Refold: Refining Protein Inverse Folding with Efficient Structural Matching and Fusion
Protein inverse folding aims to design an amino acid sequence that will fold into a given backbone structure, serving as a central task in protein design. Two main paradigms have been widely explored. Template-based methods exploit database-derived s…
Authors: Yiran Zhu, Changxi Chi, Hongxin Xiang
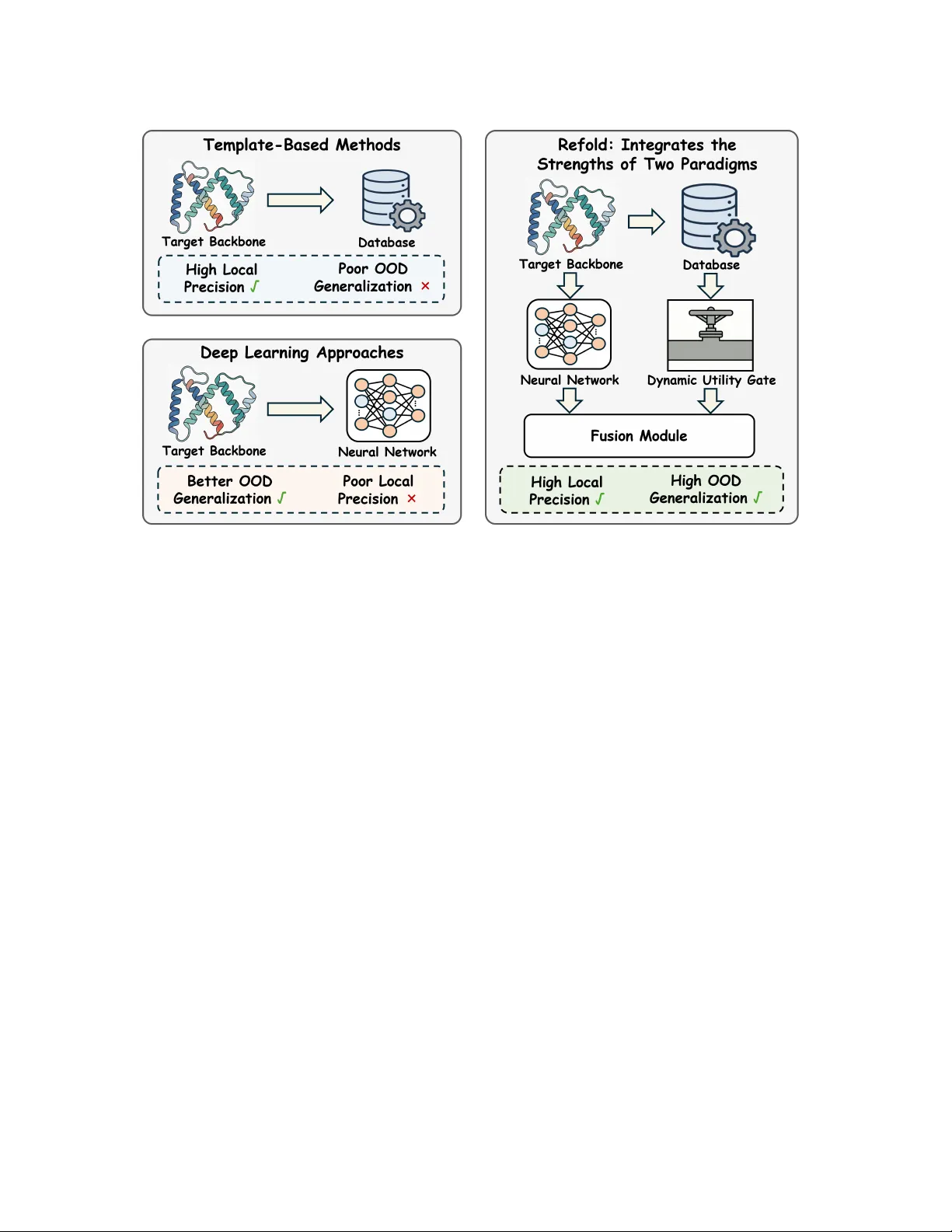
Refold: Refining Protein Inv erse Folding with Eicient Structural Matching and Fusion Yiran Zhu AIMS Lab, HKUST (GZ) Guangzhou, China North China Electric Power University Baoding, China ciaran_study@yeah.net Changxi Chi W estlake University Hangzhou, China chichangxi@westlake.edu.cn Hongxin Xiang Hunan University Changsha, China xianghx@hnu.edu.cn W enjie Du University of Science and T echnology of China Hefei, China duwenjie@mail.ustc.edu.cn Xiaoqi W ang Northwest Polytechnical University Xi’an, China xqw@nwpu.edu.cn Jun Xia ∗ AIMS Lab, HKUST (GZ) Guangzhou, China HKUST Hong Kong SAR, China junxia@hkust- gz.edu.cn Abstract Protein inverse folding aims to design an amino acid se quence that will fold into a given backbone structure, ser ving as a cen- tral task in protein design. T w o main paradigms have been widely explored. T emplate-based methods exploit database-derived struc- tural priors and can achieve high local precision when close struc- tural neighbors are available, but their dependence on database coverage and match quality often degrades performance on out- of-distribution (OOD) targets. Deep learning approaches, in con- trast, learn general structure-to-sequence regularities and usually generalize b etter to new backb ones. Howev er , they struggle to capture ne-graine d local structure, which can cause uncertain residue predictions and missed local motifs in ambiguous regions. W e introduce Refold, a novel framework that synergistically in- tegrates the strengths of database-derived structural priors and deep learning prediction to enhance inv erse folding. Refold obtains structural priors from matched neighbors and fuses them with model predictions to rene residue probabilities. In practice, low- quality neighbors can intr oduce noise, potentially degrading model performance. W e address this issue with a Dynamic Utility Gate that controls prior injection and falls back to the base prediction when the priors are untrustworthy . Comprehensive evaluations on standard benchmarks demonstrate that Refold achieves state- of-the-art native sequence recovery of 0.63 on both CA TH 4.2 and CA TH 4.3. Also, analysis indicates that Refold delivers larger gains on high-uncertainty regions, reecting the complementarity b e- tween structural priors and deep learning predictions. The code is available at https://anonymous.4open.science/r/Refold- anon. Ke ywords Protein inverse folding, Database-derived structural priors, Struc- tural matching and fusion, Dynamic Utility Gate ∗ Corresponding author . KDD ’26, Jeju, Korea 2026. ACM ISBN 978-x-xxxx-xxxx-x/Y YYY/MM https://doi.org/10.1145/nnnnnnn.nnnnnnn 1 Introduction Designing amino acid sequences that fold into prescribed three- dimensional backbones is a foundational challenge in computa- tional biology , commonly referred to as protein inverse folding. The task assigns an amino acid to each backbone p osition while satisfying physical and chemical constraints such as steric com- patibility , hydrophobic interactions, and electrostatics. Accurate inverse folding is essential for understanding protein function and enabling protein design. Traditional template-based methods rely on database-derived structural priors curated in large structural repositories such as the Protein Data Bank (PDB) [ 1 , 15 ]. Given a target backbone, they perform structural matching to retrieve similar folds or local frag- ments [ 7 ] and transfer template-implied constraints (e .g., residue preferences, conserved motifs, or geometry-compatible congura- tions) [ 12 ], often followed by packing/renement under physics- inspired scoring. When matched structural neighbors exist, these priors can deliver high local precision. However , their dep endence on database coverage and match quality can yield po or out-of- distribution (OOD) generalization when neighbors are absent or only remotely related, degrading designs for novel or weakly char- acterized folds. This limitation has motivate d deep learning ap- proaches that learn transferable structure-to-sequence regularities from large-scale data, albeit typically without explicitly leveraging matched-neighbor priors at inference time. In recent years, de ep learning has emerged as the dominant para- digm for protein inverse folding. Models such as ProteinMPNN [ 2 ], PiFold [ 5 ], and K WDesign [ 3 ] employ neural networks to model structure-to-sequence regularities from large-scale data. Through end-to-end training on diverse protein backbones, these methods achieve strong nativ e sequence recovery and improved OOD gen- eralization. Howev er , lacking access to database-derived structural priors at inference time, these models are for ced to depend entirely on learned patterns. This limitation be comes critical in structurally ambiguous regions, where weak local cues often lead to uncertain residue predictions and missed local motifs. KDD ’26, August 9–13, 2026, Jeju, K orea Yiran Zhu et al. Figure 1: T emplate-based methods use database-derived structural priors to achieve high local precision but often show po or OOD generalization. Deep learning approaches generalize better to new backbones yet struggle with ne-grained local structure. Refold integrates these complementary strengths to improve both OOD generalization and local precision. T o unify the high local precision of template-based methods and the OOD generalization of deep learning approaches, we inte- grate these tw o paradigms into a framework, Refold. Given a query backbone, Refold performs ecient structural matching against a reference database to identify a set of matche d neighbors, aligns them to the query , and derives structural priors. These priors are then fused with the mo del’s pr edictions to rene residue probabili- ties. Additionally , when the matche d neighbors are of low quality , injecting these priors can overwrite correct predictions and nega- tively aect model performance. T o address this issue, w e introduce a Dynamic Utility Gate that adaptively controls the inje ction of structural priors and falls back to the original prediction when the priors are unreliable . As a result, Refold achie ves state-of-the- art native sequence recovery on standard benchmarks, delivering substantial improvements particularly in structurally ambiguous regions where baseline models struggle. In addition, our framew ork is designed as a plug-and-play mo dule that can be seamlessly inte- grated into diverse inverse folding base models. W e highlight the core contributions of this work as follows. • As illustrated in Fig. 1, w e propose Refold, a plug-and-play framework that bridges the gap between template-based methods and deep learning approaches. By eectively inte- grating these complementary strengths, Refold signicantly improves both local precision and OOD generalization. • W e propose an ecient structural matching-and-fusion al- gorithm that fuses the structural priors with model predic- tions to rene residue probabilities. W e further introduce a D ynamic Utility Gate that adaptively controls the inje c- tion of structural priors to mitigate the adverse impact of low-quality matches. • Extensive evaluations on standard benchmarks show that Refold delivers consistent gains and achiev es state-of-the-art native sequence recovery . As revealed in Sec. 4.3, the gains are most signicant at high-uncertainty residues, empirically validating our solution to local structural ambiguities. 2 Related W ork 2.1 T emplate-Based Methods Early template-based pipelines leverage database-derived struc- tural priors from the PDB [ 1 ] and perform structural matching to retrieve similar folds or local fragments [ 7 ], transferring template- implied constraints that guide sequence optimization on a xed backbone. While they can achieve high local precision when close structural neighbors exist, their dependence on database coverage and match quality can limit generalization to out-of-distribution (OOD) backbones, where suitable matches are absent or only re- motely related [12]. 2.2 Deep Learning Approaches Recent data-driven approaches range from early autoregressive models like StructTrans [ 9 ] and GVP [ 10 ] to state-of-the-art graph- based metho ds like ProteinMPNN [ 2 ], PiFold [ 5 ], and KWDesign [ 3 ]. While these models eectively captur e global dependencies via mes- sage passing, they encode structural knowledge purely into model Refold KDD ’26, August 9–13, 2026, Jeju, K orea Figure 2: O v er view of Refold. (a) W e perform structural matching to search matched neighbors 𝑘 for the target backb one S . (b) These neighbors are organize d into a Stacked Neighbor Alignment matrix A , where row 0 ser ves as a base anchor derived from the base logits argmax ( 𝑧 base ) . A Similarity- W eighted Fusion Module distills A into reference logits 𝑧 ref and linearly fuses them with 𝑧 base to form 𝑝 fused . (c) T o mitigate the adverse impact of low-quality neighbors, a Dynamic Utility Gate guide d by the Inference- Time Statistics mo dulates the fusion, falling back to the base prediction when structural priors are unreliable. parameters. Lacking inference-time access to matched neighbors, they often suer fr om predictive uncertainty in regions with am- biguous local geometries. 2.3 Utilization of Structural Priors Incorporating structural priors has proven eective in specialized domains, such as antibody design [ 19 ] and molecule generation [ 18 ]. Howev er , extending this to general inverse folding is hinder ed by high structural diversity and noise. Refold addresses this via a Dynamic Utility Gate that selectively integrates neighbor priors only when the y pr ovide reliable improv ements, eectively bridging template-based precision with deep learning generalization. 3 Method As illustrated in Fig. 2, Refold is a plug-and-play framework that augments base models via structural matching and fusion. It aligns database-derived priors with the query backbone and employs a Dynamic Utility Gate to selectively fuse these priors with base predictions, thereby pr eventing performance degradation fr om low- quality matches. 3.1 Problem Formulation Protein inverse folding aims to design an amino acid se quence y = ( 𝑦 1 , . . . , 𝑦 𝐿 ) that is compatible with a prescribed 3D backbone structure S of length 𝐿 . Given S , we seek to maximize the condi- tional likelihood 𝑃 ( y | S ) . W e build on a pre-trained inverse folding base model 𝑓 base (e.g., ProteinMPNN), which operates on the geometric representation of S and outputs residue-wise base logits 𝑧 base ∈ R 𝐿 × 20 . W e inv estigate two training strategies regarding the base model parameters: (1) Refold (Frozen), where w e freeze 𝑓 base throughout to strictly isolate the contribution of structural priors; and (2) Refold ( Joint), where 𝑓 base is jointly ne-tuned to maximize the synergistic integration for state-of-the-art performance. In both settings, we utilize the base output as the foundational prediction: 𝑧 base = 𝑓 base ( S ) , (1) where the logits correspond to the 20 canonical amino acids. How- ever , since these base predictions r ely solely on learned parameters, structurally ambiguous regions often result in diuse distributions with high entropy . This characteristic reects the predictive uncer- tainty of the base model, leading to sub optimal recovery of comple x local motifs. This limitation necessitates augmenting the base logits with structural priors derived from matched neighbors. 3.2 Constructing the Stacke d Neighbor Alignment T o augment the base model with structural priors, we search a reference database to nd a set of matched neighbors for the quer y backbone S . W e employ Foldseek [ 17 ] to identify the top- 𝐾 struc- tural candidates based on geometric similarity . For each matched neighbor 𝑘 , we generate a residue-level alignment mapping its amino acid sequence to the query indices 1 , . . . , 𝐿 . W e further quan- tify the quality of each neighbor using a global similarity score 𝑠 𝑘 (specically , the TM-score). T o handle structural discrepancies, any query position that lacks a corresponding residue in the matched neighbor is lled with a spe cial GAP token. Matched neighbors often exhibit fragmente d co verage and local misalignments. W e pr eser ve their spatial details by organizing these priors into a Stacked Neighbor Alignment matrix A ∈ V ( 𝐾 + 1 ) × 𝐿 . Each column corresponds to a residue position on the quer y back- bone. Rows 𝑟 = 1 , . . . , 𝐾 store the aligned residue tokens from the KDD ’26, August 9–13, 2026, Jeju, K orea Yiran Zhu et al. Figure 3: Schematic of the Similarity- W eighted Fusion Module. From the base logits 𝑧 base , we construct a Stacked Neighb or Alignment matrix A : row 0 serves as the base anchor argmax ( 𝑧 base ) , while rows 1 , . . . , 𝐾 contain aligned matched neighbor tokens. The module embeds A , applies row-wise smoothing to mitigate local noise, and aggregates structural priors via attention weighted by the reliability bias 𝛽 . This process produces reference logits 𝑧 ref , which are then residually fused with the base logits to yield 𝑧 fused = 𝑧 base + 𝜆 · 𝑧 ref . matched neighbors, while row 𝑟 = 0 serves as a base anchor . Con- cretely , we instantiate this anchor via greedy de coding of the base logits: 𝑦 0 , 𝑗 = argmax 𝑎 ∈ V 𝑧 base , 𝑗 ( 𝑎 ) , (2) where V denotes the set of 20 canonical amino acids. This anchor provides a fully dened refer ence sequence, stabilizing downstream aggregation when structural priors are sparse or fragmented. Con- sequently , each entry in A is a discrete token from V or GAP . Recognizing that neighbor quality varies, we compute a reliabil- ity bias 𝛽 ∈ R 𝐾 + 1 to guide the aggregation of structural priors. Let 𝑠 = ( 𝑠 1 , . . . , 𝑠 𝐾 ) denote the global similarity scores (TM-scores) of the neighbors. W e dene: 𝛽 ( 𝑟 ) = 𝛽 0 , 𝑟 = 0 , somax ( 𝑠 ) 𝑟 − 1 , 𝑟 = 1 , . . . , 𝐾 , (3) where 𝛽 0 is a learned scalar representing the baseline condence. In the subse quent Fusion Module, 𝛽 serves as a bias term in the attention mechanism. This formulation encourages the model to prioritize high-similarity neighbors while retaining the capacity to reject irrelevant structural priors in favor of the base anchor . 3.3 Similarity- W eighted Fusion Mo dule Given the Stacked Neighbor Alignment A and the reliability bias 𝛽 from Sec. 3.2, we now describ e a Similarity- W eighte d Fusion Module designed to distill these structural priors into residue-wise reference logits 𝑧 ref . These reference logits ser v e as a corrective signal that is subsequently combine d with the base logits 𝑧 base , subject to modulation by the Dynamic Utility Gate described in the following subsection. Fig. 2 provides a schematic ov er view of this fusion process. The module operates in three logical stages. First, it spatially renes each neighbor row to encode local context and mitigate alignment noise. Next, it aggregates structural priors across all neighbors at each quer y position, utilizing the reliability bias to weigh their contributions. Finally , it projects the aggregated featur es into the target vocabular y space to produce 𝑧 ref and integrates them with the base predictions via a residual connection. 3.3.1 Row-wise Smo othing. W e rst embed the discrete tokens in the alignment matrix A using a shared emb edding layer E , obtain- ing a dense tensor 𝑋 0 ∈ R ( 𝐾 + 1 ) × 𝐿 × 𝑑 . W e then apply a depthwise- separable 1-D convolution along the sequence axis within each r ow independently , using a kernel size of 5 and a stride of 1. Positions corresponding to GAP tokens are masked to ensure that missing residues do not propagate noise into the convolved features. This row-wise smoothing aggr egates short-range context within each neighbor sequence and helps mitigate small alignment osets re- sulting from the structural matching process. 3.3.2 Similarity-Biased A ggregation. Next, we transpose the smo othed features to 𝑋 ∈ R 𝐿 × ( 𝐾 + 1 ) × 𝑑 so that each target residue position 𝑗 can aggregate structural priors across the 𝐾 + 1 rows. For each position, we compute multi-head attention over the neighb or di- mension. Queries, keys, and values are derived via standard linear projections of 𝑋 . GAP tokens in the key/value ro ws are masked to prevent the model from attending to missing data. W e augment the attention scores with the reliability bias 𝛽 : ℓ 𝑗 ( 𝑟 → 𝑟 ′ ) = 𝑄 𝑗 ( 𝑟 ) · 𝐾 𝑗 ( 𝑟 ′ ) √ 𝑑 ℎ + 𝛼 · 𝛽 ( 𝑟 ′ ) , (4) where 𝑟 ′ indexes the neighb or rows, 𝑑 ℎ is the per-head dimensional- ity , and 𝛼 ≥ 0 is a learnable scaling factor . The attention outputs are aggregated to produce 𝐻 ∈ R 𝐿 × 𝑑 , which is then projected through a feed-for war d layer to the reference logits 𝑧 ref ∈ R 𝐿 × 20 . If a target position 𝑗 lacks valid neighbors (i.e., all tokens are GAP ), we explicitly set 𝑧 ref , 𝑗 = 0 , ensuring that the fusion branch is deactivated for that residue. Refold KDD ’26, August 9–13, 2026, Jeju, K orea 3.3.3 Linear Fusion. Finally , w e integrate the structural priors with the base model’s predictions via a linear combination of logits. Let 𝑧 base and 𝑧 ref denote the residue-wise logits produced by the base model and the fusion module, respectively . W e dene the fused logits 𝑧 fused and the corresponding probabil- ity distribution 𝑝 fused as: 𝑧 fused = 𝑧 base + 𝜆 · 𝑧 ref , 𝑝 fused = somax ( 𝑧 fused ) , (5) where 𝜆 is a learnable scalar controlling the str ength of the r eference signal. This additive form acts as a residual renement, allowing the robust base pr e dictions to serve as a foundation while the reference path injects sparse, specic corrections. When 𝑧 ref provides little position-wise signal—for instance, when its magnitude is small or valid neighbors are absent—the fused distribution 𝑝 fused naturally converges to the distribution implied by 𝑧 base . In practice, this sim- ple formulation is stable and avoids the additional assumptions required by more complex fusion schemes. 3.4 Training and Robust Gated Inference 3.4.1 Training the Fusion Module. W e adopt a two-stage training strategy to ensure stability . In the rst stage , for Refold (Fr ozen), the base model 𝑓 base remains frozen, and we optimize only the fusion module parameters 𝜃 , including the shared embedding E , the row- wise convolution, and the attention layers. For Refold ( Joint), we unfreeze 𝑓 base to enable end-to-end joint optimization of both the base model and fusion parameters. The Dynamic Utility Gate is bypassed during this phase. W e minimize the cross-entropy loss between the fuse d distribution 𝑝 fused and the ground-truth se quence y : L ( 𝜃 ) = − 𝐿 𝑖 = 1 log 𝑝 fused ( 𝑦 𝑖 | S ) . (6) Training on the full dataset encourages the fusion module to learn robust aggregation patterns ev en when structural priors are noisy or incomplete. 3.4.2 D ynamic Utility Gate. Indiscriminate fusion of structural pri- ors can degrade performance when matched neighbors are globally similar but locally incompatible with the target. T o resolve this dilemma, we introduce a Dynamic Utility Gate 𝐺 ( 𝜙 ) . In the second training stage, we freeze both the base mo del and the fusion module, optimizing only the gate to predict whether the fused prediction outperforms the base model. The gate input 𝜙 comprises inference-time statistics, including alignment coverage, the mean TM-score of matched neighbors, and divergence measures between the base distribution 𝑝 base and the fuse d distribution 𝑝 fused . W e generate binary training labels 𝑔 ∈ { 0 , 1 } on a held-out validation set. Let L CE ( 𝑝 ) denote the cross- entropy loss compute d using distribution 𝑝 . W e set 𝑔 = 1 if and only if L CE ( 𝑝 fused ) < L CE ( 𝑝 base ) , and optimize 𝐺 ( 𝜙 ) using binary cross-entropy . At inference time, the gate functions as a global switch with a position-wise fallback. If the predicted utility 𝐺 ( 𝜙 ) falls b elow a threshold 𝜏 (tuned on validation data), the model re verts to the base distribution globally . Other wise, it adopts the fuse d distribution, defaulting to the base model only at specic p ositions wher e valid reference data is absent: 𝑝 out , 𝑗 = 𝑝 base , 𝑗 , if 𝐺 ( 𝜙 ) < 𝜏 or 𝑧 ref , 𝑗 = 0 , 𝑝 fused , 𝑗 , otherwise . (7) This mechanism ensures that structural priors are utilized only when predicte d to b e benecial, robustly defaulting to the base model when the gate rejects the fusion or when no aligned neigh- bors exist for a given residue. W e summarize the inference cost of Refold (parameter and latency overhead) in Sec. 4.6. 4 Experiments 4.1 Experimental Settings In this work, lev eraging the ProInvBench framework [ 6 ], we em- ploy a non-redundant refer ence database alongside the CA TH 4.2 and CA TH 4.3 benchmarks [ 13 , 14 ]. The datasets adhere to strict topology-based splitting protocols, ensuring robust data separation and preventing potential leakage between training and evaluation phases. T o further assess OOD generalization, we additionally in- clude the TS50 and TS500 test sets [11]. Refold is designed as a universal plug-and-play framework. W e integrate the fusion mo dule into ProteinMPNN [ 2 ], utilizing it as our primary base model. W e optimize the framework using the Adam optimizer with a linear warm-up schedule on an N VIDIA A800 GP U . W e train the model under two training variants (Frozen and Joint) to balance eciency and performance. For detailed hyperparameters and more training congurations, please refer to the Appendix ?? . 4.2 Main Results T able 1 compares performance on CA TH 4.2 and 4.3. Refold achieves consistent improv ements across all strategies. First, Refold (Frozen) enhances the xed ProteinMPNN, increasing recovery by +0.16/+0.17 on the full CA TH 4.2/4.3 sets and reducing p erple xity by 2.08/2.17. The marke d reduction in perplexity conrms that incorporating structural priors eectively mitigates the base model’s pr e dictiv e uncertainty . Furthermore, Refold ( Joint) establishes new state-of- the-art results via end-to-end optimization. It attains 0.63 recovery on both benchmarks ( +0.18/+0.19 gain), surpassing str ong baselines like KWDesign and demonstrating the ecacy of fusing structural priors. W e further evaluate Refold on the TS50 and TS500 b enchmarks, which provide structurally diverse targets under the standard pro- tocol. As shown in T ab. 2, Refold yields consistent improv ements on both TS50 and TS500, increasing recovery from 0.54 to 0.60 on TS50 and from 0.58 to 0.63 on TS500. These gains under distribution shift suggest that structural priors from matched neighbors can eectively complement model predictions on structurally diverse targets. 4.3 Where Does Refold Help? Uncertainty Stratied Analysis A central premise of Refold is that the base model tends to be un- reliable in structurally ambiguous regions, where the backbone geometry supp orts multiple plausible residue identities. In such scenarios, the base model typically yields a diuse pr e dictiv e dis- tribution, characterized by high entropy . Consequently , if Refold KDD ’26, August 9–13, 2026, Jeju, K orea Yiran Zhu et al. T able 1: Results comparison on the CA TH dataset. “Short” refers to proteins with length 𝐿 < 100 , and “Full” corresponds to the complete test set. Sequence recovery is reported as a fraction (0–1). V alues in parentheses indicate the absolute improvement (Recovery ↑ ) or reduction (Perplexity ↓ ) compared to the base model on the specic dataset version. Best results are bolded. Model Perplexity ↓ Recovery ↑ CA TH version Short Full Short Full 4.2 4.3 GraphTrans [9] 8.39 6.63 0.28 0.36 ✓ StructGNN [9] 8.29 6.40 0.29 0.36 ✓ ESM-IF † [8] 8.18 6.44 0.31 0.38 ✓ GCA [16] 7.09 6.05 0.33 0.38 ✓ GVP [10] 7.23 5.36 0.31 0.39 ✓ GVP-large † [10] 7.68 6.17 0.32 0.39 ✓ AlphaDesign [4] 7.32 6.30 0.34 0.41 ✓ PiFold [5] 6.04 4.55 0.40 0.52 ✓ ProteinMPNN [2] 7.96 5.92 0.32 0.45 ✓ 7.23 5.94 0.38 0.44 ✓ KWDesign [3] 5.48 3.46 0.44 0.61 ✓ 5.47 3.49 0.51 0.60 ✓ Refold (Frozen) 5.38 ( -2.58) 3.84 ( -2.08) 0.49 ( +0.17) 0.61 ( +0.16) ✓ 4.61 ( -2.62) 3.77 ( -2.17) 0.53 ( +0.15) 0.61 ( +0.17) ✓ Refold ( Joint) 4.80 ( -3.16) 3.48 ( -2.44) 0.51 ( +0.19) 0.63 ( +0.18) ✓ 4.45 ( -2.78) 3.53 ( -2.41) 0.56 ( +0.18) 0.63 ( +0.19) ✓ T able 2: OOD Generalization performance on TS50 and TS500 test sets. W e employ Refold (Frozen) trained on CA TH 4.3. Best results are bolded. Model T est Set Recovery ↑ Perplexity ↓ ProteinMPNN TS50 0.54 3.93 TS500 0.58 3.53 Refold TS50 0.60 ( +0.06) 3.78 (-0.15) TS500 0.63 (+0.05) 3.51 ( -0.02) eectively leverages structural priors, w e expe ct the performance gains to be concentrate d in these high-uncertainty positions rather than being uniformly distributed. T o verify this, we compute the token-level Shannon entropy 𝐻 𝑗 of the prediction at each residue position 𝑗 . Let 𝑝 base ( · | 𝑗 ) = somax ( 𝑧 base , 𝑗 ) denote the probability distribution implied by the base logits. W e dene: 𝐻 𝑗 = − 𝑎 ∈ V 𝑝 base ( 𝑎 | 𝑗 ) log 𝑝 base ( 𝑎 | 𝑗 ) , (8) where V represents the set of 20 canonical amino acids. W e then stratify all validation tokens into three uncertainty regimes using xed entropy thresholds: lo w uncertainty ( 𝐻 𝑗 < 1 . 5 ), medium un- certainty ( 1 . 5 ≤ 𝐻 𝑗 < 2 . 3 ), and high uncertainty ( 𝐻 𝑗 ≥ 2 . 3 ). These regimes contain 101,888, 82,763, and 87,067 tokens, respectively . T able 3 reports the native se quence r ecovery acr oss the stratie d uncertainty regimes. T o strictly isolate the corrective eect of struc- tural priors from base model adaptation, we conduct this analysis using Refold (Frozen) on CA TH 4.3. W e obser v e that p erformance gains increase monotonically with the base mo del’s uncertainty . T able 3: Uncertainty-stratied recovery analysis based on base model predictive entropy 𝐻 𝑗 . Δ denotes the absolute recovery gain. Note that gains are most pronounced in high- entropy regimes. Uncert ainty Regime Base Rec. Refold Rec. Δ Low ( 𝐻 < 1 . 5 ) 0.78 0.83 +0.05 Mid ( 1 . 5 ≤ 𝐻 < 2 . 3 ) 0.34 0.55 +0.21 High ( 𝐻 ≥ 2 . 3 ) 0.18 0.45 +0.27 Overall (All) 0.44 0.61 +0.17 Specically , Refold improves recovery by + 0 . 05 on low-entropy tokens ( 0 . 78 → 0 . 83 ), while the improvements are substantially amplied on mid-entrop y ( + 0 . 21 , 0 . 34 → 0 . 55 ) and high-entropy to- kens ( + 0 . 27 , 0 . 18 → 0 . 45 ). This pattern corroborates our hyp othesis that Refold primarily recties errors in structurally ambiguous re- gions where the base model is uncertain, utilizing structural priors as decisive corrections precisely when geometric reasoning alone is unreliable. Overall, Refold improves token-level recov er y from 0.44 to 0.61 ( + 0 . 17 ), consistent with the aggr egate gains reported in the main results. 4.4 Ablation Studies W e conduct ablation studies to validate the contribution of key components in Refold. Consistent with Se c. 4.3, all ablations are performed using the Refold (Frozen) setting on CA TH 4.3 to strictly isolate the impact of each module. Refold KDD ’26, August 9–13, 2026, Jeju, K orea 0 10 20 30 40 50 r esidues 1 2 3 4 5 6 7 8 pr oteins Neg P os Neg P os P os Neg Figure 4: Site-wise recovery states ( sampled pr oteins). W e sample 𝑁 proteins and visualize the rst 50 residues. Each cell denotes a residue-level transition from the Base model to Refold: Neg (wrong → wrong), Pos ( correct → correct), Neg → Pos (wrong → correct, correction), and Pos → Neg (correct → wrong, error). Corrections (Neg → Pos) tend to appear in localize d segments, while error events (Pos → Neg) are sparse, suggesting that Refold p erforms targeted, structur e-aligned renements rather than indiscriminate perturbations. T able 4: Ablation studies on ProteinMPNN and CA TH 4.3. Configura tion Recovery Perplexity Full Model (Ours) 0.61 3.77 Source Contribution w/o Refold (Baseline) 0.44 5.94 w/o base model (Priors Only) 0.52 6.40 Mechanisms w/o TM-score Bias 0.58 4.21 Utility Ga te w/o Ga te 0.58 4.20 Aggrega tion Module Row -only A ttention 0.55 4.33 Alterna ting A ttention 0.50 4.92 Synergy of Geometric Reasoning and Structural Priors. W e rst investigate the individual contributions of the base model’s geo- metric reasoning and the matched structural priors. As shown in T ab. 4, relying solely on the base model (w/o Refold) yields a recov- ery of 0.44, whereas relying solely on structural priors (w/o Base Model) achieves 0.52. Crucially , b oth single-source baselines fall signicantly short of the full model (0.61). This performance gap demonstrates that the two information sources ar e mutually com- plementary rather than redundant. The structural matching and fusion process provides specic structural motifs that are dicult to predict from scratch, while the base model provides the neces- sary geometric reasoning to rene and contextualize these priors. Refold achieves a true synergistic eect by eectively integrating these distinct information sources, ensuring that the fused output surpasses the capability of either individual source. Ecacy of the Utility Gate. Comparing the full model to the “w/o Gate” variant, w e verify the necessity of our Dynamic Utility Gate. Removing the gate leads to a performance drop ( 0 . 61 → 0 . 58 ), conrming that indiscriminate fusion of structural priors can hurt performance. The gate eectively acts as a reliability lter , selectively suppressing low-condence priors when they conict with the robust base model. Impact of Attention Me chanisms. Regarding aggregation, remov- ing the TM-score bias leads to degradation (0.58), indicating that explicit similarity scores eectively guide the attention mechanism to prioritize high-quality neighb ors. Furthermore , our smo othed scheme outperforms naiv e Row-only or Alternating attention strate- gies, validating the design of Similarity- W eighted Fusion Mo dule . 4.5 Qualitative Analysis: Mining Lo cal Geometric Motifs W e further investigate the mechanism by which Refold leverages structural priors. Since the base model relies solely on learned weights, it lacks access to spe cic structural priors at inference time. When target regions exhibit subtle geometric ambiguity or weak local signals, the model depends entirely on its learned regularities, which often yields diuse (high-entropy) residue distributions and consequently misses highly specic native motifs. Sampled site-wise transition patterns. Before analyzing cases, we visualize token-level transitions on sampled proteins to inspect how Refold alters predictions (Fig. 4). The transition map rev eals that Refold’s improvements often emerge as localized corrections (wrong → correct), while correct → wrong transitions remain rar e. This qualitative pattern supports our claim that Refold renes pre- dictions in a targeted manner: it integrates structural priors where benecial, without indiscriminately overwriting base predictions. Case study: 2kb9.A. T o demonstrate this granular mechanism, w e analyze the test case 2kb9.A presented in T ab. 5. For this target, the structural matching process identies a matched neighbor with a TM-score of 0.757. As visualized in the token-lev el comparison, the base model fails at several positions. In these locally ambiguous regions, the base distribution becomes diuse ( high-entr opy), indicating uncertainty and increasing the likelihood of missing specic native motifs. In contrast, the matched neighbor , despite having a distinct global sequence, preser v es precise local geometric motifs at these posi- tions, carrying the correct residues T yrosine (Y) and Cysteine ( C). Refold successfully attends to these structural priors (highlighted in yellow ), correcting the prediction to the spe cic nativ e residues. KDD ’26, August 9–13, 2026, Jeju, K orea Yiran Zhu et al. T able 5: T oken-level comparison of Base, Refold, and matched neighbor (Nbr) sequences for the 2kb9.A case. Corr ect predictions are colored green, errors red, and Nbr positions highlighted in yellow indicate structure-guided corrections derived from the matched neighbor . Row 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 . . . Base M S S E G Y S G E S S S H C A P E P G C T R G T S S R P R . . . Refold M S S Y G W S G L S C D H C A P E P G C T R G T C S R P R . . . Nbr K H Q Y G W Q G L Y C D K C I P H P G C V H G I C N E P W . . . 1 5 10 15 20 0 . 5 0 . 55 0 . 6 Number of matched neighbors ( K ) Sequence Recov ery Refold Figure 5: Sensitivity analysis of the number of matched neigh- bors 𝐾 . The p erformance gain saturates around 𝐾 = 10 , indi- cating that the model aggregates structural priors eectively without requiring a large number of neighb ors. T able 6: Computational eciency analysis on an NVIDIA A800 GP U . Latency is reported per protein with a batch size of 16. Overhead denotes the relative increase in latency com- pared to the base model ProteinMPNN . Model P arams Latency Overhead ProteinMPNN 1.7M 13.41ms – Refold 3.5M 18.22ms +35.9% KWDesign 4.1B 656.24ms +4793.7% This selective behavior conrms that Refold do esn’t blindly copy the neighbor sequence; rather , it performs context-aware motif composition. It leverages the base model’s geometric reasoning to determine where structural priors are reliable, and then integrates local priors to resolve high-entropy uncertainty into motif-specic predictions. 4.6 Sensitivity and Eciency Analysis W e investigate the hyperparameter sensitivity and computational eciency of Refold to assess its practical deployability . Sensitivity to Neighbor Count ( 𝐾 ). As illustrated in Fig. 5, we ana- lyze the impact of the number of matched neighbors 𝐾 . Sequence recovery improv es rapidly as 𝐾 increases from 1 to 10 and plateaus thereafter . This suggests that the fusion module can extract su- cient structural context from a compact set of high-quality neigh- bors ( 𝐾 ≈ 10 ), avoiding the computational overhead of processing large numbers of matches. Inference Cost. W e summarize parameter and latency overhead in T able 6. Refold introduces a minimal numb er of additional train- able parameters (appro ximately 1.8M) to the ProteinMPNN base model. While this results in a moderate latency increase of + 35 . 9% compared to the lightweight base model, the total infer ence time remains at the millisecond lev el (18.22ms). In contrast, the previous state-of-the-art model K WDesign relies on a massiv e 4.1B parame- ter ar chite ctur e, resulting in an infer ence latency of 656.24ms—ov er 30 × slower than our method. These r esults demonstrate that Refold establishes a new state-of-the-art with orders of magnitude better eciency than large-scale baselines, making it highly suitable for high-throughput applications. 5 Conclusion In this paper , we presented Refold, which enhances protein inverse folding by fusing database-derived structural priors with deep learn- ing predictions. Leveraging matche d neighbors from a reference database, Refold impro ves both local precision and OOD general- ization. Moreover , the D ynamic Utility Gate prev ents performance degradation by avoiding o ver-reliance on noisy or locally incom- patible neighbors, maintaining stable gains ev en when high-quality matched neighb ors are scarce. As a result, Refold achieves state- of-the-art native sequence recovery on both CA TH 4.2 and CA TH 4.3. Looking ahead, future work could focus on more robust utility estimation to further reduce the risk of performance degradation, and on exploring alternative ways to integrate structural priors with model predictions. 6 Limitations and Ethical Considerations While Refold enhances inverse folding accuracy , the structural matching and fusion process adds a modest latency overhead ( + 35 . 9% ) compared to the lightweight base model, though it remains orders of magnitude faster than large-scale baselines. Additionally , our approach assumes a xed backb one conformation, ignoring the dynamic nature of proteins in solution. Regarding ethical consid- erations, this work relies solely on public structural data (PDB) and involves no personal or sensitive information, though we ac- knowledge potential selection bias toward proteins that are easier to determine structurally . Finally , while intended to accelerate ther- apeutic design, we recognize the potential for dual-use misuse (e.g., harmful protein design); therefor e, any generated sequences should b e treated as computational hypotheses and undergo rig- orous biosafety , toxicity , and institutional review before wet-lab synthesis. Refold KDD ’26, August 9–13, 2026, Jeju, K orea References [1] Helen M. Berman, John W estbrook, Zukang Feng, Gar y Gilliland, T alapady N. Bhat, Helge W eissig, Ilya N. Shindyalov , and Philip E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Research 28, 1 (2000), 235–242. doi:10.1093/nar/28.1.235 [2] Jonas Dauparas, Ivan Anishchenko, Nathaniel Bennett, Hua Bai, R. J. Ragotte, Lukas F. Milles, B. I. M. Wicky , A. Courbet, R. J. de Haas, N. Bethel, P. J. Y . Leung, T . F. Huddy , S. Pellock, D. Tischer , F. Chan, B. K oepnick, H. Nguyen, A. Kang, B. Sankaran, A. K. Bera, N. P . King, and David Baker . 2022. Robust deep learning- based protein sequence design using ProteinMPNN. Science 378, 6615 (2022), 49–56. doi:10.1126/science.add2187 [3] Zhangyang Gao, Cheng T an, Xingran Chen, Yijie Zhang, Jun Xia, Siyuan Li, and Stan Z. Li. 2024. K W -Design: Pushing the limit of protein design via knowledge renement. In Proceedings of the International Conference on Learning Representa- tions (ICLR) . [4] Zhangyang Gao, Cheng T an, and Stan Z. Li. 2022. AlphaDesign: A graph protein design method and benchmark on AlphaFoldDB. arXiv preprint (2022). https://arxiv .org/abs/2202.01079 [5] Zhangyang Gao, Cheng T an, and Stan Z. Li. 2023. PiFold: T oward eective and ecient protein inverse folding. In Proceedings of the International Confer ence on Learning Representations (ICLR) . https://op enr eview .net/forum?id=oMsN9T Y wJ0j [6] Zhangyang Gao, Cheng T an, Yijie Zhang, Xingran Chen, Lirong Wu, and Stan Z. Li. 2023. ProteinInvBench: Benchmarking Protein Inverse Folding on Diverse T asks, Models, and Metrics. In Thirty-seventh Conference on Neural Information Processing Systems Datasets and Benchmarks Track . https://openreview .net/ forum?id=bqXduvuW5E [7] Liisa Holm and Chris Sander . 1993. Protein structure comparison by alignment of distance matrices. Journal of Mole cular Biology 233, 1 (1993), 123–138. doi:10. 1006/jmbi.1993.1489 [8] Chloe Hsu, Robert V erkuil, Jason Liu, Zeming Lin, Brian Hie, T om Sercu, Adam Lerer , and Alexander Rives. 2022. Learning inverse folding from millions of predicted structures. In Proceedings of the 39th International Conference on Machine Learning (Proceedings of Machine Learning Research, V ol. 162) . PMLR, 8946–8970. https://proceedings.mlr .press/v162/hsu22a.html [9] John Ingraham, Vikas Garg, Regina Barzilay , and Tommi S. Jaakkola. 2019. Gener- ative Models for Graph-Based Protein Design. In Advances in Neural Information Processing Systems , V ol. 32. https://papers.nips.cc/paper/9711- generative- models- for- graph- based- protein- design [10] Bowen Jing, Stephan Eismann, Patricia Suriana, Raphael J. L. T ownshend, and Ron O. Dror . 2021. Learning from Protein Structure with Geometric V ector Percep- trons. In Proceedings of the International Conference on Learning Representations (ICLR) . https://openreview .net/forum?id=1YLJDvSx6J4 [11] Z Li, Y Y ang, J Zhan, L Dai, and Y Zhou. 2014. Direct prediction of the prole of sequences compatible with a protein structur e by neural networks with fragment- based local and energy-based nonlocal proles. Proteins: Structure, Function, and Bioinformatics (2014). doi:10.1002/prot.24620 [12] Andrej Sali and T om L. Blundell. 1993. Comparative protein modelling by satis- faction of spatial restraints. Journal of Molecular Biology 234, 3 (1993), 779–815. doi:10.1006/jmbi.1993.1626 [13] Ian Sillitoe, Nicola Bordin, Natalie Dawson, V aishali P. W aman, Paul Ashford, Harry M. Scholes, Camilla S. M. Pang, Laurel W oodridge, Clemens Rauer , Nee- ladri Sen, Mahnaz Abbasian, Sean Le Cornu, Su Datt Lam, Karel Berka, Ivana Hutarova V arekova, Radka Svobodova, Jon Lees, and Christine A. Orengo. 2021. CA TH: increased structural coverage of functional space. Nucleic Acids Research 49, D1 (2021), D266–D273. doi:10.1093/nar/gkaa1079 [14] Ian Sillito e , Natalie Dawson, T ony E. Lewis, Sayoni Das, Jonathan G. Lees, Paul Ashford, Ade yelu T olulope, Harry M. Scholes, Ilya Senatorov , Andra Bujan, Fa- tima Ceballos Rodriguez-Conde, Benjamin Dowling, Janet Thornton, and Chris- tine A. Orengo. 2019. CA TH: expanding the horizons of structure-based func- tional annotations for genome sequences. Nucleic Acids Research 47, D1 (2019), D280–D284. doi:10.1093/nar/gky1097 [15] Manfred J. Sippl. 1995. Knowledge-based potentials for proteins. Current Opinion in Structural Biology 5, 2 (1995), 229–235. doi:10.1016/0959- 440X(95)80081- 6 [16] Cheng Tan, Zhangyang Gao, Jun Xia, Bozhen Hu, and Stan Z. Li. 2022. Generative De Novo Protein Design with Global Context. arXiv preprint (2022). https://arxiv .org/abs/2204.10673 [17] Michel van Kempen, Stephanie S. Kim, Charlotte T umescheit, Milot Mirdita, Jeongjae Lee, Cameron L. M. Gilchrist, Johannes Sö ding, and Martin Steineg- ger . 2024. Fast and accurate protein structure search with Foldseek. Nature Biotechnology (2024). doi:10.1038/s41587- 023- 01773- 0 Epub 2023-05-08. [18] Runzhong W ang, Rui-Xi W ang, Mrunali Manjrekar, and Connor W . Coley . 2025. Neural Graph Matching Improves Retrie val A ugmented Generation in Molecular Machine Learning. In Proceedings of the 42nd International Conference on Machine Learning (ICML) . [19] Zichen W ang, Yaokun Ji, Jianing Tian, and Shuangjia Zheng. 2024. Retrieval Augmented Diusion Model for Structure-informed Antibody Design and Opti- mization. arXiv preprint arXiv:2410.15040 (2024). Accepted to ICLR 2025.
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment