Differential gene expression analysis via two-component mixture models with a semiparametric skew-normal scale mixture alternative
Two-component mixture models are particularly useful for identifying differentially expressed genes, but their performance can deteriorate markedly when the alternative distribution departs from parametric assumptions or symmetry. We propose a semipa…
Authors: Sangkon Oh, Geoffrey J. McLachlan
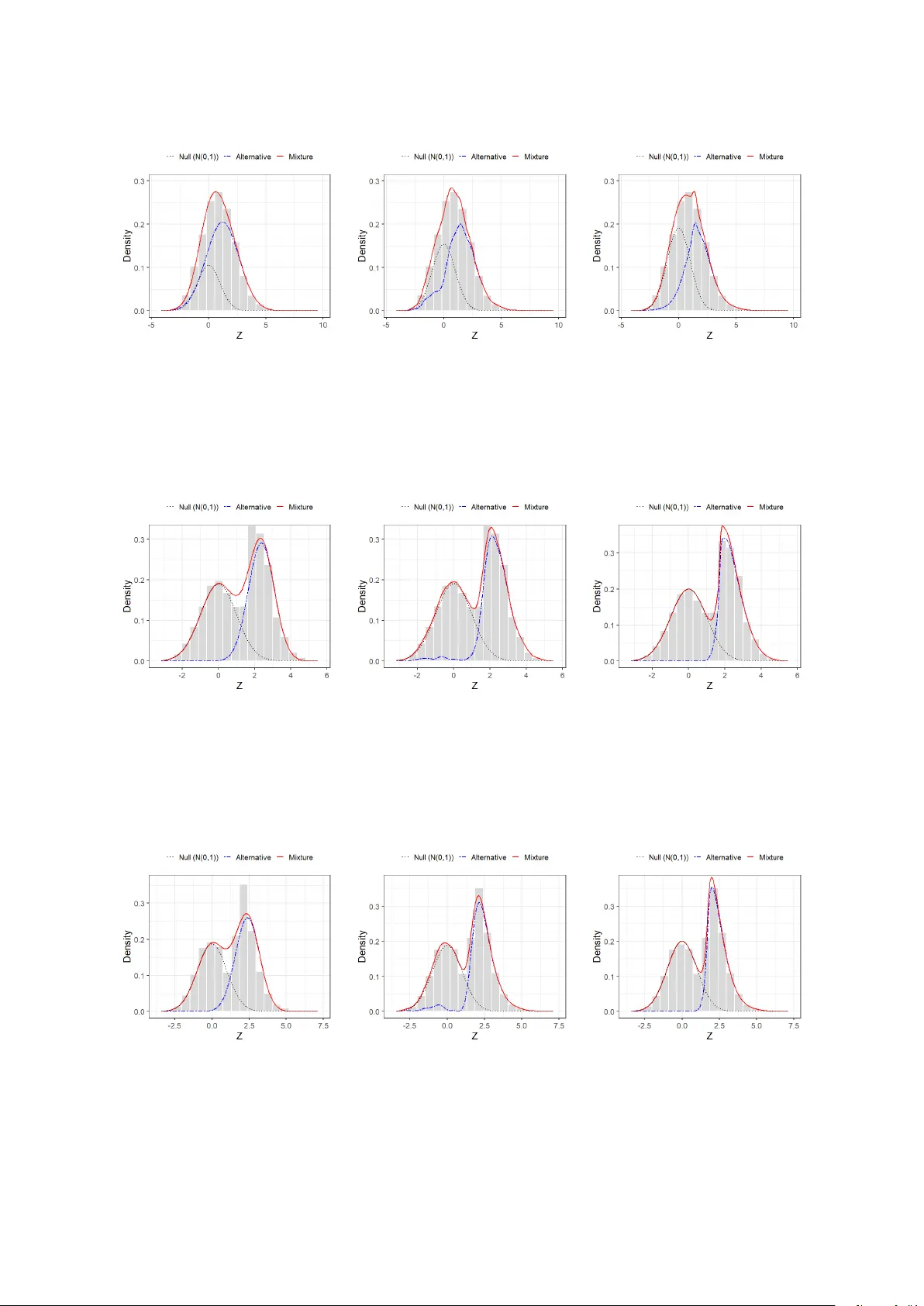
Differen tial gene expression analysis via t w o-comp onen t mixture mo dels with a semiparametric sk ew-normal scale mixture alternativ e Sangk on Oh 1 and Geoffrey J. McLac hlan 2* 1 Departmen t of Statistics and Data Science, Puky ong National Univ ersity 1 E-mail: ohsangk on@pkn u.ac.kr 2* Sc ho ol of Mathematics and Ph ysics, Univ ersit y of Queensland 2* E-mail: g.mclac hlan@uq.edu.au Abstract Tw o–comp onen t mixture mo dels are particularly useful for identifying differ- en tially expressed genes, but their p erformance can deteriorate markedly when the alternative distribution departs from parametric assumptions or symmetry . W e prop ose a semiparametric mixture mo del in whic h the n ull comp onen t is stan- dard normal and the alternativ e follo ws a skew–normal scale mixture with an un- sp ecified scale mixing distribution. This formulation accommo dates sk ewness and hea vy tails, providing a flexible and computationally tractable to ol for differen tial gene–expression analysis without restrictiv e distributional assumptions. W e estab- lish identifiabilit y and consistency of the mo del and develop an efficient estimation algorithm that incorp orates nonparametric maximum likelihoo d estimation of the scale distribution. Numerical studies sho w notable impro vemen ts o v er existing para- metric and nonparametric approac hes for mo deling the alternativ e distribution, and applications to colon cancer and leukemia datasets demonstrate reduced false dis- co very and false negativ e rates. Keywor ds: Differen tial gene expression, Microarra y data, Nonparametric maxim um like- liho od estimation, Semiparametric mixture mo dels 1 In tro duction Gene expression analysis pla ys a critical role in elucidating the biological mechanisms under- lying complex diseases, iden tifying key biomarker genes, and facilitating the developmen t of diagnostic, preven tive, and therap eutic strategies. In this context, the identification of differ- en tially expressed genes has b ecome a cornerstone of mo dern genomics, particularly with the adv ent of high-throughput technologies such as microarra ys and RNA sequencing. These plat- forms enable the simultaneous quan tification of expression lev els for tens of thousands of genes, but they also introduce substantial s tatistical c hallenges due to the scale and m ultiplicity of h yp othesis testing inv olv ed. 1 T o address this issue, a wide arra y of statistical metho ds has b een developed, among which p enalized v ariable-selection techniques ha ve gained significant prominence. Since the in tro duc- tion of the least absolute shrink age and selection op erator (LASSO) by Tibshirani (1996), nu- merous extensions and domain-sp ecific applications ha v e b een prop osed for gene expression analysis, including those by Ghosh and Chinnaiy an (2005), Zhang et al. (2006), Ma et al. (2007), and Guo et al. (2017). These approac hes are effectiv e in managing high-dimensional data by promoting sparsit y and identifying informative gene subsets. How ev er, they also exhibit several limitations, as the selected features may not generalize reliably across mo dels, and the rep eated mo del fitting required in ultra-high-dimensional settings can lead to substan tial computational burdens. Alternativ ely , motiv ated in part by the t wo-component Gaussian mixture model for observ ed log ratios prop osed b y Lee et al. (2000), Efron et al. (2001) and Efron and Tibshirani (2002) in tro duced the lo cal false disco very rate (FDR), a refinement of the classical tail-area–based FDR originally developed b y Benjamini and Ho c hberg (1995). Subsequently , Efron (2004) and Efron (2005) proposed transforming observ ed statistics into z -scores to facilitate the application of lo cal FDR metho ds for identifying differentially expressed genes. Under the assumption that gene expression levels are normally distributed and identically distributed for infected and un- infected cells, the z -scores corresp onding to non-differentially expressed genes follow a standard normal distribution. Building up on these ideas, McLachlan et al. (2006) introduced a tw o- comp onent Gaussian mixture mo del that incorp orates b oth z -score transformation and lo cal FDR estimation. In this framew ork, the z -score distribution is mo deled as g ( z ) = π f 0 ( z ) + (1 − π ) f 1 ( z ) , (1) where f 0 ( z ) denotes the n ull distribution corresp onding to non-differen tially expressed genes, f 1 ( z ) represents the alternativ e distribution for differen tially expressed genes, and π is the prop ortion of genes that are not differentially expressed. The null distribution f 0 ( z ) is typically kno wn or assumed to b e standard normal, while the alternativ e distribution f 1 ( z ) is also modeled parametrically as a normal distribution. This form ulation enables probabilistic inference at the gene level and provides a coherent basis for ranking genes according to their p osterior probabilities of b eing differentially expressed. Despite their computational conv enience, parametric assumptions for the alternativ e distri- bution f 1 ( z ) in (1) hav e inherent limitations, particularly when the true distribution deviates from the assumed form. T o address this issue, Bordes et al. (2006) prop osed a semiparametric t wo-component mixture mo del in whic h the null comp onen t is sp ecified, while the alterna- tiv e component is left completely unspecified but constrained to be symmetric. Their w ork established k ey identifiabilit y results under symmetry assumptions and dev elop ed consistent estimation pro cedures based on moment equations and symmetrization techniques. Subsequent studies, including Ma and Y ao (2015), P ommeret and V andek erkho ve (2019), and Milhaud et al. (2022), further extended this semiparametric framew ork. These contributions enhanced mo d- eling flexibility by relaxing rigid parametric assumptions on the alternative distribution while main taining iden tifiability . Ho wev er, the assumption that the alternativ e density is symmetric can still b e o verly re- strictiv e in practice. T o o v ercome this limitation, w e prop ose a new semiparametric mixture mo deling framew ork in which the n ull distribution is parametrically sp ecified, while the al- ternativ e distribution is mo deled using a semiparametric skew–normal sc ale mixture (SNSM) distribution. This semiparametric SNSM formulation pro vides enhanced flexibilit y by accom- mo dating a broad class of distributions—including b oth symmetric and asymmetric unimo dal forms—without requiring a mo del selection pro cedure. Estimation pro ceeds via a semipara- 2 metric maxim um likelihoo d metho d, where the parameters of the skew-normal comp onent are treated parametrically , and the mixing distribution is estimated nonparametrically using the nonparametric maxim um lik eliho o d estimator (NPMLE) framework of Lindsay (1995). Al- though not all distributions strictly fall within the class of skew-normal scale mixtures, the flexibilit y of the prop osed semiparametric SNSM mo del is exp ected to mitigate p otential issues arising from mo del missp ecification. Recent studies, including Lee and Seo (2024), Seo and Oh (2024), and Oh et al. (2026), hav e successfully applied the semiparametric SNSM framework in v arious con texts—suc h as finite mixture mo dels, robust regression, and accelerated failure time mo deling—demonstrating its effectiv eness in capturing diverse distributional characteris- tics without imp osing rigid parametric assumptions. The remainder of this paper is organized as follows. Section 2 reviews the prepro cessing steps used to obtain z -scores from microarray data, summarizes existing tw o-comp onent mixture ap- proac hes for differential expression analysis, and introduces the semiparametric SNSM distribu- tion. Section 3 presents the proposed semiparametric tw o-comp onent mixture model—featuring a semiparametric SNSM alternativ e that accommo dates b oth symmetric and asymmetric dis- tributions—establishes iden tifiability and consistency , and details the estimation pro cedure. Section 4 rep orts a comprehensive simulation study comparing the prop osed metho d with ex- isting comp etitors. Section 5 applies the metho dology to real gene-expression datasets. Finally , Section 6 concludes with a discussion of future researc h directions. 2 Literature Review 2.1 Data Prepro cessing Let X = ( x ij ) N × M denote the gene expression matrix, where N is the n umber of genes and M is the num b er of tissue samples. Each ro w x i · = ( x i 1 , . . . , x iM ) represents the expression profile of gene i , and eac h sample j b elongs to one of G exp erimental groups, lab eled by g j ∈ { 1 , . . . , G } . F or each gene i , w e test the null hypothesis that its mean expression lev el do es not differ across groups. When G = 2, the equality of means can b e assessed using the p o oled t w o-sample t -statistic: t i = ¯ x i 1 − ¯ x i 2 s ip p 1 /m 1 + 1 /m 2 , s 2 ip = ( m 1 − 1) s 2 i 1 + ( m 2 − 1) s 2 i 2 m 1 + m 2 − 2 , where ¯ x ig and s 2 ig denote the sample mean and v ariance of gene i in group g ( g = 1 , 2), and m g is the num b er of samples in group g (so that m 1 + m 2 = M ). Under the null h yp othesis, t i follo ws a t -distribution with ν = m 1 + m 2 − 2 degrees of freedom. F or multi-group settings ( G > 2), the one-w a y ANOV A F -statistic is used analogously to test for equalit y of group means. As the n um b er of h yp othesis tests increases, the risk of t yp e I errors rises sharply . Therefore, it is essential to perform simultaneous inference that accounts for m ultiple testing. Efron (2004), Efron (2005), and McLac hlan et al. (2006) prop osed transforming eac h observed test statistic t i in to a standard normal v ariable Z i to enable unified mo deling across all genes. Let F 0 ( · ) denote the cumulativ e distribution function (CDF) of t i under the null hypothesis. Based on t i , McLac hlan et al. (2006) prop osed computing the tw o-sided p -v alue as P i = 1 − F 0 ( t i ) + F 0 ( − t i ) , (2) whic h is then transformed into a one-sided upp er-tail z -score via the probit (Gaussian quantile) transformation: z i = Φ − 1 (1 − P i ) , (3) 3 where Φ − 1 ( · ) denotes the quantile function of the standard normal distribution. Under the as- sumption that gene expression lev els are normally and identically distributed across groups, the z -scores corresp onding to non-differentially expressed genes follow a standard normal distribu- tion. That is, under the true n ull h yp othesis, z i ∼ N (0 , 1), while larger p ositive v alues of z i indicate stronger evidence of differential expression for gene i . 2.2 Tw o-Comp onen t Mixture Mo del The transformed z -scores provide a standardized scale for assessing statistical evidence of dif- feren tial expression. T o mov e b ey ond con v entional thresholding of p -v alues or z -scores, Efron et al. (2001) and Efron and Tibshirani (2002) prop osed mo deling the empirical distribution of test statistics using a t w o-comp onen t mixture mo del as in (1). This framew ork enables the com- putation of p osterior probabilities, such as the local FDR, defined as the conditional probability that a gene is null given its observed z -score: γ ( z ) = π f 0 ( z ) g ( z ) , (4) where g ( z ) is the marginal densit y of z , f 0 ( z ) is the n ull density (typically standard normal), and π is the prior probabilit y that a gene is not differentially expressed. A widely used sp ecification for g ( z ) is the tw o-comp onen t Gaussian mixture mo del prop osed b y McLac hlan et al. (2006): g ( z ) = π ϕ ( z ; 0 , 1) + (1 − π ) ϕ ( z ; µ 1 , σ 2 1 ) , (5) where ϕ ( z ; µ, σ 2 ) denotes the normal density with mean µ and v ariance σ 2 , and the alternative distribution is c haracterized b y µ 1 > 0 to reflect right-tailed enrichmen t. While this Gaussian sp ecification is computationally conv enient, it imp oses strong parametric assumptions on the alternativ e distribution f 1 ( z ), which is typically unknown and may deviate from normality in real biological data. Such missp ecification can bias lo cal FDR estimation and compromise inference accuracy . T o address these limitations, Bordes et al. (2006) introduced a semiparametric tw o-comp onent mixture model in whic h the n ull distribution f 0 ( z ) is sp ecified, while the alternative distribution f 1 ( z ) is left unsp ecified but assumed to b e symmetric ab out an unknown lo cation. This sym- metry assumption is mild yet sufficient to ensure identifiabilit y of the mixture comp onen ts, and allo ws for mo deling a broader range of symmetric but non-Gaussian distributions. Estimation metho ds based on pro jection or momen t equations can be applied to consistently recov er the mixture structure without fully sp ecifying f 1 ( z ). Recen t extensions hav e further broadened the applicability of this framework. F or instance, P ommeret and V andekerkho ve (2019) and Milhaud et al. (2022) generalized the semiparametric mixture mo del to tw o-sample settings, allo wing for direct comparison b etw een tw o unknown alternativ e distributions. These dev elopments reinforce the semiparametric mixture approach as a p ow erful and interpretable alternativ e to b oth fully parametric and nonparametric mo dels, particularly in large-scale gene expression analysis where the n ull distribution is w ell understo o d but the alternative remains elusive. 2.3 Semiparametric Sk ew-Normal Scale Mixture Distribution In the semiparametric tw o-comp onent mixture mo del considered in this work, the n ull density f 0 ( z ) is well represen ted by a standard normal distribution, reflecting the theoretical b ehavior 4 of z -scores under the null h yp othesis. In contrast, the alternative density f 1 ( z ) typically de- parts from normality , exhibiting features such as skewness, heavy tails, or scale heterogeneity . T o flexibly accommo date these characteristics, we mo del f 1 ( z ) using a semiparametric SNSM distribution, whic h provides a unified framework capable of represen ting b oth symmetric and asymmetric families as well as a wide range of tail b ehaviors. The starting p oin t is the skew-normal distribution introduced by Azzalini (1985). F or a lo cation parameter µ , scale parameter σ > 0, and shap e parameter λ , its density is given by f SN ( z ; µ, λ, σ ) = 2 σ ϕ z − µ σ Φ λ z − µ σ , where ϕ ( · ) and Φ( · ) denote the standard normal probabilit y densit y and cum ulativ e distribution functions, respectively . The shap e parameter λ con trols the magnitude and direction of skewness, and the mo del reduces to the Gaussian distribution when λ = 0. T o extend this distribution tow ard heavier tails, Branco and Dey (2001) introduced the SNSM family . Let G b e a mixing distribution on (0 , ∞ ); then the SNSM density takes the form f SNSM ( z ; µ, λ, G ) = Z 2 σ ϕ z − µ σ Φ λ z − µ σ dG ( σ ) , (6) whic h reduces to the skew-normal when G is degenerate. V arious sk ewed and heavy-tailed distributions arise as special cases for sp ecific choices of G , including the skew- t , skew-slash, and skew-con taminated normal families. Moreo ver, setting λ = 0 yields Gaussian scale mixtures, whic h encompass the t -distribution, Laplace, logistic, and other w ell-kno wn hea vy-tailed mo dels (Andrews and Mallows, 1974; Efron and Olshen, 1978; W est, 1987). While many existing applications imp ose a parametric form on G in (6), doing so risks mo del missp ecification if the true distribution of scales deviates from the chosen parametric family . Motiv ated by this, we treat G as an infinite-dimensional parameter and estimate it nonparametrically . Let z 1 , . . . , z N b e indep enden t observ ations from the SNSM distribution. F or fixed ( µ, λ ), the log-lik eliho o d function is ˜ ℓ N ( G ) = N X i =1 log f SNSM ( z i ; µ, λ, G ) . Because G ranges ov er all probabilit y measures on (0 , ∞ ), maximizing ˜ ℓ N ( G ) requires specialized to ols from nonparametric mixture theory . F ollowing the w ork of Lindsay (1995), one charac- terizes the NPMLE via directional deriv atives. The directional deriv ativ e of ˜ ℓ N at a candidate mixing distribution ˆ G in the direction of a p oin t mass H σ is D ˆ G ( σ ) = lim α → 0 ˜ ℓ N n (1 − α ) ˆ G + αH σ o − ˜ ℓ N ( ˆ G ) α . The NPMLE ˆ G satisfies the necessary and sufficient optimality conditions D ˆ G ( σ ) ≤ 0 for all σ > 0 , D ˆ G ( σ ∗ ) = 0 for all σ ∗ ∈ S ( ˆ G ) , (7) where S ( ˆ G ) denotes the supp ort of ˆ G . Mo dels based on the NPMLE framework hav e b een successfully applied in a wide range of settings, including linear and robust regression, heteroscedastic mo deling, surviv al analysis, and finite mixture mo deling (Seo and Lee, 2015; Xiang et al., 2016; Seo et al., 2017; Seo and Kang, 2023; Seo and Oh, 2024; Lee and Seo, 2024; Oh and Seo, 2024; Park and Seo, 2025; Oh et al., 2026). By adapting automatically to unknown distributional features without imp osing restric- tiv e parametric assumptions, these mo dels offer a flexible and robust approach to statistical mo deling across diverse applications. 5 3 Prop osed Metho d 3.1 Mo del Setup T o flexibly accommo date a broad class of alternative distributions in (1), including those exhibit- ing asymmetry or heavy-tailed b ehavior, we prop ose a semiparametric t w o-comp onen t mixture mo del. The null comp onen t is mo deled b y a standard normal density , whereas the alternativ e comp onen t is represented by a semiparametric SNSM, which provides a unified mechanism for capturing sk ewness and scale heterogeneity . Let Z denote the z -score obtained through the transformation in (3). W e assume that the densit y of Z admits the mixture represen tation as f ( z ) = π ϕ ( z ; 0 , 1) + (1 − π ) f SNSM ( z ; µ, λ, G ) , (8) where • ϕ ( z ; 0 , 1) is the standard normal density defining the n ull comp onen t; • π ∈ (0 , 1) is the prior prop ortion of null genes; • f SNSM ( z ; µ, λ, G ) denotes the SNSM alternative density; • µ > 0 is the lo cation shift under the alternative; • λ ≥ 0 is the sk ewness parameter; • G is an unsp ecified mixing distribution ov er scale parameters σ > 0. Mo del (8) generalizes the classical Gaussian mixture mo del in (5) b y replacing its para- metric normal alternative with a semiparametric SNSM distribution. This mo dification p ermits the alternativ e density to exhibit a wide range of shap es—including symmetric, sk ewed, light- tailed, and heavy-tailed b ehaviors—while retaining a parsimonious and in terpretable framew ork through the finite-dimensional parameters ( µ, λ ) and the nonparametric scale mixing distribu- tion G . In this setting, b ecause non-null genes tend to produce p ositive z -scores, imp osing µ > 0 and λ ≥ 0 ensures that the alternative comp onen t appropriately reflects the exp ected righ t-tail w eight and prev ents sign-reversal ambiguit y . The standard normal distribution b elongs to the SNSM family only in the degenerate case where µ = 0, λ = 0, and G = H 1 . Under the restriction µ > 0, the SNSM family therefore excludes the standard normal density . Consequently , the null Gaussian component and the alter- nativ e SNSM comp onent are structurally distinct, and the former cannot b e represented within the latter. This structural distinction suggests that the null comp onen t should b e uniquely determined in the mixture representation. The identifiabilit y of mo del (8) is not immediate due to the presence of the infinite-dimensional mixing distribution G . T o address this issue, w e impose a set of regularity conditions that ensure a clear separation b et w een the null and alternative comp onents and preven t degeneracy in the scale mixture represen tation. In particular, Assumption (A1) establishes linear indep endence b et w een the null Gaussian density and the SNSM alternativ e class. Assumptions (A2)–(A3) imp ose mild regularity conditions on the mixing distribution G , ensuring that the scale param- eter remains b ounded aw ay from zero and that the lik eliho o d function is w ell-defined. These latter conditions are primarily required for establishing consistency of the maximum likelihoo d estimator and for guaranteeing the well-posedness of the estimation problem. Assumptions. 6 (A1) The null Gaussian density ϕ ( · ; 0 , 1) is linearly indep enden t of the non-null SNSM class F + SNSM = { f SNSM ( · ; µ, λ, G ) : µ > 0 , λ ≥ 0 , G ∈ G } . That is, if a 0 ϕ ( · ; 0 , 1) + k X j =1 a j f j ( · ) = 0 a.e. , for some k ∈ N , co efficien ts a 0 , . . . , a k ∈ R , and functions f j ∈ F + SNSM , then a 0 = a 1 = · · · = a k = 0 . (A2) The supp ort of G is con tained in [ ℓ, ∞ ) for some constant ℓ > 0. (A3) The mixing distribution G satisfies Z ∞ ℓ log σ dG ( σ ) < ∞ . The following theorem establishes identifiabilit y of the parameters ( π , µ, λ, G ) under the separation condition in Assumption (A1). Theorem 1. Supp ose Assumption (A1) holds. L et µ, ˜ µ > 0 , λ, ˜ λ ≥ 0 , and G, ˜ G b e pr ob ability me asur es on (0 , ∞ ) . If π ϕ ( z ; 0 , 1) + (1 − π ) f SNSM ( z ; µ, λ, G ) = ˜ π ϕ ( z ; 0 , 1) + (1 − ˜ π ) f SNSM ( z ; ˜ µ, ˜ λ, ˜ G ) for almost al l z ∈ R , then ( π , µ, λ, G ) = ( ˜ π , ˜ µ, ˜ λ, ˜ G ) . Pr o of. A pro of is giv en in App endix A. Remark 1. When λ = 0 and G is de gener ate at a fixe d sc ale value, mo del (8) r e duc es to the standar d two-c omp onent Gaussian mixtur e mo del with distinct me ans, as given in (5) . Thus, The or em 1 enc omp asses the classic al two-c omp onent Gaussian mixtur e as a sp e cial c ase. Remark 2. Be c ause the z -sc or es in (3) assign lar ger p ositive values to str onger evidenc e against the nul l, we r estrict attention to the right-taile d setting with µ > 0 . Mor e gener al ly, left-taile d alternatives c an also b e ac c ommo date d by al lowing µ < 0 and aligning the dir e ction of skewness thr ough the c onstr aint λ ≥ 0 when µ > 0 and λ ≤ 0 when µ < 0 . The general consistency of maximum lik eliho o d estimators in semiparametric mixture mo d- els w as demonstrated by Kiefer and W olfo witz (1956). By verifying their regularity conditions for mo del (8), we obtain the following consistency result for the estimators of ( π , µ, λ, G ). Theorem 2. Supp ose that Assumptions (A1) – (A3) hold. Then the MLE ( ˆ π , ˆ µ, ˆ λ, ˆ G ) ⊤ is c on- sistent for ( π , µ, λ, G ) ⊤ . Pr o of. A pro of is giv en in the App endix B. 7 3.2 Exp ectation–Conditional Maximization Algorithm Let z i ∈ R denote the z -score for gene i , for i = 1 , . . . , N . The observ ed log-lik eliho o d corre- sp onding to the tw o-comp onent mixture mo del in (8) is ℓ ( θ , G ) = N X i =1 log n π ϕ ( z i ; 0 , 1) + (1 − π ) f SNSM ( z i ; µ, λ, G ) o , (9) where θ = ( π , µ, λ ). Maximizing (9) is challenging due to the infinite-dimensional parameter G . W e therefore adopt the exp ectation–conditional maximization (ECM) algorithm of Meng and Rubin (1993). In tro duce the latent v ariable γ ∈ { 0 , 1 } , where γ = 1 if Z arises from the null comp onent and γ = 0 otherwise. The complete log-likelihoo d is then ℓ c ( θ , G ) = N X i =1 γ i log { π ϕ ( z i ; 0 , 1) } + N X i =1 (1 − γ i ) log { (1 − π ) f SNSM ( z i ; µ, λ, G ) } . (10) E-step. Given current estimates ( θ ( t ) , G ( t ) ), the conditional exp ectation of (10) is Q ( θ , G | θ ( t ) , G ( t ) ) = E h ℓ c ( θ , G ) | z 1 , . . . , z N , θ ( t ) , G ( t ) i , where the p osterior probability that observ ation z i b elongs to the null comp onent is γ ( t ) i = Pr( U i = 1 | z i , θ ( t ) , G ( t ) ) = π ( t ) ϕ ( z i ; 0 , 1) π ( t ) ϕ ( z i ; 0 , 1) + (1 − π ( t ) ) Z 2 σ ϕ z i − µ ( t ) σ Φ λ ( t ) z i − µ ( t ) σ dG ( t ) ( σ ) . Substituting γ ( t ) i in to the exp ectation gives Q ( θ , G ) = N X i =1 γ ( t ) i log π + log ϕ ( z i ; 0 , 1) + N X i =1 (1 − γ ( t ) i ) log(1 − π ) + N X i =1 (1 − γ ( t ) i ) log Z 2 σ ϕ z i − µ σ Φ λ z i − µ σ dG ( σ ) . CM-step 1: Up dating G (NPMLE). Conditioning on θ ( t ) , the up date of G requires maximizing ˜ ℓ N ( G ) = N X i =1 (1 − γ ( t ) i ) log Z 2 σ ϕ z i − µ ( t ) σ Φ λ ( t ) z i − µ ( t ) σ dG ( σ ) . This is a conv ex but infinite-dimensional optimization problem. W e emplo y directional-deriv ative based algorithms suc h as the vertex direction method (B¨ ohning, 1985), v ertex exchange method (B¨ ohning, 1986), intra-simplex direction metho d (Lesp erance and Kalbfleisch, 1992), or con- strained Newton metho d (CNM; W ang, 2007). The directional deriv ativ e at G ( t ) in the direction of H σ is D G ( t ) ( σ ) = N X i =1 (1 − γ ( t ) i ) 2 σ ϕ z i − µ ( t ) σ Φ λ ( t ) z i − µ ( t ) σ Z 2 s ϕ z i − µ ( t ) s Φ λ ( t ) z i − µ ( t ) s dG ( t ) ( s ) − N X i =1 (1 − γ ( t ) i ) . Up dates pro ceed along directions with D G ( t ) ( σ ) > 0 un til the NPMLE G ( t +1) satisfies the optimalit y conditions of Lindsa y (1995). 8 CM-step 2: Up dating ( π , µ, λ ) . With G ( t +1) fixed, the mixing prop ortion is up dated as π ( t +1) = 1 N N X i =1 γ ( t ) i . The remaining parameters are up dated by solving ( µ ( t +1) , λ ( t +1) ) = arg max µ,λ N X i =1 (1 − γ ( t ) i ) log Z 2 σ ϕ z i − µ σ Φ λ z i − µ σ dG ( t +1) ( σ ) , whic h can b e efficiently p erformed using the Bro yden–Fletcher–Goldfarb–Shanno (BFGS) algo- rithm (Bro yden, 1970; Fletc her, 1970; Goldfarb, 1970; Shanno, 1970). The E-step and the tw o CM-steps are iterated until con v ergence of the observ ed log-likelihoo d. 4 Sim ulation Study W e conduct a comprehensive Mon te Carlo sim ulation study to ev aluate the p erformance of three approac hes for distinguishing differen tially expressed genes from non–differen tially expressed ones: (i) the parametric t w o-comp onen t Gaussian mixture mo del; (ii) the nonparametric semi- parametric tw o-comp onent metho d, whic h mo dels the alternative densit y nonparametrically un- der a symmetry constraint; and (iii) the prop osed semiparametric SNSM mixture mo del, whic h allo ws sk ewness and hea vy-tailed b ehavior through an unsp ecified scale mixing distribution. Throughout this section, these three approac hes are referred to as the parametric, nonparamet- ric, and semiparametric metho ds, resp ectively , reflecting the degree of assumptions placed on the alternativ e distribution. The aim of the sim ulation study is to examine how these three metho ds p erform under a broad range of alternativ e distributions differing in symmetry , skewness, and tail weigh t, and under v arying degrees of sparsity in the non-null comp onen t and different total num b ers of genes. Eac h syn thetic dataset consists of indep endent observ ations Z i ∼ π N (0 , 1) + (1 − π ) f 1 , i = 1 , . . . , N , where the null comp onent is standard normal and f 1 denotes the alternative (non-null) distri- bution. T o capture a div erse set of realistic scenarios, w e consider six representativ e c hoices for f 1 : Case I: f 1 = N ( µ, 1) (symmetric, ligh t-tailed) , Case I I: f 1 = 0 . 9 N ( µ, 1) + 0 . 1 N ( µ, 2) (symmetric, mo derately heavy-tailed) , Case I I I: f 1 = Laplace( µ, 1) (symmetric, hea vy-tailed) , Case IV: f 1 = t( µ, ν = 10) (symmetric, hea vy-tailed) , Case V: f 1 = SN( µ, 1 , λ = 5) (asymmetric) , Case VI: f 1 = Sk ew - t ( µ, 1 , λ = 5 , ν = 10) (asymmetric and heavy-tailed) . The alternative lo cation is set to µ = 1 . 645, corresp onding to the 95% quantile of the standard normal distribution, providing a mo derately separated but non-extreme signal strength on the z-scale. 9 T o in vestigate the effects of sparsit y and dimensionality , we v ary the n ull prop ortion and the total num b er of genes as π ∈ { 0 . 3 , 0 . 5 , 0 . 7 } , N ∈ { 1000 , 5000 } . Larger v alues of π corresp ond to sparser settings, with π = 0 . 7 indicating that most genes follo w the n ull distribution, while π = 0 . 3 yields a comparatively dense non-n ull setting. F or eac h combination of ( f 1 , π , N ), w e generate 200 indep endent replications to obtain stable Mon te Carlo p erformance measures. Because the true comp onent membership is known in simulation, clustering accuracy is ev aluated by classifying each observ ation as n ull or non-null according to its p osterior proba- bilit y , using the maximum a p osteriori (MAP) rule with threshold γ ( z ) = 0 . 5. P erformance is then quan tified using the Adjusted Rand Index (ARI; Hub ert and Arabie, 1985) and Adjusted Mutual Information (AMI; Vinh et al., 2010), computed across the 200 replications. Both ARI and AMI correct for chance agreemen t, with higher v alues indicating b etter alignmen t b etw een estimated and true classifications. Across all sim ulation settings, the prop osed semiparametric approach consisten tly attains the highest ARI and AMI v alues, demonstrating sup erior accuracy in recov ering the true n ull and non-n ull classifications. Under symmetric and light-tailed alternatives (Case I), the three metho ds p erform similarly , though the prop osed estimator retains a slight but systematic ad- v antage. In symmetric y et heavy-tailed settings (Cases II–IV), the parametric Gaussian mixture deteriorates substantially due to tail missp ecification, and while the nonparametric approach impro ves up on the parametric metho d, it remains clearly inferior to the prop osed estimator, whic h more effectiv ely accounts for scale heterogeneity through its flexible mixing distribution. Under asymmetric alternatives (Cases V–VI), the sup eriority of the prop osed metho d is most striking, as neither the parametric nor the symmetric nonparametric mo del can accommo date sk ewness. Increasing the sample size from N = 1000 to N = 5000 improv es the p erformance of all metho ds, but the semiparametric estimator contin ues to dominate, underscoring its robust- ness and adaptability across symmetric, asymmetric, light-tailed, and heavy-tailed alternatives. Figures 1–6 present the estimated null, alternativ e, and mixture densities obtained from the parametric, nonparametric, and prop osed semiparametric approac hes across the six simulation scenarios. In the normal setting (Case I), all three metho ds recov er the ov erall mixture shap e reasonably well. As the alternative distribution b ecomes hea vie r–tailed (Cases I I–IV), the para- metric metho d exhibits pronounced distortions in the tails, reflecting its sensitivity to missp eci- fication, while the nonparametric metho d improv es up on the parametric fit but remains limited in accommo dating tail heterogeneity . In contrast, the prop osed metho d successfully adapts to v ariations in tail weigh t and shap e b y learning the underlying scale mixing distribution. Un- der asymmetric alternativ es (Cases V–VI), the limitations of the comp eting methods become more apparent: the parametric metho d enforces symmetry and fails to capture right–sk ewed structure, and the nonparametric metho d cannot represent directional asymmetry . The pro- p osed semiparametric approach, how ever, accurately reco vers b oth skewness and heavy–tailed b eha vior, pro viding the closest match to the true alternative density across all scenarios. 5 Real Data Analysis In real gene–expression studies, the true null or non–null status of each gene is unkno wn, and therefore external clustering metrics such as ARI or AMI cannot b e computed. Instead, follo wing the empirical Ba yes framework of Efron et al. (2001) and Efron and Tibshirani (2002), w e assess p erformance through p osterior–probability–based error summaries. Under the tw o–comp onent 10 (a) P arametric metho d (b) Nonparametric method (c) Semiparametric method Figure 1: Estimated densit y from eac h metho d with one sim ulated sample of Case I when N = 5000 (a) P arametric metho d (b) Nonparametric method (c) Semiparametric method Figure 2: Estimated densit y from eac h metho d with one simulated sample of Case I I when N = 5000 (a) P arametric metho d (b) Nonparametric method (c) Semiparametric method Figure 3: Estimated densit y from each metho d with one sim ulated sample of Case I I I when N = 5000 11 (a) P arametric metho d (b) Nonparametric method (c) Semiparametric method Figure 4: Estimated densit y from each metho d with one simulated sample of Case I V when N = 5000 (a) P arametric metho d (b) Nonparametric method (c) Semiparametric method Figure 5: Estimated densit y from each metho d with one simulated sample of Case V when N = 5000 (a) P arametric metho d (b) Nonparametric method (c) Semiparametric method Figure 6: Estimated densit y from each metho d with one simulated sample of Case V I when N = 5000 12 T able 1: ARI and AMI under each simulation scenario when N = 1000 (Boldface indicates the b est p erformance in each setting) Case π P arametric metho d Nonparametric metho d Semiparametric metho d ARI AMI ARI AMI ARI AMI I 0 . 3 0.3717 0.2336 0.3649 0.2292 0 . 3757 0 . 2471 0 . 5 0.3348 0.2627 0.3144 0.2463 0 . 3363 0 . 2642 0 . 7 0.3799 0.2426 0.3522 0.2287 0 . 3901 0 . 2468 I I 0 . 3 0.2036 0.1249 0.3178 0.1977 0 . 3569 0 . 2303 0 . 5 0.1808 0.1493 0.2699 0.2132 0 . 3191 0 . 2521 0 . 7 0.3426 0.2325 0.3476 0.2253 0 . 3852 0 . 2412 I I I 0 . 3 0.3896 0.2495 0.4260 0.2845 0 . 4345 0 . 2987 0 . 5 0.3081 0.2473 0.3941 0.3095 0 . 4049 0 . 3180 0 . 7 0.3053 0.2275 0.3950 0.2671 0 . 4390 0 . 2926 I V 0 . 3 0.0035 0.0021 0.0659 0.0429 0 . 3579 0 . 2252 0 . 5 0.1661 0.1581 0.2111 0.1821 0 . 3374 0 . 2644 0 . 7 0.3778 0.2459 0.3442 0.2301 0 . 3868 0 . 2449 V 0 . 3 0.8287 0.7312 0.8534 0.7604 0 . 8805 0 . 7868 0 . 5 0.7976 0.7434 0.7427 0.6898 0 . 8312 0 . 7567 0 . 7 0.7903 0.6839 0.7467 0.6439 0 . 7964 0 . 6769 V I 0 . 3 0.7892 0.6846 0.8401 0.7420 0 . 8753 0 . 7779 0 . 5 0.7672 0.7158 0.7434 0.6882 0 . 8281 0 . 7510 0 . 7 0.7798 0.6778 0.7299 0.6291 0 . 7986 0 . 6787 mixture mo del in (1), the lo cal FDR is defined as the p osterior probability that a gene with transformed statistic z b elongs to the null comp onent, as given in (4). F or eac h gene i , we denote by ˆ γ i the estimated p osterior null probability obtained from the resp ectiv e metho d. Given a threshold c , we declare gene i to b e non–null whenever ˆ γ i ≤ c . Let N r = N X i =1 I [0 ,c ] ( ˆ γ i ) denote the total n umber of selected genes. F ollowing McLachlan et al. (2006), the estimated FDR is [ FDR( c ) = N X i =1 ˆ γ j I [0 ,c ] ( ˆ γ i ) N r , whic h estimates the exp ected prop ortion of n ull genes among the declared discov eries. Con- v ersely , the false negative rate (FNR) measures the prop ortion of truly non–null genes that fail to b e selected. Its empirical Bay es estimator is [ FNR( c ) = N X i =1 (1 − ˆ γ i ) I ( c, ∞ ) ( ˆ γ i ) N X i =1 (1 − ˆ γ i ) , 13 T able 2: ARI and AMI under each simulation scenario when N = 5000 (Boldface indicates the b est p erformance in each setting) Case π P arametric metho d Nonparametric metho d Semiparametric metho d ARI AMI ARI AMI ARI AMI I 0 . 3 0 . 3907 0.2452 0.3789 0.2380 0.3869 0 . 2541 0 . 5 0 . 3454 0 . 2666 0.3383 0.2612 0.3403 0.2650 0 . 7 0.3918 0.2456 0.3470 0.2252 0 . 3926 0 . 2471 I I 0 . 3 0.1369 0.0812 0.3503 0.2155 0 . 3640 0 . 2318 0 . 5 0.1069 0.0864 0.2956 0.2280 0 . 3263 0 . 2531 0 . 7 0.3385 0.2287 0.3585 0.2287 0 . 3820 0 . 2358 I I I 0 . 3 0.4079 0.2581 0.4273 0.2869 0 . 4328 0 . 2975 0 . 5 0.3590 0.2870 0.3742 0.2935 0 . 4115 0 . 3224 0 . 7 0.2201 0.1727 0.4256 0.2869 0 . 4403 0 . 2917 I V 0 . 3 0.0000 0.0000 0.0202 0.0164 0 . 3841 0 . 2399 0 . 5 0.1773 0.1845 0.2453 0.2164 0 . 3460 0 . 2673 0 . 7 0 . 3933 0 . 2548 0.3774 0.2462 0.3931 0.2467 V 0 . 3 0.8271 0.7287 0.8687 0.7758 0 . 8804 0 . 7846 0 . 5 0.7958 0.7402 0.7385 0.6805 0 . 8316 0 . 7551 0 . 7 0.7911 0.6836 0.7443 0.6387 0 . 7984 0 . 6773 V I 0 . 3 0.7937 0.6890 0.8600 0.7625 0 . 8766 0 . 7776 0 . 5 0.7699 0.7169 0.7740 0.7112 0 . 8298 0 . 7507 0 . 7 0.7798 0 . 6757 0.6308 0.5403 0 . 7971 0.6750 whic h a verages the p osterior probabilities of b eing non–null ov er the unselected genes. These p osterior–probability–based summaries pro vide practical and interpretable to ols for comparing the comp eting mixture–modeling approaches in real datasets, where the true null and non–n ull lab els are unobserv ed. In this context, metho ds achieving low er v alues of FDR and FNR are considered to exhibit sup erior differen tial–expression detection p erformance. 5.1 Colon Cancer Data Alon et al. (1999) obtained microarray gene–expression me asuremen ts from colon cancer and normal colon tissues using Affymetrix oligonucleotide arrays. The dataset contains expression lev els for more than 6500 genes across 40 tumor samples and 22 normal samples. F ollowing the filtering strategy used in McLachlan et al. (2002), McLachlan et al. (2006), and Pommeret and V andekerkho ve (2019), we restrict the analysis to the N = 2000 genes with the highest minimal in tensity across samples to reduce the impact of low–expression noise. Using the prepro cessing pro cedure describ ed in Section 2.1, we compute for each gene a p o oled tw o–sample t –statistic comparing tumor and normal tissues, and then transform these statistics into z –scores via the mapping in (3). Genes yielding z = −∞ whic h o ccurs when the observ ed test statistic provides essentially no evidence against the null hypothesis are excluded from downstream analysis. After this filtering step, a total of 971 genes remain. F or these trans- formed z –scores, we apply the three mixture–mo deling approaches examined in the simulation study—the parametric metho d, the nonparametric metho d, and the prop osed semiparametric metho d—to identify differentially expressed genes and to compare their b ehavior on real data. 14 Figures 7–8 summarize the results. Across all thresholds c , the prop osed semiparametric metho d yields the lo west FDR and FNR, indicating more reliable iden tification of differen- tially expressed genes. The parametric and nonparametric metho ds show differing behavior: the parametric Gaussian mixture achiev es uniformly low er FDR than the nonparametric estimator, whereas the FNR curv es exhibit a threshold-dep endent pattern. F or c < 0 . 3, the parametric metho d has smaller FNR, while for c > 0 . 3 the nonparametric metho d p erforms b etter. The estimated alternative densities in Figure 8 provide additional insigh t. The nonparamet- ric density is irregular and unstable, while the parametric mo del enforces a symmetric normal form that limits its abilit y to capture subtle departures from normalit y . The proposed semipara- metric method pro duces a smo oth and stable estimate that aligns closely with the empirical distribution, suggesting that the underlying alternativ e distribution in this dataset is neither strongly sk ew ed nor hea vy-tailed. (a) FDR (b) FNR Figure 7: Estimated FDR and FNR for the colon cancer dataset across a range of thresh- olds (a) P arametric metho d (b) Nonparametric method (c) Semiparametric method Figure 8: Estimated n ull, alternativ e, and mixture densities for the colon cancer dataset 15 5.2 Leuk emia Data Golub et al. (1999) analyzed microarray gene–expression profiles from patien ts with acute leuk emia, distinguishing Acute Lymphoblastic Leukemia (ALL) from Acute Myeloid Leukemia (AML) using Affymetrix high–densit y oligonucleotide arra ys. The dataset consists of M = 72 tissue samples (47 ALL and 25 AML) and N = 7129 genes. F ollo wing the prepro cessing pip eline of Dudoit et al. (2002), we apply filtering to remov e uninformative genes and then p erform log–transformation and column–wise and ro w–wise standardization, yielding 3731 re- tained genes. Using the pro cedure in Section 2.1, w e further restrict attention to 1853 informa- tiv e genes. F or these transformed z –scores, w e fit the parametric, nonparametric, and prop osed semiparametric mixture mo dels to compute p osterior probabilities and estimate FDR and FNR. Figures 9–10 display the results for the leukemia dataset. F or the FDR curves, all three metho ds b ehav e similarly when the threshold c is small, but clear differences emerge once c exceeds approximately 0 . 2. Bey ond this p oin t, the prop osed semiparametric metho d ac hieves the lo west FDR across the en tire range, while the parametric metho d yields slightly low er FDR than the nonparametric approach. A different pattern is observ ed for the FNR: for relativ ely large thresholds ( c > 0 . 4), the nonparametric method attains the smallest FNR, whereas for more stringent thresholds ( c < 0 . 4), the prop osed semiparametric metho d achiev es the low est FNR, outp erforming b oth comp etitors. T aken together, these results indicate that the proposed metho d provides the most fav orable balance b et ween false discov eries and false negativ es across practically relev ant choices of c . The estimated alternativ e densities in Figure 10 further clarify these differences. The non- parametric estimator sho ws substan tial v ariabilit y , particularly in the tails, indicating sensitivit y to sampling fluctuations, whereas the parametric Gaussian mixture enforces a symmetric nor- mal form that cannot accommo date the evident asymmetry and heavy–tailed b ehavior present in the data. By contrast, the prop osed semiparametric approach yields a smo oth and stable densit y estimate that successfully captures b oth sk ewness and tail hea viness, consistent with the more flexible structure of the SNSM formulation. (a) FDR (b) FNR Figure 9: Estimated FDR and FNR for the leuk emia dataset across a range of thresholds 16 (a) P arametric metho d (b) Nonparametric method (c) Semiparametric method Figure 10: Estimated n ull, alternativ e, and mixture densities for the leuk emia dataset 6 Discussion This pap er introduces a semiparametric tw o-comp onent mixture mo del for large–scale infer- ence in gene-expression studies, in whic h the null distribution is fixed as standard normal and the alternative comp onent is mo deled by a semiparametric skew-normal scale mixture with an unsp ecified scale mixing distribution. By com bining a finite–dimensional parametrization for lo cation and sk ewness with a nonparametric maxim um likelihoo d estimator for the scale dis- tribution, the prop osed mo del provides substan tially greater flexibility than fully parametric approac hes while a v oiding the instabilit y that often affects fully nonparametric metho ds. The numerical study demonstrates that the prop osed metho d ac hiev es consistently higher accuracy in separating null and non–null genes across a wide range of symmetric, sk ewed, ligh t–tailed, and heavy–tailed alternatives. When the parametric Gaussian mixture is missp ec- ified, the p erformance gains of the prop osed metho d are particularly pronounced, and it also a voids the instability inheren t in fully nonparametric estimators. Analyses of the colon can- cer and leukemia datasets further supp ort these findings: the prop osed approac h yields low er false discov ery and false negative rates across practically relev ant thresholds, and the estimated alternativ e densities exhibit mo derate deviations from normalit y that are captured without unnecessary complexity . Overall, the prop osed semiparametric mixture mo del provides a flexi- ble, robust, and computationally tractable framew ork for differential gene–expression analysis, offering impro v ed p erformance without imp osing restrictive distributional assumptions. As a direction for future w ork, it would b e of interest to relax the assumption of a stan- dard normal null distribution. Allo wing for an empirical or nonparametric null comp onen t, for example under mild symmetry constraints, within the prop osed framework could further im- pro ve robustness in identifying differential gene expression while maintaining iden tifiability and computational tractability . Such an extension woul d broaden the applicability of the prop osed mo del b eyond the strictly sp ecified marginal setting considered here. It w ould also b e v aluable to incorporate cov ariate information directly into the mixture struc- ture. In many gene-expression studies, gene-sp ecific or sample-sp ecific co v ariates may provide additional information relev an t to differen tial expression. Extending the prop osed semiparamet- ric alternativ e to co v ariate-dep endent mixture form ulations (e.g., Nguy en and McLac hlan, 2016; Oh and Seo, 2023; Oh and Seo, 2024; Chamroukhi et al., 2024; Hw ang et al., 2025) ma y further enhance flexibility and detection p ow er while preserving the stabilit y of the alternativ e den- sit y sp ecification. Suc h developmen ts would allow the framework to accommo date structured heterogeneit y b ey ond the marginal mixture setting considered here. 17 References Alon, U., Bark ai, N., Notterman, D. A., Gish, K., Ybarra, S., Mac k, D., and Levine, A. J. (1999). Broad patterns of gene expression revealed b y clustering analysis of tumor and normal colon tissues prob ed by oligonucleotide arrays. Pr o c e e dings of the National A c ademy of Scienc es , 96(12):6745–6750. Andrews, D. F. and Mallows, C. L. (1974). Scale mixtures of normal distributions. Journal of the R oyal Statistic al So ciety: Series B (Metho dolo gic al) , 36(1):99–102. Azzalini, A. (1985). A class of distributions whic h includes the normal ones. Sc andinavian journal of statistics , 12(2):171–178. Benjamini, Y. and Hoch b erg, Y. (1995). Controlling the false discov ery rate: a practical and p o w erful approach to m ultiple testing. Journal of the R oyal statistic al so ciety: series B (Metho dolo gic al) , 57(1):289–300. Bordes, L., Delmas, C., and V andekerkho v e, P . (2006). Semiparametric estimation of a t w o- comp onen t mixture mo del where one comp onent is kno wn. Sc andinavian journal of statistics , 33(4):733–752. Branco, M. D. and Dey , D. K. (2001). A general class of m ultiv ariate sk ew-elliptical distributions. Journal of Multivariate Analysis , 79(1):99–113. Bro yden, C. G. (1970). The conv ergence of a class of double-rank minimization algorithms: 2. the new algorithm. IMA journal of applie d mathematics , 6(3):222–231. B¨ ohning, D. (1985). Numerical estimation of a probability measure. Journal of statistic al planning and infer enc e , 11:57–69. B¨ ohning, D. (1986). A v ertex-exchange-method in D-optimal design theory . Metrika , 33:337– 347. Chamroukhi, F., Pham, N. T., Hoang, V. H., and McLachlan, G. J. (2024). F unctional mixtures- of-exp erts. Statistics and Computing , 34(3):98. Dudoit, S., F ridlyand, J., and Sp eed, T. P . (2002). Comparison of discrimination metho ds for the class ification of tumors using gene expression data. Journal of the A meric an statistic al asso ciation , 97(457):77–87. Efron, B. (2004). Large-scale simultaneous hypothesis testing: the choice of a null hypothesis. Journal of the Americ an Statistic al Asso ciation , 99(465):96–104. Efron, B. (2005). Lo cal false discov ery rates. Efron, B. and Olshen, R. A. (1978). How broad is the class of normal scale mixtures? The A nnals of Statistics , 6(5):1159–1164. Efron, B. and Tibshirani, R. (2002). Empirical ba yes metho ds and false discov ery rates for microarra ys. Genetic epidemiolo gy , 23(1):70–86. Efron, B., Tibshirani, R., Storey , J. D., and T usher, V. (2001). Empirical bay es analysis of a microarra y exp erimen t. Journal of the Americ an statistic al asso ciation , 96(456):1151–1160. 18 Fletc her, R. (1970). A new approac h to v ariable metric algorithms. The c omputer journal , 13(3):317–322. Ghosh, D. and Chinnaiyan, A. M. (2005). Classification and selection of biomarkers in genomic data using lasso. BioMe d R ese ar ch International , 2005(2):147–154. Goldfarb, D. (1970). A family of v ariable-metric metho ds derived b y v ariational means. Math- ematics of c omputation , 24(109):23–26. Golub, T. R., Slonim, D. K., T ama yo, P ., Huard, C., Gaasen b eek, M., Mesirov, J. P ., Coller, H., Loh, M. L., Downing, J. R., Caligiuri, M. A., et al. (1999). Molecular classification of cancer: class discov ery and class prediction by gene expression monitoring. scienc e , 286(5439):531– 537. Guo, S., Guo, D., Chen, L., and Jiang, Q. (2017). A l1-regularized feature selection metho d for lo cal dimension reduction on microarray data. Computational biolo gy and chemistry , 67:92–101. Hub ert, L. and Arabie, P . (1985). Comparing partitions. Journal of Classific ation , 2(1):193–218. Hw ang, Y., Seo, B., and Oh, S. (2025). Mixture of partially linear exp erts. Stat , 14(2):e70062. Kiefer, J. and W olfo witz, J. (1956). Consistency of the maximum likelihoo d estimator in the presence of infinitely many inciden tal parameters. The Annals of Mathematic al Statistics , 27(4):887–906. Lee, H. and Seo, B. (2024). Finite mixture of semiparametric m ultiv ariate sk ew-normal distri- butions. Communic ations in Statistics-Simulation and Computation , 53(11):5659–5679. Lee, M.-L. T., Kuo, F. C., Whitmore, G., and Sklar, J. (2000). Importance of replication in microarra y gene expression studies: statistical metho ds and evidence from rep etitive cdna h ybridizations. Pr o c e e dings of the National A c ademy of Scienc es , 97(18):9834–9839. Lesp erance, M. L. and Kalbfleisch, J. D. (1992). An algorithm for computing the nonparametric MLE of a mixing distribution. Journal of the Americ an Statistic al Asso ciation , 87:120–126. Lindsa y , B. G. (1995). Mixtur e Mo dels: The ory, Ge ometry and Applic ations . Institute of Math- ematical Statistics and American Statistical Asso ciation. Ma, S., Song, X., and Huang, J. (2007). Sup ervised group lasso with applications to microarray data analysis. BMC bioinformatics , 8(1):60. Ma, Y. and Y ao, W. (2015). Flexible estimation of a semiparametric tw o-comp onen t mixture mo del with one parametric comp onent. Ele ctr onic Journal of Statistics , 9(1):444–474. McLac hlan, G. J., Bean, R. W., and Jones, L. B.-T. (2006). A simple implementation of a normal mixture approac h to differen tial gene expression in m ulticlass microarrays. Bioinformatics , 22(13):1608–1615. McLac hlan, G. J., Bean, R. W., and Peel, D. (2002). A mixture mo del-based approach to the clustering of microarray expression data. Bioinformatics , 18(3):413–422. Meng, X.-L. and Rubin, D. B. (1993). Maxim um likelihoo d estimation via the ecm algorithm: A general framework. Biometrika , 80(2):267–278. 19 Milhaud, X., P omm eret, D., Salhi, Y., and V andekerkho ve, P . (2022). Semiparametric t w o- sample admixture comp onen ts comparison test: The symmetric case. Journal of Statistic al Planning and Infer enc e , 216:135–150. Nguy en, H. D. and McLachlan, G. J. (2016). Laplace mixture of linear exp erts. Computational Statistics & Data Analysis , 93:177–191. Oh, S., Lee, H., Kang, S., and Seo, B. (2026). Adaptive accelerated failure time mo deling with a semiparametric sk ew ed error distribution. Computational Statistics & Data Analysis , 219:108357. Oh, S. and Seo, B. (2023). Merging comp onents in linear gaussian cluster-w eighted mo dels. Journal of Classific ation , 40:25–51. Oh, S. and Seo, B. (2024). Semiparametric mixture of linear regressions with nonparametric gaussian scale mixture errors. A dvanc es in Data Analysis and Classific ation , 18(1):5–31. P ark, S.-Y. and Seo, B. (2025). P enalized maxim um likelihoo d estimation with nonparametric gaussian scale mixture errors. Computational Statistics & Data Analysis , 211:108206. P ommeret, D. and V andekerkho ve, P . (2019). Semiparametric densit y testing in the contami- nation mo del. Ele ctr onic Journal of Statistics , 13:4743–4793. Seo, B. and Kang, S. (2023). Accelerated failure time mo deling via nonparametric mixtures. Biometrics , 79(1):165–177. Seo, B. and Lee, T. (2015). A new algorithm for maxim um lik eliho o d estimation in normal scale- mixture generalized autoregressive conditional heteroskedastic mo dels. Journal of Statistic al Computation and Simulation , 85:202–215. Seo, B., Noh, J., Lee, T., and Y o on, Y. J. (2017). Adaptiv e robust regression with con tin uous gaussian scale mixture errors. Journal of the Kor e an Statistic al So ciety , 46(1):113–125. Seo, B. and Oh, S. (2024). Adaptive learning in robust linear regression with a semiparametric sk ew-normal scale mixture distribution. Stat , 13(4):e70026. Shanno, D. F. (1970). Conditioning of quasi-newton metho ds for function minimization. Math- ematics of c omputation , 24(111):647–656. Tibshirani, R. (1996). Regression shrink age and selection via the lasso. Journal of the R oyal Statistic al So ciety Series B: Statistic al Metho dolo gy , 58(1):267–288. Vinh, N. X., Epps, J., and Bailey , J. (2010). Information theoretic measures for clusterings comparison: V ariants, prop erties, normalization and correction for c hance. The Journal of Machine L e arning R ese ar ch , 11:2837–2854. W ang, Y. (2007). On fast computation of the non-parametric maxim um likelihoo d estimate of a mixing distribution. Journal of the R oyal Statistic al So ciety, Series B, Metho dolo gic al , 69:185–198. W est, M. (1987). On scale mixtures of normal distributions. Biometrika , 74(3):646–648. Xiang, S., Y ao, W., and Seo, B. (2016). Semiparametric mixture: Contin uous scale mixture approac h. Computational Statistics and Data Analysis , 103:413–425. 20 Zhang, H. H., Ahn, J., Lin, X., and Park, C. (2006). Gene selection using supp ort vector mac hines with non-con v ex p enalt y . bioinformatics , 22(1):88–95. App endix A: Pro of of Theorem 1 Assume that there exist ( ˜ π , ˜ µ, ˜ λ, ˜ G ) suc h that π ϕ ( z ; 0 , 1) + (1 − π ) f SNSM ( z ; µ, λ, G ) = ˜ π ϕ ( z ; 0 , 1) + (1 − ˜ π ) f SNSM ( z ; ˜ µ, ˜ λ, ˜ G ) . (11) Rearranging terms gives ( π − ˜ π ) ϕ ( z ; 0 , 1) = (1 − ˜ π ) f SNSM ( z ; ˜ µ, ˜ λ, ˜ G ) − (1 − π ) f SNSM ( z ; µ, λ, G ) . Since b oth f SNSM ( · ; µ, λ, G ) , f SNSM ( · ; ˜ µ, ˜ λ, ˜ G ) ∈ F + SNSM , Assumption (A1) implies that the co efficient of ϕ ( · ; 0 , 1) m ust b e zero. Hence π = ˜ π . T aking characteristic functions on b oth sides of (11), we obtain: ψ z ( t ) = π exp( − t 2 / 2) + (1 − π ) exp( iµt ) ψ G,λ ( t ) , and ˜ ψ z ( t ) = ˜ π exp( − t 2 / 2) + (1 − ˜ π ) exp( i ˜ µt ) ψ ˜ G, ˜ λ ( t ) , where i = √ − 1 and ψ G,λ ( t ) denotes the characteristic function of the zero-lo cation SNSM distribution with ( λ, G ). Since π = ˜ π , equating b oth characteristic functions yields exp( iµt ) ψ G,λ ( t ) = exp( i ˜ µt ) ψ ˜ G, ˜ λ ( t ) , ∀ t ∈ R . (12) Since ψ G,λ (0) = ψ ˜ G, ˜ λ (0) = 1 and both characteristic functions are contin uous, there exists δ > 0 suc h that ψ G,λ ( t ) = 0 and ψ ˜ G, ˜ λ ( t ) = 0 for all | t | < δ . Thus, for | t | < δ , from (12) we obtain ψ G,λ ( t ) ψ ˜ G, ˜ λ ( t ) = exp { i ( ˜ µ − µ ) t } . (13) The right-hand side of (13) is the characteristic function of a p oin t-mass distribution at ( ˜ µ − µ ), that is, a pure lo cation shift. Hence, (13) implies that if X ∼ SNSM(0 , λ, G ) and Y ∼ SNSM(0 , ˜ λ, ˜ G ), then X d = Y + ( ˜ µ − µ ). In particular, the zero-location parameterization is fixed by construction (i.e., the lo cation parameter is not absorb ed into ( λ, G )). Therefore, t wo distributions in the subfamily { SNSM(0 , λ, G ) } cannot differ by a nonzero translation. Hence, w e m ust ha ve ˜ µ − µ = 0, that is, µ = ˜ µ. Since µ = ˜ µ , it follows from (12) that ψ G,λ ( t ) = ψ ˜ G, ˜ λ ( t ) , ∀ t ∈ R . 21 W e now prov e that ψ G,λ ( t ) = ψ ˜ G, ˜ λ ( t ) implies G = ˜ G and λ = ˜ λ . Note that ψ G,λ ( t ) = Z exp − t 2 σ 2 2 dG ( σ ) + i Z exp − t 2 σ 2 2 e Γ( λ ) σ t √ 2 dG ( σ ) , ψ ˜ G, ˜ λ ( t ) = Z exp − t 2 σ 2 2 d ˜ G ( σ ) + i Z exp − t 2 σ 2 2 e Γ( ˜ λ ) σ t √ 2 ! d ˜ G ( σ ) , where Γ( λ ) = λ/ √ 1 + λ 2 and e ( z ) denotes the complementary error function: e ( z ) = − 2 i √ π Z iz 0 exp( − t 2 ) dt. Equating the real and imaginary parts of ψ G,λ ( t ) and ψ ˜ G, ˜ λ ( t ), w e obtain: Z exp − t 2 σ 2 2 dG ( σ ) = Z exp − t 2 σ 2 2 d ˜ G ( σ ) , (14) Z exp − t 2 σ 2 2 e Γ( λ ) σ t √ 2 dG ( σ ) = Z exp − t 2 σ 2 2 e Γ( ˜ λ ) σ t √ 2 ! d ˜ G ( σ ) . (15) Equation (14) implies G = ˜ G b y the uniqueness of the Laplace transform. Substituting into (15) and using the injectivit y of e ( · ), w e obtain Γ( λ ) = Γ( ˜ λ ), and hence λ = ˜ λ . Therefore, iden tifiability holds. App endix B: Pro of of Theorem 2 Let us define the following metric: d ( π , µ, λ, G ) , ( ˆ π , ˆ µ, ˆ λ, ˆ G ) = | π − ˆ π | + | µ − ˆ µ | + | λ − ˆ λ | + Z | G ( σ ) − ˆ G ( σ ) | e −| σ | dτ ( σ ) , where τ denotes the Leb esgue measure on R + . Kiefer and W olfo witz (1956) established that under fiv e assumptions—including contin uity (Assumption 2), identifiabilit y (Assumption 4), and integrabilit y (Assumption 5)—the MLE con verges in probabilit y: d ( π , µ, λ, G ) , ( ˆ π , ˆ µ, ˆ λ, ˆ G ) p → 0 . The semiparametric SNSM densit y in (8) trivially satisfies Assumptions 1, 2, and 3. Assumption 4 (iden tifiabilit y) is established in Theorem 1. Since the supp ort of G is restricted to [ ℓ, ∞ ), the densit y in (8) is uniformly b ounded. Thus, it remains to verify Assumption 5, which requires: − E [log { π ϕ ( z ) + (1 − π ) f SNSM ( z ; µ, λ, G ) } ] < ∞ . Because π ∈ (0 , 1), we hav e − log π < ∞ and − log(1 − π ) < ∞ . Moreov er, the normal and sk ew-normal densities ha v e finite first momen ts, implying − E [log ϕ ( z )] < ∞ and Z ∞ −∞ f ( z ; µ, λ, σ ) [log | z | ] + dz < ∞ . By the condition R ∞ ℓ log σ dG ( σ ) < ∞ , it follows that E [log | Z | ] + < ∞ , 22 and th us − Z ∞ −∞ log { f ( z ; µ, λ, G ) } f ( z ; µ, λ, G ) dz < ∞ , b y Lemma in Section 2 of Kiefer and W olfowitz (1956). This verifies Assumption 5 and completes the pro of. 23
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment