (or , for the ω -conotoxin O-superfamily , compiled from SwissProt and aligned with MAFFT [ 12 ]). Alignments were cleaned by removing columns with > 50% gaps and sequences with > 30% gaps in the remaining columns. The cleaned alignment was one-hot encoded ( 20 amino acid channels per position, gaps mapped to zero vectors) yielding a 20 L × K matrix, reduced by PCA (retaining 95% of v ariance) to a d × K matrix, and column-normalized to unit norm. ULA chains were run with step size α = 0 . 01 for T = 5 , 000 steps, initialized near random stored patterns with small Gaussian perturbation; samples were collected after a burn-in of 2,000 steps with thinning e very 100 steps. W e tested fi ve Pfam families (Kunitz domains (PF00014, K = 99 , L = 53 ), SH3 domains (PF00018, K = 55 , L = 48 ), WW domains (PF00397, K = 420 , L = 31 ), Homeobox domains (PF00046, K = 136 , L = 57 ), and Forkhead domains (PF00250, K = 246 , L = 87 )) and the ω -conotoxin O-superfamily ( K = 74 , L = 26 , curated from SwissProt). For each f amily , we defined a binary functional split based on a single marker position reflecting external biological knowledge, not a property discovered by the method: for Kunitz, the P1 position (primary contact residue with serine proteases) w as identified as the column with the highest L ys frequency and sequences with K or R at P1 were designated as the functional subset ( K des = 32 ); for SH3, the conserved T rp in the hydrophobic binding groo ve was the marker ( K des = 33 ); for WW , the specificity-determining position was the highest-entropy column in the middle third of the alignment ( K des = 69 ); for Homeobox, Gln at position 50 in the recognition helix (the primary determinant of T AA T -core DN A-binding specificity) was the marker ( K des = 102 ); for Forkhead, His or Asn at the H3 recognition helix position (residues that make base-specific contacts in the major groove) was the marker ( K des = 122 ); and for ω -conotoxin, 23 confirmed or predicted N-type Cav2.2 blockers were designated as strong binders based on pharmacological characterization [ 9 ]. For multiplicity-weighted runs, the multiplicity vector w as set to r k = ρ for designated patterns and r k = 1 for background patterns, and β ∗ ( r ) was found by sweeping 50–60 logarithmically spaced β v alues in [0 . 1 , 500] and locating the inflection point of the weighted attention entrop y . T wenty to fifty independent chains were run per condition, yielding 620–1,550 sequences after burn-in and thinning. Phenotype fidelity was measured as the fraction of generated sequences displaying the designated marker residue at the marker position; attention diagnostics recorded the mean softmax weight on designated patterns during sampling; sequence di versity was computed as one minus the mean pairwise sequence identity (estimated from 300–500 random pairs); and amino acid composition fidelity was measured by KL di ver gence between generated and reference amino acid frequencies. The Fisher separation index S was computed from pairwise cosine similarities between unit-norm PCA vectors within and between functional groups as defined in Section 4.3 . Structural validation used ESMFold [ 5 ] to predict structures for 50 sequences per source, with pLDDT extracted from the B-factor column and TM-scores computed against e xperimental reference structures (1BPI chain A for Kunitz, 1OMG chain A for ω -conotoxin) using TM-align [ 20 ]; these results were independently cross-validated with AlphaFold2-ptm via ColabF old [ 16 , 17 ] (1 model, 3 recycles, MMseqs2 MSA, no relaxation) run on Google Colab with a T4 GPU. Sequence plausibility was assessed using ESM2-650M pseudo-perplexity computed via masked marginal scoring ov er all positions [ 5 ]. All experiments were implemented in Julia 1.12 using NNlib.jl (softmax), Multi variateStats.jl (PCA), 9 and Distributions.jl; source code and e xperiment scripts are a v ailable at https://github.com/ varnerlab/SA- Binding- Generation- Study . Acknowledgments The author thanks the Cornell Center for Advanced Computing for computational resources. A uthor contributions. J.D.V . concei ved the study , dev eloped the theory , wrote the code, performed the experiments, and wrote the paper . Competing interests. The author declares no competing interests. Data av ailability . All Pfam seed alignments are publicly av ailable from InterPro. Source code and experiment scripts are av ailable at https://github.com/varnerlab/ SA- Binding- Generation- Study . References [1] Ke vin K. Y ang, Zachary W u, and Frances H. Arnold. Machine-learning-guided directed ev olution for protein engineering. Natur e Methods , 16:687–694, 2019. [2] Ali Madani, Ben Krause, Eric R. Greene, Subu Subramanian, Benjamin P . Mohr , James M. Holton, Jose Luis Olmos, Caiming Xiong, Zhi Zhong Sun, Richard Socher, James S. Fraser , and Nikhil Naik. Large language models generate functional protein sequences across di v erse families. Nature Biotechnology , 41:1099–1106, 2023. [3] Sarah Alamdari, Nitya Thakkar, Rianne v an den Ber g, Alex X. Lu, Nicolo Fusi, A va P . Amini, and K evin K. Y ang. Protein generation with e volutionary dif fusion: sequence is all you need. bioRxiv , 2023. doi: 10.1101/2023.09.11.556673. [4] Justas Dauparas, Iv an Anishchenko, Nathaniel Bennett, Hua Bai, Robert J. Ragotte, Lukas F . Milles, Basile I. M. W icky , Alexis Courbet, Rob J. de Haas, Ne ville Bethel, Philip J. L. Leung, T imothy F . Huddy , Sam Pellock, Doug Tischer , Frederick Chan, Brian Koepnick, Hannah Nguyen, Alex Kang, Banumathi Sankaran, Asim K. Bera, Neil P . King, and David Baker . Robust deep learning-based protein sequence design using ProteinMPNN. Science , 378:49–56, 2022. [5] Zeming Lin, Halil Akin, Roshan Rao, Brian Hie, Zhongkai Zhu, W enting Lu, Nikita Smetanin, Robert V erkuil, Ori Kabeli, Y aniv Shmueli, Allan dos Santos Costa, Maryam Fazel-Zarandi, T om Sercu, Sal Candido, and Alexander Ri v es. Evolutionary-scale prediction of atomic-lev el protein structure with a language model. Science , 379(6637):1123–1130, 2023. [6] Simon C Potter , Aurélien Luciani, Sean R Eddy , Y oungmi Park, Rodrigo Lopez, and Robert D Finn. HMMER web server: 2018 update. Nucleic acids r esear ch , 46:W200–W204, 2018. [7] Jeffre y D. V arner . Training-free generation of protein sequences from small family alignments via stochastic attention. arXiv pr eprint arXiv:2603.14717 , 2026. doi: 10.48550/arXiv .2603. 14717. [8] Michael Lasko wski and Izydor Kato. Protein inhibitors of proteinases. Annual Revie w of Biochemistry , 49:593–626, 1980. [9] Christina I. Schroeder and Richard J. Le wis. ω -conotoxins GVIA, MVIIA and CVID: SAR and clinical potential. Marine Drugs , 4(3):193–214, 2006. [10] George P . Miljanich. Ziconotide: neuronal calcium channel blocker for treating se vere chronic pain. Current Medicinal Chemistry , 11(23):3029–3040, 2004. [11] Sang Ok Song and Jef fre y V arner . Modeling and analysis of the molecular basis of pain in sensory neurons. PLOS ONE , 4(9):e6758, 2009. 10 [12] Kazutaka Katoh and Daron M. Standle y . MAFFT multiple sequence alignment software version 7: improvements in performance and usability . Molecular Biology and Evolution , 30(4): 772–780, 2013. [13] J. I. Kim, M. T akahashi, A. Ohtake, A. W akamiya, and K. Sato. T yr13 is essential for the acti vity of omega-conotoxin MVIIA and GVIA, specific N-type calcium channel blockers. Biochemical and Biophysical Researc h Communications , 206(2):449–454, 1995. [14] K. Sato, N. G. Park, T . Kohno, T . Maeda, J. I. Kim, R. Kato, and M. T akahashi. Role of basic residues for the binding of ome ga-conotoxin GVIA to N-type calcium channels. Biochemical and Biophysical Researc h Communications , 194(3):1292–1296, 1993. [15] R. J. Le wis, S. Dutertre, I. V etter , and M. J. Christie. Conus venom peptide pharmacology . Pharmacological Reviews , 64(2):259–298, 2012. [16] John Jumper et al. Highly accurate protein structure prediction with AlphaFold. Natur e , 596: 583–589, 2021. [17] Milot Mirdita, K onstantin Schütze, Y oshitaka Moriw aki, Lim Heo, Serge y Ovchinniko v , and Martin Steinegger . ColabFold: making protein folding accessible to all. Natur e Methods , 19: 679–682, 2022. [18] M. Heyne, J. Shirian, I. Cohen, Y . Pele g, E. S. Radisky , N. Papo, and J. M. Shifman. Climbing up and do wn binding landscapes through deep mutational scanning of three homologous protein– protein complex es. J ournal of the American Chemical Society , 143(41):17261–17275, 2021. [19] Hubert Ramsauer , Bernhard Schäfl, Johannes Lehner , Philipp Seidl, Michael W idrich, Thomas Adler , Lukas Gruber , Markus Holzleitner , Milena Pavlo vi ´ c, Geir Kjetil Sandve, et al. Hopfield networks is all you need. International Conference on Learning Representations , 2021. [20] Y ang Zhang and Jeffrey Sk olnick. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Researc h , 33(7):2302–2309, 2005. [21] K. Sato, C. Raymond, N. Martin-Moutot, T . Sasaki, A. Ohtake, K. Minami, C. V an Renterghem, J. I. Kim, M. T akahashi, and M. J. Seagar . Binding of Ala-scanning analogs of omega-conotoxin MVIIC to N- and P/Q-type calcium channels. FEBS Letters , 469(2–3):147–150, 2000. [22] C. I. Schroeder , K. J. Nielsen, D. A. Adams, M. Loughnan, L. Thomas, P . F . Alewood, R. J. Lewis, and D. J. Craik. Effects of L ys2 to Ala2 substitutions on the structure and potency of omega-conotoxins MVIIA and CVID. Biopolymers , 98(4):345–356, 2012. [23] C. I. Schroeder , M. L. Smythe, R. J. Lewis, and D. J. Craik. Effects of chirality at T yr13 on the structure–activity relationships of ome ga-conotoxins from Conus magus. Biochemistry , 38(5): 1440–1446, 1999. 11 T able 1: Cross-family summary of all pr otein families tested. For each family: source database, number of sequences ( K ), aligned length ( L ), PCA dimensionality ( d , capturing 95% v ariance), designated subset size ( K des ), phenotype mark er , natural designated fraction ( f nat ), Fisher separation index ( S ), observed phenotype fraction under hard curation ( f hard obs ), and calibration gap ( ∆ at ρ = 500 ). All six families were tested with both multiplicity weighting and hard curation. Family Source K L d K des Marker f nat S f hard obs ∆ WW PF00397 420 31 186 69 Spec. loop 0.16 0.11 1.00 0.64 Forkhead PF00250 246 87 172 122 H/N at H3 0.50 0.17 1.00 0.27 Kunitz PF00014 99 53 80 32 P1 K/R 0.32 0.20 1.00 0.39 SH3 PF00018 55 48 46 33 T rp 0.60 0.34 1.00 0.01 Homeobox PF00046 136 57 101 102 Gln at pos. 50 0.75 0.42 1.00 0.04 ω -Conotoxin SwissProt 74 26 34 23 T yr13 0.31 0.78 0.98 0.15 5 10 15 20 25 30 Number of input binders 0.36 0.39 0.42 0.45 0.48 Pairwise diversity Sequence Diversity 5 10 15 20 25 30 Number of input binders 0.01 0.02 0.03 0.04 0.05 0.06 0.07 KL(AA) AA KL Divergence Figure 1: Diversity and composition fidelity improve with mor e input binders; phenotype fidelity is perfect at every sample size. Subsampling K des ∈ { 3 , 5 , 8 , 10 , 15 , 20 , 25 , 32 } sequences from the Kunitz strong-binder set (3 replicates each; shaded bands sho w ± 1 s.d.). P1 K/R fraction was 1.0 at e very sample size (not sho wn). Left: Pairwise sequence div ersity increases from 0.36 ( K des = 3 ) to 0.50 ( K des = 32 ). Right: KL di ver gence of amino acid composition decreases from 0.047 to 0.009 ov er the same range. Three input binders suf fice for perfect phenotype transfer; more binders improve the compositional and div ersity quality of the generated library . 12 1 0 1 1 0 0 1 0 1 1 0 2 I n v e r s e t e m p e r a t u r e 0.0 0.2 0.4 0.6 0.8 1.0 N o r m a l i z e d a t t e n t i o n e n t r o p y H ( ) / l o g K H = l o g K ( u n i f o r m ) l o g K d e s / l o g K i n c r e a s i n g s h i f t s * r i g h t w a r d = 1 ( K e f f = 9 9 , * = 4 . 4 ) = 5 ( K e f f = 5 9 . 4 , * = 4 . 8 ) = 2 0 ( K e f f = 3 8 . 8 , * = 6 . 0 ) = 1 0 0 ( K e f f = 3 3 . 3 , * = 7 . 5 ) = 1 0 0 0 ( K e f f = 3 2 . 1 , * = 9 . 3 ) Figure 2: Phase transition shifts rightward with increasin g multiplicity ratio ρ . Attention entropy H ( β ) normalized by log K (the uniform limit) as a function of in verse temperature β (log scale) for ρ ∈ { 1 , 5 , 20 , 100 , 1000 } on the Kunitz family ( K = 99 , K des = 32 ). At ρ = 1 (standard SA), the entropy starts at H / log K = 1 (uniform attention over all patterns); as ρ increases, the multiplicity bias pre-concentrates the attention distribution, lowering the starting entropy toward log K des / log K ≈ 0 . 75 (dotted line). Each curve sho ws a sigmoidal drop to near-zero entropy (retriev al phase); dots and dashed vertical lines mark the inflection point β ∗ ( ρ ) . As ρ increases from 1 to 1000, K eff decreases from 99 to 32.1 and β ∗ shifts rightward from 4.4 to 9.3, consistent with fewer ef fecti v e attractors requiring higher in verse temperature to produce an ordered phase. 0.0 0.2 0.4 0.6 0.8 1.0 E f f e c t i v e d e s i g n a t e d f r a c t i o n f e f f 0.0 0.2 0.4 0.6 0.8 1.0 O b s e r v e d p h e n o t y p e f r a c t i o n f o b s = 1 = 5 0 0 = 0 . 6 2 = 0 . 2 7 = 1 = 5 0 0 = 0 . 3 9 = 1 = 5 0 0 = 1 = 5 0 0 = 0 . 1 5 perfect calibration A W W ( S = 0 . 1 1 ) F o r k h e a d ( S = 0 . 1 7 ) K u n i t z ( S = 0 . 2 0 ) S H 3 ( S = 0 . 3 4 ) H o m e o b o x ( S = 0 . 4 2 ) - C o n o t o x i n ( S = 0 . 7 8 ) 0.2 0.4 0.6 0.8 F i s h e r s e p a r a t i o n i n d e x S 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 C a l i b r a t i o n g a p = f e f f f o b s WW F orkhead K unitz SH3 Homeobox - C n t x 0 . 7 1 1 . 8 S R 2 = 0 . 8 1 (Pfam families) B Figure 3: Calibration trajectories rev eal family-dependent gaps pr edicted by PCA-space separa- tion. (A) Observ ed phenotype fraction f obs versus effecti ve designated fraction f eff as the multiplicity ratio ρ increases from 1 to 500 for six protein families. The dashed diagonal marks perfect calibration ( f obs = f eff ). Homeobox ( S = 0 . 42 ) and SH3 ( S = 0 . 34 ) domains track the diagonal closely; Kunitz ( S = 0 . 20 ) and Forkhead ( S = 0 . 17 ) curv e belo w; WW domains ( S = 0 . 11 ) sho w the largest departure; ω -conotoxin ( S = 0 . 78 ) shows a moderate gap despite high PCA separation. Double-headed arrows indicate the calibration gap at ρ = 500 . (B) Fisher separation inde x S versus calibration gap ∆ at ρ = 500 . A linear fit to the fi ve Pfam families (filled markers; ∆ ≈ 0 . 72 − 1 . 8 S , R 2 = 0 . 80 ) shows that the gap is predicted by the geometric separation of functional subsets in PCA space: families with S > 0 . 3 hav e gaps near zero, while families with S < 0 . 2 hav e gaps of 0 . 27 – 0 . 64 . The ω -conotoxin point (open diamond) follo ws the monotonic trend. 13 T able 2: Per -family ρ sweep: attention weights track f eff exactly; decoded phenotype fidelity depends on PCA separation. Selected ρ values for each family . f eff : effecti ve designated fraction from multiplicity weights; ¯ a des : mean softmax attention on designated patterns; f obs : hard-decoded phenotype fraction (930 sequences); D : mean pairwise diversity . In all families ¯ a des ≈ f eff (energy conditioning is exact), but f obs con ver ges fastest for Homeobox ( S = 0 . 42 ) and slowest for WW ( S = 0 . 11 ), reflecting the PCA-space calibration gap. Family ( K , K des , S ) ρ f eff ¯ a des f obs ∆ D WW (PF00397) (420, 69, 0.11) 1 0.164 0.156 0.126 0.04 0.682 10 0.663 0.653 0.224 0.44 0.667 100 0.952 0.951 0.302 0.65 0.653 1000 0.995 0.996 0.371 0.62 0.618 Forkhead (PF00250) (246, 122, 0.17) 1 0.496 0.490 0.556 − 0.06 0.480 10 0.908 0.904 0.638 0.27 0.447 100 0.990 0.988 0.710 0.28 0.417 1000 0.999 0.999 0.730 0.27 0.398 Kunitz (PF00014) (99, 32, 0.20) 1 0.323 0.319 0.389 − 0.07 0.563 10 0.827 0.827 0.513 0.31 0.543 100 0.979 0.981 0.587 0.39 0.523 1000 0.998 0.998 0.609 0.39 0.512 SH3 (PF00018) (55, 33, 0.34) 1 0.600 0.621 0.863 − 0.26 0.587 10 0.938 0.944 0.915 0.02 0.583 100 0.993 0.994 0.980 0.01 0.532 1000 0.999 0.999 0.991 0.01 0.519 Homeobox (PF00046) (136, 102, 0.42) 1 0.750 0.758 0.935 − 0.19 0.525 10 0.968 0.969 0.955 0.01 0.522 100 0.997 0.997 0.957 0.04 0.521 1000 1.000 1.000 0.957 0.04 0.521 ω -Cntx (SwissProt) (74, 23, 0.78) 1 0.311 0.299 0.456 − 0.15 0.557 10 0.819 0.809 0.650 0.17 0.516 100 0.978 0.972 0.769 0.21 0.470 500 0.996 0.994 0.847 0.15 0.435 T able 3: Structural, linguistic, and functional validation on the Kunitz domain. pLDDT : ESM- Fold predicted confidence (0–100; > 70 = confident). TM: TM-score against 1BPI chain A ( > 0 . 5 = same fold). PPL: ESM2-650M masked-marginal pseudo-perplexity (lo wer = more plausible). P1 K/R: fraction with L ys/Ar g at the primary trypsin contact. KL: amino acid composition di v ergence ( × 10 3 ). D : mean pairwise di versity . Structural and LM values are mean ± s.d. across 50 sequences; sequence- lev el metrics are mean ± s.d. across 5 replicates. SA-generated sequences are indistinguishable from natural sequences on all structural and linguistic metrics; SA strong-conditioning achie v es perfect phenotype transfer , a capability una vailable to unconditional baselines. Source pLDDT TM PPL P1 K/R KL ( × 10 3 ) D Stored (natural) 89 . 3 ± 3 . 2 0 . 83 ± 0 . 03 4 . 97 ± 0 . 98 0.32 0 0.63 SA (full family) 90 . 4 ± 1 . 2 0 . 84 ± 0 . 01 4 . 78 ± 0 . 64 0 . 41 ± 0 . 01 6 . 9 ± 0 . 1 0.56 SA (strong binders) 90 . 7 ± 1 . 1 0 . 83 ± 0 . 01 4 . 72 ± 0 . 60 1 . 00 ± 0 . 00 19 . 5 ± 0 . 4 0.50 SA (weak binders) 91 . 0 ± 1 . 2 0 . 85 ± 0 . 01 4 . 73 ± 0 . 72 0 . 00 ± 0 . 00 12 . 4 ± 0 . 3 0.53 HMM emit (HMMER3) 60 . 4 ± 13 . 1 0 . 60 ± 0 . 10 16 . 1 ± 4 . 2 0 . 15 ± 0 . 03 29 . 7 ± 11 . 3 0.56 Bootstrap (resample) — — — 0 . 30 ± 0 . 04 1 . 0 ± 0 . 3 0.56 14 T able 4: AlphaF old2-ptm cr oss-validation of structural quality . Independent structure predictions via ColabFold (AlphaF old2-ptm, 1 model, 3 recycles, MMseqs2 MSA) for 50 sequences per source. pLDDT : mean predicted confidence ( 0 – 100 ). TM: TM-score against experimental reference (1BPI chain A for Kunitz, 1OMG chain A for ω -conotoxin). AF2 confirms the ESMFold results: SA- generated Kunitz sequences match or exceed stored sequences on both metrics; conotoxin TM-scores are lower overall, consistent with the 26-residue peptide length, b ut SA strong-seeded sequences achiev e the highest pLDDT and TM-scores in both families. Family Source n pLDDT TM-score Kunitz Stored (natural) 50 93 . 5 ± 4 . 0 0 . 83 ± 0 . 03 SA (strong binders) 50 94 . 6 ± 1 . 4 0 . 84 ± 0 . 01 SA (full family) 50 95 . 0 ± 1 . 3 0 . 85 ± 0 . 01 ω -Conotoxin Stored (natural) 50 67 . 9 ± 9 . 8 0 . 35 ± 0 . 11 SA (strong binders) 50 78 . 5 ± 4 . 1 0 . 47 ± 0 . 02 SA (full family) 50 73 . 9 ± 10 . 4 0 . 39 ± 0 . 12 T able 5: ω -Conotoxin Cav2.2 pharmacophor e inheritance under curated vs. full-family seeding. T yr13%: fraction with tyrosine at the primary pharmacophore position (col. 13). K+R%: basic residue fraction (electrostatic complementarity). KL: amino acid composition div ergence. Novelty: mean cosine distance to nearest stored pattern. Strong-binder seeding amplifies T yr13 from 82.6% to 98.3% and preserves the basic-residue signature exactly , while full-family seeding dilutes both signals. Condition N T yr13 (%) K+R (%) KL Novelty Input: full family 74 33.8 9.9 0 0 Input: strong binders 23 82.6 19.7 0 0 Generated: full-seeded 1550 46.9 12.0 0.0075 0.60 Generated: strong-seeded 1550 98.3 20.1 0.0083 0.46 T able 6: SA-generated ω -conotoxin sequences preserve experimentally validated binding determinants. Published structure–acti vity relationship (SAR) data for ω -conotoxin binding to Cav2.2 identify critical residues whose mutation abolishes or substantially reduces channel block [ 13 , 14 , 21 , 22 , 15 , 23 ]. For each SAR-critical position, the table reports the fraction of se- quences that retain the wild-type residue (or a conservati ve K/R substitution for basic positions). SA strong-seeded generation preserves the primary pharmacophore (T yr13: 98.3%) and all six cysteines ( ≥ 97 . 4% ), while enriching basic residues at functionally important loop 2 positions relati ve to the input. Pos. WT Role Mutation ef fect Input SA strong SA full 13 T yr Primary pharmacophore Ala: abolishes activity 0.83 0.98 0.47 2 L ys Loop 2 stabilization Ala: 40 × loss (GVIA) 0.96 0.98 0.46 10 Arg Loop 2 binding Critical for interaction 0.70 0.82 0.35 11 Leu Loop 2 binding Critical for interaction 0.17 0.24 0.11 1 Cys Disulfide framework Required for fold 1.00 1.00 1.00 8 Cys Disulfide framework Required for fold 1.00 1.00 1.00 15 Cys Disulfide framew ork Required for fold 1.00 0.97 1.00 16 Cys Disulfide framew ork Required for fold 0.87 1.00 1.00 20 Cys Disulfide framew ork Required for fold 0.70 1.00 1.00 25 Cys Disulfide framew ork Required for fold 0.52 1.00 1.00 21 Arg Electrostatic Ala: reduced potency 0.44 0.66 0.14 4 L ys P/Q selectivity Ala: important for P/Q 0.52 0.61 0.28 15 T able 7: AlphaFold2 multimer complex pr ediction validation f or SA-generated ω -conotoxins. Complex-le vel scores for sequences docked with Cav2.2 pore domain via ColabFold in multimer mode. For each group we report mean v alues and standard de viation of pTM, iPTM, and interface pLDDT . Group n Mean pTM Mean iPTM Mean Interface pLDDT SA strong-conditioned 10 0 . 221 ± 0 . 007 0 . 105 ± 0 . 008 37 . 2 ± 5 . 4 SA full-conditioned 10 0 . 219 ± 0 . 008 0 . 104 ± 0 . 007 38 . 2 ± 4 . 0 Natural controls 5 0 . 228 ± 0 . 004 0 . 108 ± 0 . 007 40 . 7 ± 3 . 5 Figure 4: Binding-loop amino acid frequencies and generation residuals for the ω -conotoxin family (positions 9–17). Left column: Per-position amino acid frequency heatmaps for the input sequences (strong Cav2.2 binders, n = 23 , panel a; full O-superf amily , n = 74 , panel c). Right column: Residual heatmaps (generated − input) showing the change in frequency at each position– residue pair after SA generation. Panel (b): strong-seeded residuals are near-zero throughout ( | ∆ f | < 0 . 15 ), confirming that the T yr pharmacophore at position 13, the conserved Cys frame work (positions 14–16), and the variable-loop di v ersity (positions 9–12) are reproduced with high fidelity . Panel (d): full-seeded residuals are slightly larger , reflecting the broader sequence div ersity of the input. Blue: enrichment; red: depletion; white: no change. 16 Figure 5: Sequence-level analysis of SA-generated Kunitz domain sequences conditioned on strong trypsin binders. (A) Alignment of the family consensus with the five highest-confidence SA strong-binder-conditioned sequences (ranked by ESMFold pLDDT). Positions are colored by conservation in the stored MSA: highly conserved ( > 90% , red), conserved ( 70 – 90% , orange), moderate ( 50 – 70% , yellow), and v ariable ( < 50% , blue). Cysteines forming the three disulfide bonds are highlighted in gold; the P1 position is highlighted in purple. Dots indicate matches to the consensus; letters indicate substitutions. All fi v e sequences preserve e very highly conserv ed position ( 11 / 11 ), all six c ysteines ( 6 / 6 ), and carry K or R at P1, while introducing 19 – 26 substitutions concentrated at variable sites. (B) Per -position Shannon entropy for the stored MSA (gray), SA full-family generation (blue), and SA strong-binder-conditioned generation (red). The full-family entropy correlation is v ery high ( r = 0 . 988 ), confirming that SA recapitulates the position-specific conservation pattern of the natural f amily . The strong-conditioned entrop y correlation is slightly lower ( r = 0 . 932 ), with the largest de viation at the P1 position (column 25): conditioning collapses P1 div ersity to K/R, reducing entropy at this site while preserving the entrop y profile elsewhere. 17 Figure 6: Sequence-le vel analysis of SA-generated ω -conotoxin sequences conditioned on strong Cav2.2 binders. (A) Alignment of the family consensus with the five highest-confidence SA strong- binder-conditioned sequences (rank ed by ESMFold pLDDT). Positions are colored by conserv ation in the stored MSA: highly conserved ( > 90% , red), conserved ( 70 – 90% , orange), moderate ( 50 – 70% , yellow), and v ariable ( < 50% , blue). Cysteines forming the three disulfide bonds are highlighted in gold; the T yr13 pharmacophore position is highlighted in green. All fiv e sequences preserve e very highly conserved position ( 6 / 6 ), all six cysteines ( 6 / 6 ), and carry T yr at the pharmacophore position, while introducing 5 – 11 substitutions concentrated at variable sites. (B) Per -position Shannon entropy for the stored MSA (gray), SA full-seeded generation (blue), and SA strong-seeded generation (red). The full-seeded entropy correlation is near-perfect ( r = 0 . 997 ), confirming faithful recapitulation of position-specific conserv ation. The strong-seeded correlation is lo wer ( r = 0 . 745 ), reflecting the fact that conditioning on 23 strong Ca v2.2 binders collapses di versity not only at the T yr13 pharmacophore but also at co-v arying loop residues that contrib ute to channel binding specificity . 18 A N C B N C C N C D N C 50 60 70 80 90 100 pLDDT K unitz Domain - C o n o t o x i n Figure 7: SA-generated sequences f old into nati ve-like structures f or both pr otein families. For each family , the single highest TM-score SA strong-conditioned sequence (colored by per- residue pLDDT confidence) is superimposed onto the e xperimental reference structure (gray , semi- transparent) and shown in two orthogonal vie ws (front and 90 ◦ rotation) to rev eal the full three- dimensional architecture. (A) K unitz domain, front vie w . (B) Kunitz domain, 90 ◦ rotation. The SA-generated variant aligns to the BPTI crystal structure (1BPI chain A) with TM = 0 . 94 , reproducing the double-loop architecture, three-stranded antiparallel β -sheet, and α -helical turn; pLDDT > 90 throughout (deep blue) indicates high-confidence prediction. (C) ω -Conotoxin, front view . (D) ω - Conotoxin, 90 ◦ rotation. The SA-generated v ariant aligns to the MVIIA NMR structure (1OMG chain A) with TM = 0 . 53 , consistent with the small peptide length (26 residues) where TM-score normalization is noisy; the cystine-knot topology and loop architecture are preserved, with pLDDT ∼ 80 (cyan/green) reflecting the intrinsic flexibility of the inter -cysteine loops. Colorbar: pLDDT confidence scale (red = 50 , blue = 100 ). Structures predicted by ESMFold [ 5 ]; alignment by TM-align [ 20 ]. 19 SI A ppendix S1 Sequence Encoding, PCA Projection, and Multiplicity Conditioning Stochastic attention (SA) operates on continuous vectors on the unit sphere, while protein sequences are discrete objects ov er a 20-letter amino acid alphabet. Bridging the two requires three transforma- tions (one-hot encoding, PCA dimensionality reduction, and unit-norm projection), and the rev erse path (in verse PCA followed by ar gmax decoding) recovers discrete sequences from continuous samples. Here we describe the full encoding–decoding pipeline, deriv e the multiplicity-weighted conditioning formulas used in the main text, and discuss ho w the multiplicity vector r interacts with the PCA representation. Giv en a cleaned alignment of K sequences, each of length L amino acids over the standard 20- letter alphabet A = { A , R , N , . . . , V } , we encoded each sequence as a binary vector in R 20 L by concatenating per-position indicator v ectors. F or sequence k ( k = 1 , . . . , K ) with amino acid a ( k ) ℓ at alignment position ℓ ( ℓ = 1 , . . . , L ): x k = e a ( k ) 1 , e a ( k ) 2 , . . . , e a ( k ) L ⊤ ∈ { 0 , 1 } 20 L , (8) where e a ∈ { 0 , 1 } 20 is the standard basis vector for amino acid a (i.e., a one-hot indicator with a 1 in the position corresponding to a and 0 else where). The full one-hot dimensionality is d full = 20 L , which ranged from 520 ( ω -conotoxin, L =26 ) to 1,060 (Kunitz, L =53 ) across the four families. This space is over -dimensioned relativ e to the number of stored patterns K ( d full /K ranged from 7.0 to 10.7), which creates a problem for the modern Hopfield energy [ 19 ]: the similarity score e k = m ⊤ k ξ between a stored pattern m k and a query state ξ has variance 1 /d for a random query on the unit sphere S d − 1 , so when d is large all scores concentrate near zero and the in v erse temperature β must be set extremely high to dif ferentiate among patterns, making Langevin sampling inef ficient. PCA remov es this redundancy by projecting onto the d ≪ d full directions of actual v ariation in the alignment. W e computed PCA from the one-hot encoded alignment by first centering the data: ¯ x = K − 1 P K k =1 x k (the family’ s position-specific amino acid frequency profile), ˜ X = X − ¯ x1 ⊤ K , and then computing the economy SVD ˜ X = UΣV ⊤ , where U ∈ R d full × r contains the prin- cipal directions of variation, Σ = diag( σ 1 , . . . , σ r ) the singular values in decreasing order , and r = rank( ˜ X ) ≤ min( d full , K ) − 1 . W e retained the smallest dimension d such that P d j =1 σ 2 j / P r j =1 σ 2 j ≥ 0 . 95 (at least 95% of total v ariance). Across the four families, d ranged from 34 ( ω -conotoxin) to 186 (WW), yielding compression ratios of 3 . 3 × to 21 × (T able 1 ). Each sequence was then projected as z k = W ⊤ d ( x k − ¯ x ) ∈ R d , where W d = U : , 1: d is the projection matrix, and normalized to unit L 2 norm: m k = z k / ∥ z k ∥ 2 ∈ S d − 1 . This normalization ensures that similarity scores lie in [ − 1 , 1] and pre vents patterns with larger PCA norms from dominating the softmax attention. The columns m 1 , . . . , m K form the memory matrix ˆ X ∈ R d × K used throughout the paper . The Langevin sampler (Eq. 1 ) produces continuous states ξ ∈ R d , which are decoded back to amino acid sequences by inv erse PCA reconstruction ˆ x = ¯ x + W d ξ ∈ R d full followed by per-position argmax: a ℓ = arg max a ∈A ˆ x 20( ℓ − 1)+ a for ℓ = 1 , . . . , L . This decoding is deterministic and produced valid amino acids at 100% of positions across all f amilies and conditions. The multiplicity-weighted Hopfield energy (Eq. 2 ) assigns a weight r k > 0 to each stored pattern m k . F or the binary functional split used throughout this paper , we set r k = ρ for the K des designated patterns and r k = 1 for the K bg = K − K des background patterns, where ρ ≥ 1 is the multiplicity ratio. The weighted score function (Proposition 1 ) adds log ρ to the pre-softmax logits of designated patterns and log 1 = 0 to background patterns. T o deriv e the effect of ρ on the attention distribution, consider the idealized case where the query ξ has equal similarity to all stored patterns, i.e., m ⊤ k ξ = c for all k . The softmax attention weight on pattern k simplifies to a k = r k / P j r j , and the total attention weight on all designated patterns is: f eff ( ρ ) = X k ∈ des a k = K des ρ K des ρ + K bg . (9) At ρ = 1 , f eff = K des /K (the natural proportion); as ρ → ∞ , f eff → 1 . T o find the multiplicity ratio needed for a target ef fecti ve fraction f ∈ (0 , 1) , we in vert Eq. ( 9 ) by multiplying both sides by 20 the denominator and solving for ρ : f ( K des ρ + K bg ) = K des ρ, f K bg = K des ρ (1 − f ) , ρ ( f ) = f K bg K des (1 − f ) . (10) This is Eq. ( 4 ) in the main text. The formula is e xact under the equal-similarity assumption; in practice, we confirmed empirically that the mean attention weight on designated patterns tracked f eff ( ρ ) with < 0 . 3% deviation across all ρ values and all families (T able 2 ), v alidating the approximation as operationally exact. The effecti ve number of patterns under multiplicity weighting, K eff ( r ) = ( P k r k ) 2 / P k r 2 k = ( K des ρ + K bg ) 2 / ( K des ρ 2 + K bg ) , equals K when ρ = 1 and con v erges to K des as ρ → ∞ ; this quantity governs the shift in the phase transition temperature β ∗ . A key property of multiplicity-weighted conditioning is that r does not alter the PCA encoding. The memory matrix ˆ X is constructed once from the full f amily alignment and remains fix ed regardless of ρ ; the multiplicity weights enter only through the softmax logits. This separation has three consequences. First, the PCA subspace is independent of ρ : the principal directions W d , the centering vector ¯ x , and the retained dimension d are all determined by the alignment alone, so the calibration gap ∆ = f eff − f obs is identical for all ρ values on a given family . Second, ρ controls attention, not geometry: increasing ρ adds log ρ to the designated logits, shifting attention weight tow ard designated patterns, but this shift must still pass through the PCA reconstruction and ar gmax decoding to produce discrete residues, which is where the calibration g ap arises. Third, ρ shifts the phase transition: the logit biases pre-concentrate the attention distribution at β = 0 , reducing the weighted entrop y to H r (0) = log K eff ( r ) < log K , so reaching the retriev al-phase inflection requires a higher in v erse temperature β ∗ ( ρ ) ≥ β ∗ (1) . W e observed this empirically: β ∗ increased from 4.4 ( ρ = 1 , K eff = 99 ) to 9.3 ( ρ = 1 , 000 , K eff = 32 . 1 ) on the Kunitz domain (Fig. 2 ). By contrast, hard curation (where the memory matrix is b uilt from the designated subset only) does change the PCA: the centering vector , principal directions, and retained dimension are all computed from K des sequences, capturing subset-specific v ariation that may be orthogonal to the leading components of the full-family PCA. This is why hard curation achie ved 100% phenotype transfer re gardless of the Fisher separation index S , while multiplicity weighting was limited by the full-family PCA geometry . S2 Supplementary T ables and Figures T ables S1 – S5 report selected values from the ρ sweep on each of the fiv e Pfam benchmark f amilies. f eff is the effecti ve designated fraction computed from the multiplicity vector; f obs is the hard-decoded marker fraction a veraged o ver 930 generated sequences; ¯ a des is the mean softmax attention weight on designated patterns; and D is the mean pairwise div ersity . T able S6 reports phenotype fraction and di versity as a function of β /β ∗ for three fixed ρ values on the Kunitz domain. Figure S1 shows the joint distrib ution of ESMF old pLDDT and TM-score for all 250 predicted Kunitz structures: SA-generated sequences cluster tightly in the high-pLDDT , high-TM-score re gion while HMM- emitted sequences scatter broadly across lower v alues. The entropy curves for the full ρ sweep ( ρ ∈ { 1 , 2 , 5 , 10 , 20 , 50 , 100 , 200 , 500 , 1000 } ) on the Kunitz family produce a monotonically rightw ard- shifting f amily of sigmoid curves with β ∗ ( ρ ) increasing from 4.35 ( ρ = 1 ) to 9.26 ( ρ = 1000 ), consistent with β ∗ ∝ log K eff predicted by the mean-field argument in Section 4.1 (Fig. 2 ). T able S1: Kunitz domain (PF00014) ρ sweep. K = 99 , K des = 32 . ρ f eff f obs ¯ a des D 1 0.323 0.406 0.324 0.563 5 0.705 0.481 0.712 0.556 10 0.827 0.539 0.832 0.547 50 0.962 0.598 0.963 0.526 100 0.980 0.615 0.981 0.520 200 0.990 0.623 0.991 0.519 500 0.996 0.631 0.997 0.515 1000 0.998 0.634 0.998 0.514 21 T able S2: SH3 domain (PF00018) ρ sweep. K = 55 , K des = 33 . ρ f eff f obs ¯ a des D 1 0.600 0.852 0.614 0.580 5 0.882 0.897 0.890 0.575 10 0.938 0.906 0.941 0.575 50 0.983 0.965 0.983 0.567 100 0.991 0.977 0.991 0.559 200 0.996 0.984 0.996 0.554 500 0.999 0.989 0.999 0.513 1000 0.999 0.992 0.999 0.510 Docking-statistics note For the ω -conotoxin docking validation (T able 7 ), group-level comparisons (SA strong-conditioned, SA full-conditioned, and natural ω -conotoxin controls) were ev aluated using exact two-sided per- mutation tests on mean complex-le vel scores (iPTM, pTM, confidence, and interface-pLDDT), with sample sizes n SA , strong = 10 , n SA , full = 10 , and n control = 5 . No pairwise contrast reached significance at α = 0 . 05 (all p > 0 . 05 ; smallest observed p = 0 . 0726 for confidence, SA full vs controls). 22 T able S3: WW domain (PF00397) ρ sweep. K = 420 , K des = 69 . ρ f eff f obs ¯ a des D 1 0.164 0.131 0.152 0.690 5 0.496 0.184 0.487 0.689 10 0.663 0.226 0.661 0.676 50 0.901 0.277 0.901 0.657 100 0.947 0.303 0.947 0.649 200 0.973 0.327 0.974 0.643 500 0.990 0.360 0.991 0.642 1000 0.995 0.371 0.995 0.642 T able S4: Homeobox domain (PF00046) ρ sweep. K = 136 , K des = 102 . ρ f eff f obs ¯ a des D 1 0.750 0.935 0.758 0.525 5 0.938 0.954 0.939 0.521 10 0.968 0.955 0.969 0.522 50 0.993 0.957 0.994 0.521 100 0.997 0.957 0.997 0.521 200 0.998 0.957 0.998 0.521 500 0.999 0.957 0.999 0.521 1000 1.000 0.957 1.000 0.521 T able S5: Forkhead domain (PF00250) ρ sweep. K = 246 , K des = 122 . ρ f eff f obs ¯ a des D 1 0.496 0.556 0.490 0.480 5 0.831 0.610 0.826 0.464 10 0.908 0.638 0.904 0.447 50 0.980 0.670 0.979 0.431 100 0.990 0.710 0.988 0.417 200 0.995 0.711 0.994 0.416 500 0.998 0.730 0.998 0.399 1000 0.999 0.730 0.999 0.398 T able S6: β sweep on Kunitz domain ( K = 99 , K des = 32 ). ρ β/β ∗ f obs D 10 0.50 0.471 0.581 10 1.00 0.539 0.547 10 1.50 0.573 0.525 10 2.00 0.597 0.508 10 3.00 0.613 0.484 50 0.50 0.506 0.573 50 1.00 0.582 0.537 50 1.50 0.631 0.514 50 2.00 0.653 0.499 50 3.00 0.700 0.469 200 0.50 0.535 0.564 200 1.00 0.613 0.527 200 1.50 0.648 0.503 200 2.00 0.677 0.488 200 3.00 0.706 0.463 23 T able S7: Pairwise exact permutation p-v alues for ω -conotoxin docking comparisons (all H 0 : equal means). Metric SA strong vs SA full SA str ong vs controls SA full vs contr ols iPTM 1.0000 0.7502 0.4752 pTM 0.7873 0.1285 0.1092 Confidence 0.6560 0.1245 0.0726 Interface pLDDT 0.6556 0.2238 0.2934 Figure S1: pLDDT and TM-score ar e positiv ely correlated; SA sequences cluster in the high- quality region. Each point represents one predicted structure (50 per source). Stored (gray) and SA-generated (blue, red, green) sequences cluster tightly abo ve pLDDT > 85 and TM-score > 0 . 8 (upper right quadrant). HMM sequences (orange) scatter broadly , with many falling belo w the pLDDT > 70 confidence threshold (vertical dashed line) and the TM-score > 0 . 5 same-fold threshold (horizontal dashed line). 24
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment