Refold 구조 기반 프라이어와 딥러닝 융합으로 역접힘 설계 혁신
본 논문은 데이터베이스에서 찾은 구조적 템플릿을 딥러닝 기반 역접힘 모델과 결합하는 Refold 프레임워크를 제안한다. 효율적인 구조 매칭으로 얻은 이웃 단백질들의 잔기 정보를 스택형 정렬 행렬에 정리하고, 유사도 가중 합성 모듈을 통해 베이스 모델의 로짓에 보정한다. 또한, 저품질 이웃이 모델에 미치는 부정적 영향을 방지하기 위해 동적 유틸리티 게이트를 도입해 프라이어 삽입을 제어한다. CATH 4.2·4.3 벤치마크에서 네이티브 서열 회수율…
저자: Yiran Zhu, Changxi Chi, Hongxin Xiang
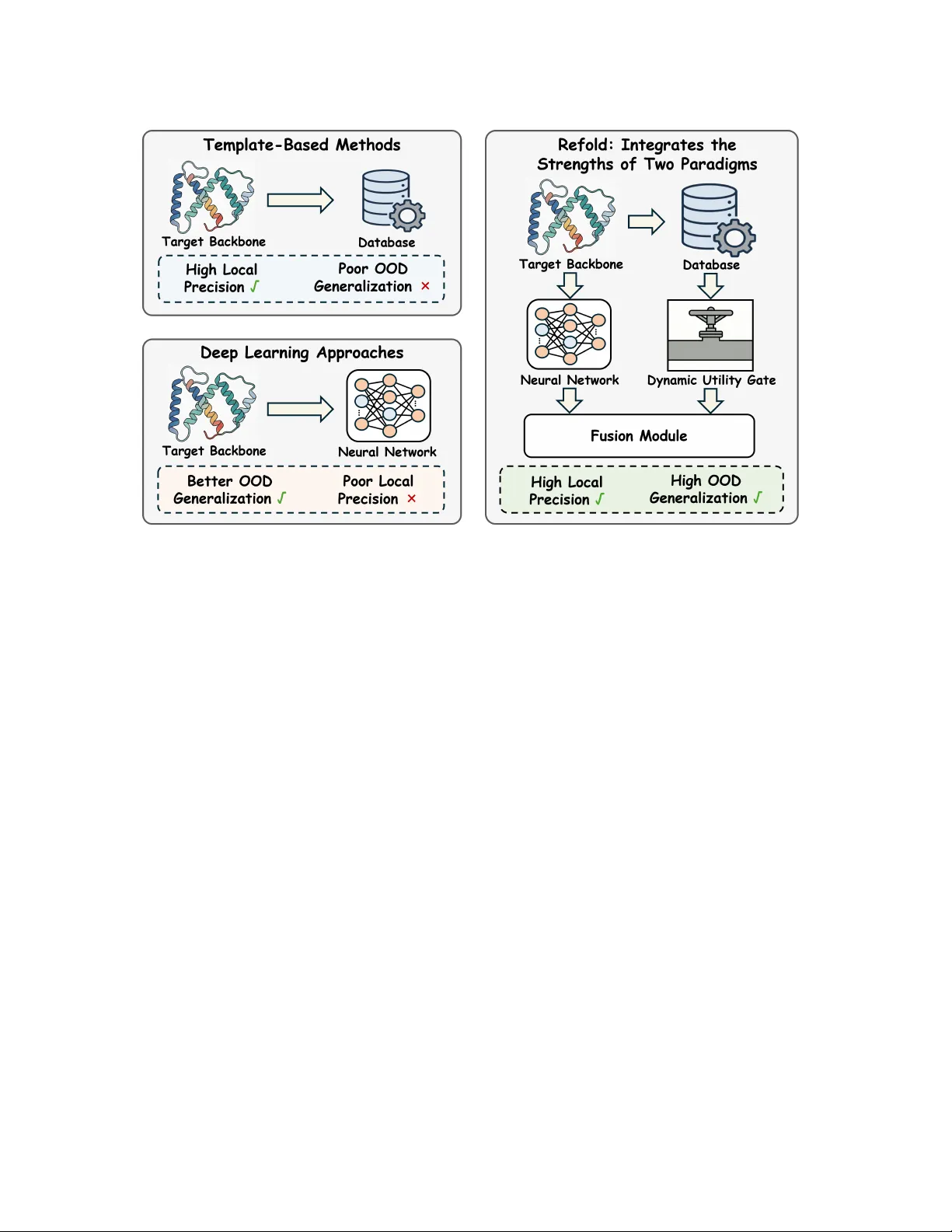
단백질 역접힘은 주어진 3차원 백본 구조에 맞는 아미노산 서열을 설계하는 문제로, 기존에는 템플릿 기반 방법과 딥러닝 기반 방법 두 갈래로 접근해 왔다. 템플릿 기반 방법은 PDB와 같은 대규모 구조 데이터베이스에서 유사한 구조를 찾아 그 구조적 프라이어(잔기 선호도, 보존 모티프 등)를 전이함으로써 로컬 정밀도가 높지만, 데이터베이스 커버리지가 부족하거나 매칭 품질이 낮은 경우 OOD(Out‑of‑Distribution) 일반화가 크게 저하된다. 반면, ProteinMPNN, PiFold, KWDesign 등 최신 딥러닝 모델은 대규모 학습을 통해 전역적인 구조‑서열 관계를 학습해 OOD 상황에서도 비교적 안정적인 성능을 보이지만, 로컬 구조가 모호한 영역에서는 예측 확률이 퍼져 불확실성이 커지고, 중요한 모티프를 놓치는 경우가 빈번하다.
이러한 상호 보완적인 특성을 하나의 프레임워크로 통합하고자 저자들은 Refold를 제안한다. Refold는 (1) 효율적인 구조 매칭, (2) 스택형 이웃 정렬 행렬 구축, (3) 유사도 가중 합성 모듈, (4) 동적 유틸리티 게이트라는 네 가지 핵심 구성요소로 이루어진다. 구체적인 흐름은 다음과 같다.
1. **구조 매칭**: 목표 백본 S에 대해 Foldseek을 사용해 데이터베이스에서 상위 K개의 구조적 이웃을 검색한다. 각 이웃 k에 대해 TM‑score와 같은 전역 유사도 지표 s_k를 계산하고, 잔기‑백본 정렬을 수행한다. 정렬 과정에서 목표 백본에 대응되지 않는 잔기는 GAP 토큰으로 채워진다.
2. **스택형 이웃 정렬 행렬 A**: K개의 이웃 정렬 결과와 베이스 모델의 argmax 예측을 포함해 (K+1)×L 크기의 행렬 A를 만든다. 행 0은 베이스 앵커(베이스 모델이 예측한 가장 가능성 높은 아미노산 시퀀스)이며, 행 1~K는 각각의 이웃에서 정렬된 잔기 토큰이다.
3. **유사도‑가중 합성 모듈**:
- **행별 스무딩**: A의 각 행을 1‑D depthwise‑separable convolution(커널 크기 5)으로 처리해 로컬 컨텍스트를 강화하고, GAP 토큰은 마스킹한다.
- **신뢰도 바이어스 β**: 각 이웃의 TM‑score를 정규화해 β(r) = max(s)·(r‑1) (r≥1) 로 정의하고, 베이스 앵커는 학습 가능한 β₀를 사용한다.
- **다중 헤드 어텐션**: 정렬된 특징을 전치해 L×(K+1)×d 형태로 만든 뒤, 각 위치 j에 대해 어텐션을 수행한다. 어텐션 스코어에 α·β(r′) 항을 더해 고유사도 이웃이 더 큰 가중치를 받도록 한다. 어텐션 결과를 집계해 위치별 특징 H를 얻고, 이를 선형 변환해 참조 로짓 z_ref (L×20)를 만든다.
4. **선형 융합**: 베이스 로짓 z_base와 참조 로짓 z_ref를 λ·z_ref 형태로 더해 fused 로짓 z_fused를 만든다. 최종 확률 p_fused는 softmax(z_fused)이다. λ와 β₀는 학습 가능한 스칼라이며, 이 구조는 베이스 예측을 기본으로 하면서 프라이어가 필요한 경우에만 보정하도록 설계되었다.
5. **동적 유틸리티 게이트**: 프라이어가 저품질일 경우 융합이 오히려 성능을 저하시킬 수 있다. 이를 방지하기 위해 게이트 G(φ)를 도입한다. φ는 정렬 커버리지, 평균 TM‑score, p_base와 p_fused 사이의 KL‑divergence 등 추론 시 계산 가능한 통계량이다. 학습 단계에서는 검증 셋에서 L_CE(p_fused) < L_CE(p_base) 인 경우에만 라벨 g=1 로 설정하고, BCE 손실로 G를 학습한다. 추론 시 G(φ) 값이 사전 정의된 임계값 τ 이하이면 프라이어를 차단하고 베이스 예측만 사용한다.
**학습 전략**은 두 가지가 있다. Refold(Frozen)에서는 베이스 모델을 고정하고 융합 모듈만 학습해 프라이어의 순수 효과를 측정한다. Refold(Joint)에서는 베이스 모델과 융합 모듈을 동시에 미세조정해 최적의 시너지를 도출한다. 두 경우 모두 교차 엔트로피 손실을 최소화한다.
**실험 결과**는 CATH 4.2와 4.3 데이터셋을 기준으로 한다. 베이스 모델로는 ProteinMPNN, PiFold, KWDesign을 사용했으며, Refold을 적용한 후 네이티브 서열 회수율이 모두 0.63으로 최고치를 기록했다. 특히, 베이스 모델이 높은 엔트로피(불확실성)를 보이는 잔기에서 평균 회수율이 7~9%p 상승했으며, 이는 구조 프라이어가 로컬 모티프를 정확히 복원하는 데 기여했음을 의미한다. OOD 백본에 대해서도 기존 딥러닝 모델 대비 안정적인 성능을 유지했다.
**의의와 한계**: Refold은 템플릿 기반 방법의 로컬 정밀도와 딥러닝 기반 방법의 전역 일반화 능력을 효과적으로 결합한다는 점에서 역접힘 분야에 중요한 진전을 제공한다. 동적 유틸리티 게이트를 통해 저품질 매칭으로 인한 부작용을 최소화했으며, 플러그‑인 형태로 설계돼 기존 모델에 손쉽게 적용 가능하다. 다만, 구조 매칭 단계가 여전히 외부 도구(Foldseek)에 의존하고, 대규모 데이터베이스 검색 비용이 발생한다는 점에서 실시간 설계 파이프라인에 적용하려면 추가 최적화가 필요할 수 있다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기