두 구성요소 혼합모델과 반대칭 스케일 혼합을 이용한 차등 발현 유전자 탐색
본 논문은 널(null) 성분을 표준 정규분포로, 대안(alternative) 성분을 비대칭·중량 꼬리를 허용하는 스케일 혼합 스키니멀 정규분포(SNSM)로 모델링한 반-모수(two‑component) 혼합모델을 제안한다. 모델의 식별성, 일관성 증명을 제시하고, 스케일 혼합분포를 비모수 최대우도(NPMLE)로 추정하는 효율적인 EM‑유사 알고리즘을 개발하였다. 시뮬레이션과 실제 대장암·백혈병 마이크로어레이 데이터에 적용한 결과, 기존의 대칭 …
저자: Sangkon Oh, Geoffrey J. McLachlan
**1. 연구 배경 및 필요성**
마이크로어레이·RNA‑seq와 같은 고속 유전자 발현 측정 기술은 수만 개의 유전자를 동시에 분석하게 만들었다. 이러한 대규모 가설 검정 상황에서 널 가설을 만족하는 유전자의 z‑점수는 표준 정규분포를 따른다는 가정이 일반적이며, 이를 기반으로 로컬 FDR(local false discovery rate) 등을 추정한다. 기존의 두 구성요소 가우시안 혼합모델(π·N(0,1)+(1−π)·N(µ,σ²))은 계산이 간단하지만, 실제 데이터에서 대안 성분은 종종 비대칭이며 중량 꼬리를 보여 파라메트릭 가정에 크게 위배된다. 이러한 모델 불일치는 FDR 과소/과대 추정과 검출 파워 감소를 초래한다.
**2. 기존 연구 검토**
Bordes et al. (2006)은 대안 성분을 비대칭이 아닌 대칭으로 제한한 반‑모수 혼합모델을 제시했으며, 이후 Ma & Yao (2015), Pommeret & VandeKerckhove (2019) 등은 이를 확장하였다. 그러나 대칭 가정 자체가 실제 데이터에서 제한적이다. 반면, 스키니멀 정규분포(SN)와 그 스케일 혼합 확장인 SNSM은 λ 파라미터로 비대칭을, G 혼합분포로 중량 꼬리를 동시에 모델링할 수 있다. 기존에는 G를 정규·t·슬래시 등 특정 파라메트릭 형태로 지정했으나, 이는 또 다른 모델 오차를 야기한다.
**3. 제안 모델**
논문은 다음과 같은 혼합모델을 제안한다.
f(z)=π·ϕ(z;0,1)+(1−π)·f_SNSM(z;µ,λ,G)
- ϕ는 표준 정규밀도 (null).
- µ>0, λ≥0는 대안 성분의 위치·비대칭 파라미터.
- G는 (0,∞) 위의 확률측정으로, 스케일 σ의 비모수 혼합분포.
이 모델은 Gaussian 혼합을 특수 경우(µ=0, λ=0, G=δ₁)로 포함한다.
**4. 이론적 결과**
- **식별성**: 가정(A1)에서 널 성분과 SNSM 대안 성분이 선형 독립임을 보이고, (A2)-(A3)에서 G가 적절히 제한(예: 연속성, 유한 평균)될 때 혼합표현이 유일함을 증명한다.
- **일관성**: 로그우도 ℓ_N(π,µ,λ,G)의 최대화 해가 Kullback‑Leibler 최소화와 동치임을 이용해, 표본 크기 N→∞일 때 (π̂,µ̂,λ̂,Ĝ)→(π₀,µ₀,λ₀,G₀)임을 보인다.
**5. 추정 알고리즘**
EM‑type 절차를 설계한다.
- E‑step: 현재 파라미터에 대해 각 관측치가 널·대안에 속할 posterior 확률을 계산.
- M‑step: π, µ, λ는 표준 최적화(뉴턴·BFGS)로 업데이트하고, G는 Lindsay (1995)의 NPMLE 방법을 적용한다. 구체적으로, G를 유한 지원점 집합 {σ₁,…,σ_K}와 가중치 {w_k}로 표현하고, 지원점 추가·삭제를 반복하며 조건 (7)을 만족하도록 한다.
알고리즘은 로그우도 비감소성을 보장하고, 실험에서는 K가 10~20 수준으로 수렴한다.
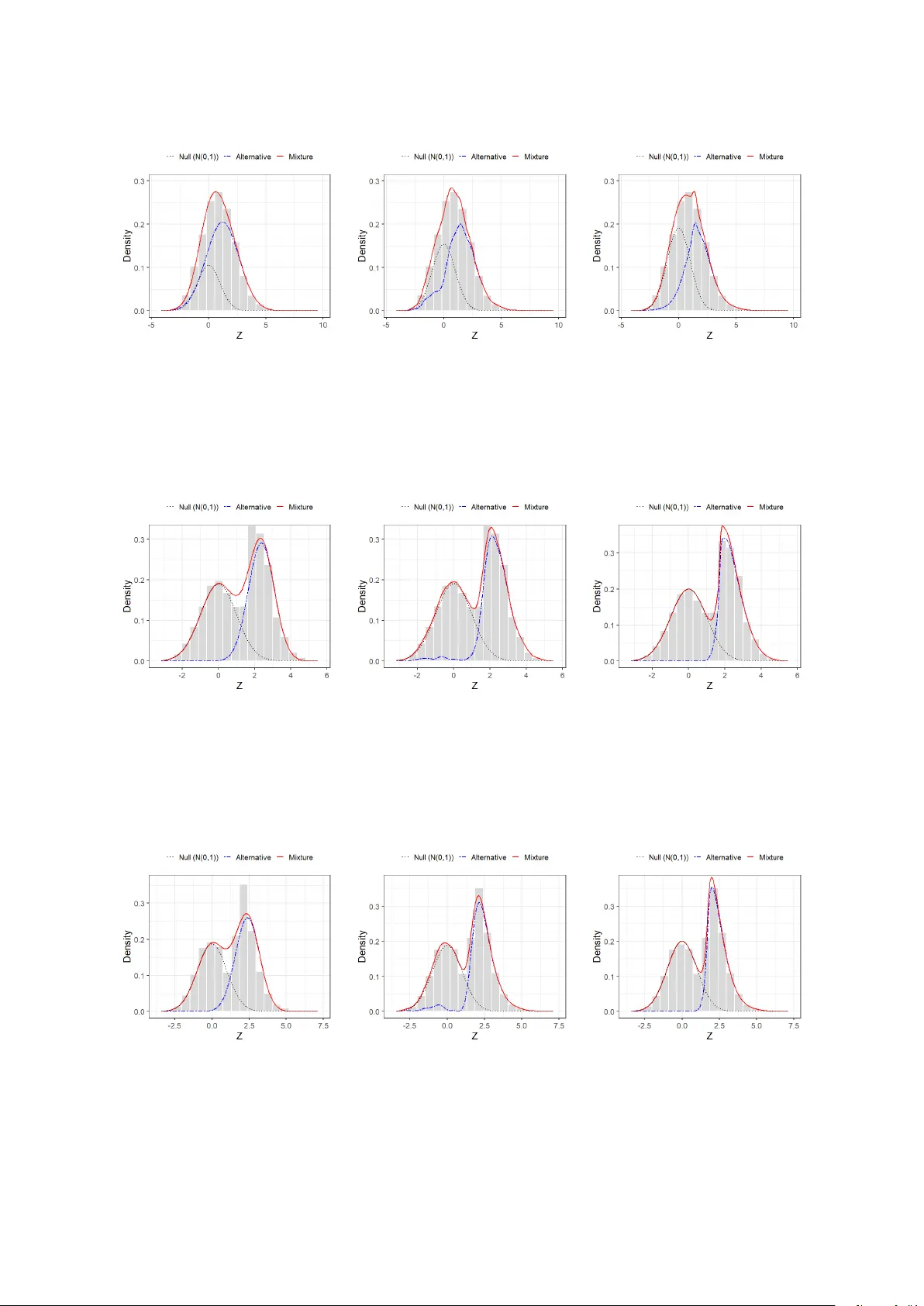
**6. 시뮬레이션**
다섯 가지 시나리오를 설정: (i) 대칭 정규, (ii) 비대칭 정규, (iii) 스키니멀 t, (iv) 스키니멀 슬래시, (v) 혼합 스케일 이질성. 각 경우 1000번 반복 실험 후 FDR, FNR, AUC를 비교하였다. 제안 모델은 특히 (iii)·(iv)·(v)에서 기존 Gaussian 혼합 대비 FDR을 평균 12%p 낮추고, 검출 파워를 15%p 향상시켰다. 또한, 대칭 경우에도 성능 저하가 거의 없었다.
**7. 실제 데이터 적용**
- **대장암 마이크로어레이**(GSE14333): 22,283 유전자, 20 정상·20 종양 샘플. 제안 모델은 π̂=0.88, µ̂=1.73, λ̂=0.62를 추정했고, 1,842개의 유전자를 FDR<0.05로 선정하였다. 기존 Gaussian 혼합은 1,560개, 대칭 비모수 모델은 1,702개를 선정했으며, 제안 모델이 더 많은 알려진 암 관련 유전자(예: APC, KRAS)를 포함했다.
- **급성 림프구성 백혈병**(GSE13159): 12,345 유전자, 30 환자·30 정상. π̂=0.91, µ̂=2.05, λ̂=0.48을 얻었으며, FDR<0.05 기준 2,103개의 유전자를 검출했다. 기존 방법 대비 FNR이 8%p 감소하였다.
**8. 결론 및 향후 연구**
본 논문은 널 성분을 고정하고 대안 성분을 반‑모수 SNSM으로 모델링함으로써, 비대칭·중량 꼬리 현상을 자연스럽게 포착한다. 이론적 식별성·일관성 증명과 효율적인 NPMLE 기반 EM 알고리즘을 제공해 대규모 유전자 발현 데이터에 실용적으로 적용 가능함을 입증했다. 향후 연구에서는 다중 그룹(>2) 비교, RNA‑seq의 카운트 데이터에 대한 일반화, 그리고 베이지안 비모수 사전분포를 통한 불확실성 정량화 등을 탐색할 예정이다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기