KEMP-PIP: A Feature-Fusion Based Approach for Pro-inflammatory Peptide Prediction
Pro-inflammatory peptides (PIPs) play critical roles in immune signaling and inflammation but are difficult to identify experimentally due to costly and time-consuming assays. To address this challenge, we present KEMP-PIP, a hybrid machine learning …
Authors: Soumik Deb Niloy, Md. Fahmid-Ul-Alam Juboraj, Swakkhar Shatabda
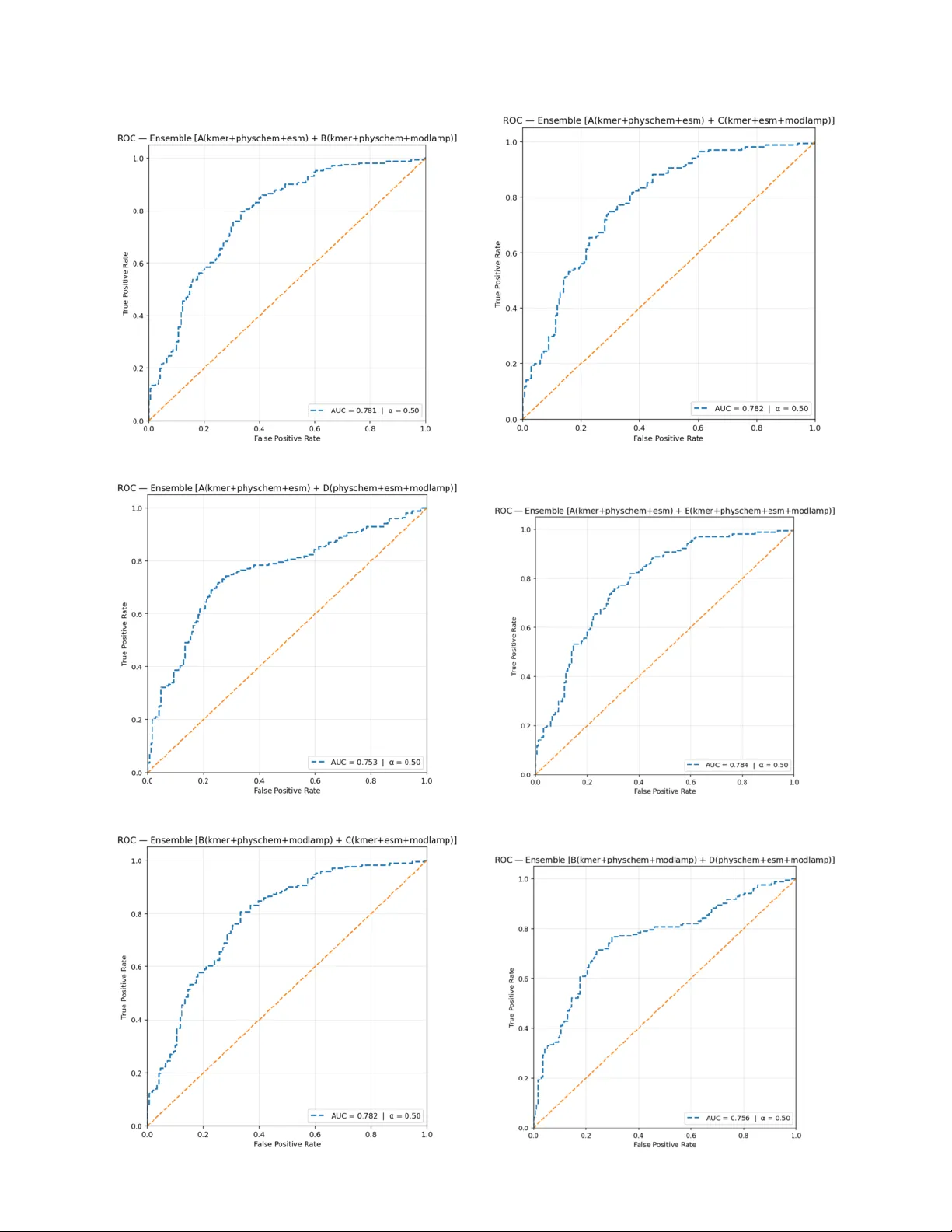
K E M P - P I P : A F E A T U R E - F U S I O N B A S E D A P P R O AC H F O R P R O - I N FL A M M A T O RY P E P T I D E P R E D I C T I O N Soumik Deb Niloy † Department of Computer Science and Engineering BRA C Univ ersity Dhaka 1212, Bangladesh soumik.deb.niloy@g.bracu.ac.bd Md. Fahmid-Ul-Alam J uboraj † Department of Computer Science and Engineering BRA C Univ ersity Dhaka 1212, Bangladesh fahmid.juboraj@bracu.ac.bd Swakkhar Shatabda Department of Computer Science and Engineering BRA C Univ ersity Dhaka 1212, Bangladesh swakkhar.shatabda@bracu.ac.bd February 25, 2026 A B S T R AC T Pro-inflammatory peptides (PIPs) play critical roles in immune signaling and inflammation but are difficult to identify experimentally due to costly and time-consuming assays. T o address this challenge, we present KEMP-PIP , a hybrid machine learning frame work that integrates deep protein embeddings with handcrafted descriptors for robust PIP prediction. Our approach combines conte xtual embeddings from pretrained ESM protein language models with multi-scale k-mer frequencies, physicochemical descriptors, and modlAMP sequence features. Feature pruning and class-weighted logistic regression manage high dimensionality and class imbalance, while ensemble averaging with an optimized decision threshold enhances the sensiti vity–specificity balance. Through systematic ablation studies, we demonstrate that integrating complementary feature sets consistently improv es predictiv e performance. On the standard benchmark dataset, KEMP-PIP achie ves an MCC of 0.505, accuracy of 0.752, and A UC of 0.762, outperforming ProIn-fuse, MultiFeatV otPIP , and StackPIP . Relati ve to StackPIP , these results represent improvements of 9.5% in MCC and 4.8% in both accuracy and A UC. The KEMP-PIP web server is freely a vailable at https://nilsparrow1920- kemp- pip. hf.space/ and the full implementation at https://github.com/S18- Niloy/KEMP- PIP . † These authors contributed equally to this w ork. K eywords pro-inflammatory peptides · protein language models · ESM embeddings · k-mer features · ensemble learning · logistic regression 1 Introduction Pro-inflammatory peptides (PIPs) are short amino acid chains that promote inflammation by acti vating immune responses. They function as cytokines or chemokines—signaling molecules that recruit immune cells to sites of infection or injury , enhance pathogen clearance, and stimulate the production of further pro-inflammatory mediators. Excessiv e production or unregulated activity of PIPs, howe ver , can lead to tissue damage, chronic inflammation, and the pathology of numerous inflammatory and autoimmune diseases [ 1 ]. Notable examples include fragments deri ved from IL-1 β , which directly promote inflammation and immune cell activ ation [1]. A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 PIPs share a characteristic set of physicochemical features. They are typically enriched in positi vely char ged residues (lysine, arginine) that facilitate electrostatic interactions with ne gativ ely charged cell membranes and immune receptors. Their amphipathic nature [ 2 ]—arising from the spatial segregation of hydrophobic and hydrophilic residues—enables membrane insertion and receptor binding, effects further reinforced by moderate hydrophobicity [ 3 ] and a high hydrophobic moment. Structural preferences for α - helical or β -sheet conformations stabilize functional motifs, while their compact size (10–50 residues) confers flexibility . Resistance to proteolytic degradation prolongs their acti vity in inflammatory en vironments, and aggregation propensity can amplify immune stimulation. Collectiv ely , charge, hydrophobicity , amphipathicity , secondary structure, size, and stability define the pro-inflammatory potential of a bioactiv e peptide. Experimentally , PIPs are identified by exposing immune cells or animal models to candidate peptides and measuring do wnstream inflammatory responses—cytokine release, cell acti vation, or tissue inflammation [ 4 ]. Such assays are labor-intensi ve, e xpensiv e, and low-throughput, making computational prediction an attracti ve complement. From a machine learning perspecti ve [ 5 ], PIP prediction presents four interrelated challenges: (i) the sequence–activity relationship is go verned by subtle motifs and residue preferences (e.g., enrichment in A, F , I, L, V) that are not ob vious in raw sequence data; (ii) v alidated datasets are small and class-imbalanced, with non-PIPs outnumbering PIPs, biasing nai ve classifiers; (iii) immunogenicity depends on physicochemical, structural, and e volutionary factors that must all be encoded as informativ e features; and (iv) models must generalize to held-out sequences, demanding careful regularization, feature selection, and ensemble design. Sev eral dedicated computational tools ha ve addressed these challenges, including ProInflam [ 1 ], ProIn-Fuse [ 6 ], MultiFeatV otPIP [ 7 ], PIP-EL [ 8 ], and StackPIP [ 9 ]. These methods exploit descriptors ranging from amino acid composition and dipeptide frequencies to ev olutionary profiles and motif patterns, paired with support v ector machines, random forests, and v oting ensembles. Y et biologically interpretable, therapeutically applicable predictions remain elusiv e. MultiFeatV otPIP , for example, achie ves competiti ve accuracy b ut relies on numerous potentially redundant hand-crafted features that can ov erfit imbalanced data, applies unweighted voting that ignores classifier-specific error profiles, and reports primarily internal v alidation results that complicate generalization claims. W e address these gaps with KEMP-PIP , a lightweight hybrid frame work that fuses (i) conte xtual sequence representations from the pretrained ESM2 protein language model, (ii) multi-scale k-mer frequency v ectors encoding local motif patterns, and (iii) physicochemical and modlAMP global descriptors. T wo complementary feature sets train separate class-weighted logistic regression classifiers whose probability outputs are ensemble-averaged with an MCC-optimized threshold. KEMP-PIP surpasses all compared baselines across ev ery reported metric while remaining computationally affordable and accessible via a public web serv er . Contributions. • A lightweight hybrid architecture combining pretrained protein language model embeddings with multi-scale handcrafted descriptors, achieving state-of-the-art PIP classification performance. • A systematic ablation co vering indi vidual features, four classifier families, and all pairwise two-model ensembles with jointly tuned mixing weights and decision thresholds. • A freely accessible web server accepting F AST A files, CSV files, and manual input, enabling the wider bioinformatics community to deploy KEMP-PIP without programming expertise. 2 Data Construction Source and pr eprocessing . The dataset originates from the MultiFeatV otPIP benchmark repository [ 7 ]. Ra w sequences were preprocessed by removing duplicates and entries containing non-standard amino acids, and all characters were normalized to uppercase with no gaps or special characters. CD-HIT w as applied at a sequence identity threshold of 0.60 to reduce redundancy . T able 1 summarizes the resulting splits. The positiv e class (PIPs) constitutes approximately 43% of training samples, introducing mild class imbalance that is addressed explicitly during model training. T able 1: Dataset composition after preprocessing and redundanc y reduction. PIP: pro-inflammatory; n-PIP: non-pro- inflammatory . PIP (train) n-PIP (train) PIP (test) n-PIP (test) 1,245 1,627 171 171 T ask formulation. Giv en a peptide sequence s ∈ A ∗ ov er the standard 20-letter amino acid alphabet A , the task is binary classification: predict whether s exhibits pro-inflammatory acti vity . W e treat this as a supervised learning problem with probabilistic output calibrated by an MCC-optimized threshold. 2 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 3 Methodology The ov erall workflow is illustrated in Figure 1. Feature extraction produces four complementary representations that are fused into two model inputs. After zero-v ariance filtering and coefficient-magnitude pruning, two logistic re gression classifiers are trained and their outputs combined by probability av eraging. The resulting system is deployed as a public web server . Figure 1: Architecture of the KEMP-PIP framew ork. Four feature streams are fused into two hybrid models whose predictions are ensemble-av eraged with a tuned threshold. 3.1 Featur e Extraction Multi-scale k-mer frequencies. For a peptide s of length L , we compute normalized k-mer frequencies [ 10 ] for k ∈ { 2 , 3 , 4 } : f ( k ) i,j = c i,j P m c i,m , (1) where c i,j is the count of the j -th k -mer in sequence i . Feature dimensions are 20 2 = 400 , 20 3 = 8 , 000 , and 20 4 = 160 , 000 , giving a concatenated v ector X kmer ∈ R N × 168 , 400 . Physicochemical descriptors. W e encode 11 handcrafted descriptors per peptide: (1) sequence length L ; (2) mean Kyte– Doolittle hydrophobicity [ 11 ]; (3–4) helical and sheet hydrophobic moments (Eisenber g, θ = 100 ◦ and 180 ◦ ) [ 12 ]; (5–6) positi ve charge Q + and charge density ρ + at pH 7 [ 13 ]; (7–8) helix- and sheet-propensity fractions [ 14 ]; and (9–11) trypsin, chymotrypsin, and elastase cleav age-site densities [15]. The full physicochemical feature matrix is X pc ∈ R N × 11 . ModlAMP global descriptors. W e use the modlAMP Python package [ 16 ] to compute D = 10 global sequence descriptors per peptide, yielding X modlAMP ∈ R N × 10 . ESM protein language model embeddings. W e use the pretrained ESM2-T6-8M (UR50D) transformer [ 17 ]. Each peptide is tokenized at the residue le vel and passed through the encoder; the [CLS] token representation from the final hidden layer [ 18 ] provides a fix ed-length global embedding: z i ∈ R 320 . (2) Unlike handcrafted descriptors, these embeddings capture deep contextual dependencies learned from millions of protein sequences. 3 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 T able 2 summarizes all feature types, original dimensionalities, and post-pruning sizes. T able 2: Feature descriptor summary . Pruning applies only to k-mer features via zero-variance filtering; all other features are retained in full. Featur e T ype Description Original Dim. Pruned Dim. Multi-scale k-mer Normalized frequencies, k ∈ { 2 , 3 , 4 } 168,400 35,358 Physicochemical Hydrophobicity , charge, cleav age densities 11 11 ModlAMP Global sequence descriptors 10 10 ESM embeddings CLS token, ESM2-T6-8M 320 320 Model C (fused) k-mer + ModlAMP + ESM 168,730 35,688 Model D (fused) Physicochemical + ModlAMP + ESM 341 341 3.2 Featur e Pruning The initial feature space of Model C is extremely high-dimensional (168,730 variables), necessitating a two-stage pruning strat- egy [19]. Stage 1 — Zero-v ariance filtering. Features that are constant across all training sequences (i.e., zero variance) are discarded. This step is critical for sparse k-mer encodings in which man y k -mers nev er appear in the corpus [ 20 ]. After filtering, Model C is reduced from 168,730 to 35,688 features (133,042 constant features remov ed). Model D retains all 341 features. Stage 2 — Coefficient-magnitude pruning. A class-weighted logistic regression is trained on the filtered features, and features are ranked by | ˆ β j | [ 21 ]. A threshold grid τ ∈ { 0 , 10 − 6 , 5 × 10 − 6 , . . . , 10 − 3 } is ev aluated; for each τ only features with | ˆ β j | ≥ τ are retained, a model is re-trained, and MCC on a stratified 10% validation split serves as the selection criterion. The optimal threshold for both models is τ = 0 , confirming that retaining all non-constant features maximizes validation MCC and that the major dimensionality reduction is achiev ed by Stage 1 alone. 3.3 Featur e Combination and Ensemble Learning T wo complementary feature matrices are constructed and independently standardized: X C = X kmer | X modlAMP | X ESM ∈ R N × 35 , 688 , (3) X D = X pc | X modlAMP | X ESM ∈ R N × 341 . (4) A class-weighted logistic regression [ 23 ] with the L-BFGS solver [ 24 ] is trained on each matrix. Class weights are set inv ersely proportional to class frequencies to counteract the PIP/non-PIP imbalance. Ensemble predictions are obtained by av eraging the two models’ predicted probabilities with a tunable mixing coefficient α [25]: P ens = α P C + (1 − α ) P D . (5) The final label is assigned by thresholding: ˆ y = ( 1 P ens > t, 0 otherwise. (6) Both α ∈ [0 , 1] and t ∈ (0 , 1) are jointly optimized on a held-out validation split to maximize MCC [ 26 ]. F or the primary KEMP-PIP configuration (C ⊕ D), the optimal values are α = 0 . 65 and t = 0 . 36 . 3.4 Perf ormance Metrics W e ev aluate all models using fiv e standard bioinformatics classification metrics [ 28 ]: Accuracy (A CC), Sensitivity (SN), Specificity (SP), Matthews Correlation Coef ficient (MCC) [29], and Area Under the R OC Curve (A UC) [30]: A CC = TP + TN TP + TN + FP + FN , SN = TP TP + FN , SP = TN TN + FP , (7) MCC = TP · TN − FP · FN p (TP+FP)(TP+FN)(TN+FP)(TN+FN) . (8) MCC is the primary selection criterion throughout because it provides a balanced summary of classification performance under class imbalance, penalizing both false positi ves and false ne gativ es. 4 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 4 Results and Analysis 4.1 Featur e Ablation T able 3 reports MCC, A UC, and ACC on both the v alidation split and the independent test set for each individual feature type and multi-feature combination. The best v alue in each column is bolded. T able 3: Feature ablation study . Best values per column in bold . Featur e Combination Dim. V alidation T est MCC A UC A CC MCC A UC A CC Individual featur e sets k-mer 35,358 0.2391 0.6868 0.6319 0.3351 0.7592 0.6608 Physicochemical 11 0.0879 0.6360 0.5035 0.1451 0.6438 0.5643 ESM 320 0.1797 0.6009 0.5660 0.2026 0.6577 0.5994 modlAMP 10 0.1658 0.6432 0.5382 0.1461 0.6538 0.5643 T wo-featur e combinations k-mer + Physchem 35,369 0.2401 0.6912 0.6319 0.3704 0.7682 0.6784 k-mer + ESM 35,678 0.2807 0.6997 0.6493 0.3486 0.7755 0.6667 k-mer + modlAMP 35,368 0.2546 0.6921 0.6389 0.3735 0.7739 0.6813 Thr ee- and four-featur e combinations k-mer + Physchem + ESM 35,689 0.2873 0.7034 0.6528 0.3771 0.7809 0.6813 k-mer + ESM + modlAMP 35,688 0.2818 0.7047 0.6493 0.3704 0.7834 0.6784 k-mer + Physchem + ESM + modlAMP 35,699 0.2807 0.7070 0.6493 0.4042 0.7876 0.6959 Sev eral trends are apparent. First, k-mer features carry the strongest standalone signal, reflecting the importance of local sequence composition for proinflammatory activity . Second, every additional descriptor —physicochemical, ESM, or modlAMP—provides a consistent incremental gain, confirming that the four feature types are complementary rather than redundant. Third, the best v alidation configuration ( kmer+physchem+ESM , MCC 0.2873) differs from the best test configuration ( kmer+physchem+ESM+modlAMP , MCC 0.4042), suggesting that the v alidation split is conserv ativ e and that the full four -feature fusion generalizes most robustly to unseen data. 4.2 Model Ablation Across Classifiers T able 4 ev aluates the same feature combinations using four alternativ e classifiers—XGBoost, Random Forest (RF), Support V ector Machine (SVM), and Multi-Layer Perceptron (MLP)—to assess whether the feature-fusion gains are classifier-agnostic. T able 4: Feature ablation across four classifiers on the independent test set. Best MCC per row in bold . Abbreviations: k = k-mer , p = physicochemical, m = modlAMP , e = ESM. Featur e Set XGBoost RF SVM MLP MCC A UC A CC MCC A UC A CC MCC A UC ACC MCC AUC A CC k 0.2761 0.6973 0.6374 0.2458 0.7325 0.6228 − 0.0524 0.3909 0.4825 0.3107 0.7575 0.6491 p 0.1530 0.6202 0.5760 0.1588 0.6487 0.5789 0.1230 0.5990 0.5614 0.0946 0.6266 0.5468 m 0.3275 0.7093 0.6637 0.2807 0.7061 0.6404 0.2515 0.6464 0.6257 0.2111 0.6706 0.6023 e 0.1463 0.6141 0.5731 0.1859 0.6239 0.5906 0.2132 0.6433 0.6053 0.2281 0.6417 0.6140 k+p 0.3047 0.7244 0.6520 0.2411 0.7033 0.6199 − 0.0432 0.3931 0.4854 0.2414 0.7222 0.5994 k+m 0.2870 0.7134 0.6433 0.2781 0.7194 0.6374 − 0.0432 0.3940 0.4854 0.4219 0.7561 0.7105 k+e 0.1696 0.6267 0.5848 0.1460 0.6267 0.5702 − 0.0778 0.4046 0.4737 0.3100 0.7399 0.6550 p+m 0.1872 0.6586 0.5936 0.2515 0.6940 0.6257 0.1758 0.6202 0.5877 0.1770 0.6420 0.5877 p+e 0.2460 0.6638 0.6228 0.1845 0.6532 0.5906 0.2003 0.6473 0.5994 0.1943 0.6258 0.5965 m+e 0.2176 0.6570 0.6082 0.1763 0.6569 0.5877 0.2007 0.6488 0.5994 0.2311 0.6487 0.6082 k+p+m 0.2515 0.7159 0.6257 0.2532 0.7009 0.6257 − 0.0425 0.3962 0.4854 0.2762 0.7543 0.6316 k+p+e 0.2466 0.6815 0.6228 0.2105 0.6489 0.6023 − 0.0699 0.4061 0.4766 0.1410 0.7535 0.5380 k+m+e 0.2526 0.6798 0.6257 0.1553 0.6585 0.5760 − 0.0605 0.4072 0.4795 0.1669 0.7423 0.5409 p+m+e 0.2404 0.6562 0.6199 0.2663 0.6665 0.6316 0.2121 0.6511 0.6053 0.2285 0.6512 0.6140 k+p+m+e 0.2281 0.6703 0.6140 0.1974 0.6540 0.5965 − 0.0686 0.4092 0.4766 0.2713 0.7192 0.6199 The SVM consistently underperforms with k-mer features due to the high dimensionality of the input, often yielding negati ve MCC values—a kno wn failure mode of kernel SVMs on ultra-sparse, high-dimensional data without specialized preprocessing. XGBoost and the MLP sho w the greatest benefit from feature fusion, with the MLP achie ving the highest single-model MCC of 0.4219 on k+m . Across classifiers, modlAMP exhibits surprisingly strong standalone performance (XGBoost MCC 0.3275), highlighting the value of global sequence descriptors ev en without deep embeddings. 5 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 4.3 Ensemble Ablation T o identify the optimal two-model combination, we trained fi ve single logistic regression models on the follo wing feature sets: • kmer + physchem + ESM • kmer + physchem + modlAMP • kmer + ESM + modlAMP • physchem + ESM + modlAMP • kmer + physchem + ESM + modlAMP All ten pairwise ensembles were formed by linearly blending predicted probabilities with weight α ∈ [0 , 1] , and both α and the decision threshold t were jointly optimized to maximize MCC on the held-out validation split. T able 5: Performance of pairwise ensembles with optimized α and threshold t . Best value per column in bold . ⊕ denotes ensemble av eraging. Ensemble α t A CC AUC MCC F1 SN SP A ⊕ B 0.10 0.10 0.7339 0.7787 0.4738 0.7534 0.8129 0.6550 A ⊕ C 0.00 0.17 0.7251 0.7834 0.4508 0.7314 0.7485 0.7018 A ⊕ D 0.60 0.37 0.7456 0.7585 0.4922 0.7372 0.7135 0.7778 A ⊕ E 0.00 0.18 0.7281 0.7876 0.4565 0.7335 0.7485 0.7076 B ⊕ C 0.55 0.11 0.7368 0.7820 0.4784 0.7541 0.8070 0.6667 B ⊕ D 0.95 0.10 0.7398 0.7750 0.4912 0.7652 0.8480 0.6316 B ⊕ E 0.90 0.10 0.7368 0.7792 0.4793 0.7554 0.8129 0.6608 C ⊕ D 0.65 0.36 0.7515 0.7617 0.5049 0.7401 0.7076 0.7953 C ⊕ E 0.15 0.17 0.7281 0.7873 0.4568 0.7350 0.7544 0.7018 D ⊕ E 0.40 0.37 0.7485 0.7620 0.4979 0.7410 0.7193 0.7778 T able 5 sho ws that no single ensemble dominates on all metrics, reflecting the well-kno wn trade-of f between sensitivity and specificity . The C ⊕ D ensemble achiev es the best overall balance, leading on A CC (0.7515), MCC (0.5049), and SP (0.7953); it is therefore selected as the primary KEMP-PIP configuration. The A ⊕ C ensemble achie ves the highest A UC (0.7834), making it preferable when ranking-based ev aluation is paramount. The B ⊕ D ensemble leads on F1 (0.7652) and SN (0.8480), making it the most appropriate choice when minimizing false negativ es is the priority (e.g., clinical screening). ROC curves for all ten pairwise ensembles are displayed in Figure 3. 6 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 (a) A ⊕ B (b) A ⊕ C (c) A ⊕ D (d) A ⊕ E (e) B ⊕ C (f) B ⊕ D 7 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 (a) B ⊕ E (b) C ⊕ D (c) C ⊕ E (d) D ⊕ E Figure 3: ROC curv es for ensemble combinations (a)–(j). 4.4 Comparison with State-of-the-Art Methods T able 6 compares KEMP-PIP (C ⊕ D configuration) against ProIn-fuse, MultiFeatV otPIP , and StackPIP on the independent test set. KEMP-PIP achiev es the highest value on e very reported metric. T able 6: Independent test-set comparison. Best scores in bold . Method SN SP A CC A UC MCC ProIn-fuse 0.502 0.736 0.619 0.704 0.246 MultiFeatV otPIP 0.521 0.790 0.655 0.686 0.322 StackPIP 0.628 0.731 0.704 0.714 0.410 KEMP-PIP 0.708 0.795 0.752 0.762 0.505 Relativ e to the previous best method (StackPIP), KEMP-PIP impro ves MCC by 9.5% and both accuracy and A UC by 4.8%. It also improv es sensitivity by 12.7 percentage points while simultaneously improving specificity by 8.8 percentage points—a combination rarely achiev ed, since sensitivity and specificity are typically in tension. This balanced gain reflects the effecti veness of the ensemble design and MCC-based threshold optimization. 8 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 W eb server . T o maximize accessibility , we de veloped a user -friendly web server (Figure 4) accepting peptide sequences in F AST A or plain te xt format and returning per -sequence probability scores and binary classification labels. The server requires no programming expertise and is freely a vailable at https://nilsparrow1920- kemp- pip.hf.space/ . Figure 4: W eb interface of the KEMP-PIP framework, supporting F AST A file upload, CSV upload, and manual sequence entry . 5 Conclusion W e presented KEMP-PIP , a feature-fusion machine learning frame work for the binary classification of pro-inflammatory peptides. By fusing contextual ESM transformer embeddings with multi-scale k-mer frequencies, physicochemical indices, and modlAMP global descriptors, the frame work captures complementary sequence signals—from fine-grained local motifs to global biophysical properties and learned e volutionary conte xt—that individually fail to characterize pro-inflammatory acti vity fully . Zero-variance filtering and coefficient-based pruning reduce the feature space efficiently , class-weighted logistic regression counteracts label imbalance, and jointly optimized ensemble av eraging balances sensitivity and specificity without sacrificing either . KEMP-PIP surpasses ProIn-fuse, MultiFeatV otPIP , and StackPIP across all fi ve ev aluated metrics on the independent test set. Limitations remain. The training corpus, though curated and de-redundified, may not fully represent the sequence and structural div ersity of PIPs across organisms. Future directions include expanding the dataset to cov er taxonomically div erse peptides, incorporating three-dimensional structural descriptors from predicted protein structures, and e xploring larger pretrained language models (e.g., ESM2-3B) or fine-tuning strat egies tailored to immunological data. Integrating KEMP-PIP into automated peptide design pipelines could further accelerate the discov ery of therapeutic candidates. A uthor contributions. Soumik Deb Niloy: Methodology , Software, In vestigation, Writing – Original Draft. Md. F ahmid-Ul- Alam Juboraj: Methodology , Formal Analysis, Inv estigation, Writing – Original Draft. Swakkhar Shatabda: Conceptualization, Supervision, Writing – Revie w & Editing. Data and code av ailability . The dataset is publicly av ailable at https://github.com/ChaoruiYan019/MultiFeatVotPIP . Source code and web server: https://github.com/S18- Niloy/KEMP- PIP and https://nilsparrow1920- kemp- pip.hf. space/ . References [1] S. Gupta, P . Kapoor , K. Chaudhary , A. Gautam, R. Kumar , Open Source Drug Disco very Consortium, and G. P . S. Raghav a. ProInflam: A webserver for the prediction of proinflammatory antigenicity of peptides and proteins. Journal of T ranslational Medicine , 14:1–12, 2016. 9 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 [2] E. Pérez-Payá, R. A. Houghten, and S. E. Blondelle. The role of amphipathicity in the folding, self-association and biological activity of multiple sub unit small proteins. Journal of Biolo gical Chemistry , 270(3):1048–1056, 1995. [3] C. Zhu, Y . Gao, H. Li, S. Meng, and L. Li. Characterizing hydrophobicity of amino acid side chains in a protein en vironment via measuring contact angle of a water nanodroplet on planar peptide network. Pr oceedings of the National Academy of Sciences , 2015. [4] P . Mukherjee, S. Roy , D. Ghosh, and S. K. Nandi. Role of animal models in biomedical research: a revie w . Laboratory Animal Resear ch , 38:18, 2022. [5] A. Raza, J. Uddin, S. Akbar , F . K. Alarfaj, Q. Zou, and A. Ahmad. Comprehensive analysis of computational methods for predicting anti-inflammatory peptides. Ar chives of Computational Methods in Engineering , 31:3211–3229, 2024. [6] M. Khatun, M. M. Hasan, W . Shoombuatong, and H. Kurata. ProIn-Fuse: Improved and robust prediction of proinflammatory peptides by fusing of multiple feature representations. Journal of Computer-Aided Molecular Design , 34(12):1229–1236, 2020. [7] C. Y an, A. Geng, Z. Pan, Z. Zhang, and F . Cui. MultiFeatV otPIP: A voting-based ensemble learning frame work for predicting proinflammatory peptides. Briefings in Bioinformatics , 25(6):bbae505, 2024. [8] B. Manav alan, T . H. Shin, M. O. Kim, and G. Lee. PIP-EL: A new ensemble learning method for impro ved proinflammatory peptide predictions. F rontier s in Immunology , 9:1783, 2018. [9] L. Y ao, F . W ang, P . Xie, J. Guan, Z. Zhao, X. He, X. Liu, Y .-C. Chiang, and T .-Y . Lee. StackPIP: An effecti ve computational framew ork for accurate and balanced identification of proinflammatory peptides. Journal of Chemical Information and Modeling , 65(14):7777–7788, 2025. [10] K. M. Jenike, L. Campos-Domínguez, M. Boddé, J. Cerca, C. N. Hodson, M. C. Schatz, and K. S. Jaron. Guide to k-mer approaches for genomics across the tree of life. arXiv:2404.01519, 2024. [11] F . W aibl, M. L. Fernández-Quintero, F . S. W edl, et al. Comparison of hydrophobicity scales for predicting biophysical properties of antibodies. F rontier s in Molecular Biosciences , 9:960194, 2022. [12] D. Eisenber g, R. M. W eiss, T . C. T erwilliger , and W . W ilcox. Hydrophobic moments and protein structure. F araday Discussions of the Chemical Society , 17:109–120, 1982. [13] B. F . Shaw , H. Arthanari, M. Naro vlyansky , et al. Neutralizing positiv e charges at the surf ace of a protein lo wers its rate of amide hydrogen exchange without altering its structure or increasing its thermostability . J ournal of the American Chemical Society , 132(49):17411–17425, 2010. [14] C. O. Mackenzie and G. Grigoryan. Protein structural motifs in prediction and design. Curr ent Opinion in Structural Biology , 44:161–167, 2017. [15] H. Sun, B. Qiao, W . Choi, et al. Origin of proteolytic stability of peptide-brush polymers as globular proteomimetics. ACS Central Science , 7(12):2063–2072, 2021. [16] A. T . Müller , G. Gabernet, J. A. Hiss, and G. Schneider . modlAMP: Python for antimicrobial peptides. Bioinformatics , 33(17):2753–2755, 2017. [17] K. Zheng, S. Long, T . Lu, J. Y ang, X. Dai, M. Zhang, Z. Nie, W .-Y . Ma, and H. Zhou. ESM all-atom: Multi-scale protein language model for unified molecular modeling. arXiv:2403.12995, 2024. [18] R. Seoh, H.-S. Chang, and A. McCallum. Encoding multi-domain scientific papers by ensembling multiple CLS tokens. arXiv:2309.04333, 2023. [19] Y . Y e, H. Zhou, J. Cai, et al. Dynamic feature pruning and consolidation for occluded person re-identification. 2022. [20] Y .-H. Xiao, L. Huang, D. Ramírez, C. Qian, and H. C. So. One-bit cov ariance reconstruction with non-zero thresholds. arXiv:2303.16455, 2023. [21] M. Sun, Z. Liu, A. Bair , and J. Z. Kolter . A simple and effecti ve pruning approach for large language models. In Pr oceedings of ICLR 2024 . arXiv:2306.11695, 2024. [22] L. Gao and L. Guan. Interpretability of machine learning: Recent advances and future prospects. arXiv:2305.00537, 2023. [23] N. Abbood, J. Ef fert, K. A. J. Bozhueyuek, and H. B. Bode. Guidelines for optimizing type S nonribosomal peptide synthetases. A CS Synthetic Biology , 12(8):2432–2443, 2023. [24] Y . Niu, Z. Fabian, S. Lee, M. Soltanolkotabi, and S. A vestimehr . mL-BFGS: A momentum-based L-BFGS for distributed large-scale neural netw ork optimization. arXiv:2307.13744, 2023. [25] Q. Y ang. Blending ensemble for classification with genetic-algorithm generated alpha factors and sentiments (GAS). arXiv:2411.03035, 2024. [26] N. Das and M. Chakraborty . Machine learning-driven threshold optimization for wa velet packet denoising in EEG-based mental state classification. IEEE Access , 2023. [27] P . Greenside, M. Hillenmeyer , and A. Kundaje. Prediction of protein-ligand interactions from paired protein sequence motifs and ligand substructures. P acific Symposium on Biocomputing , 23:20–31, 2018. 10 A P R E P R I N T - F E B R UA RY 2 5 , 2 0 2 6 [28] X. Fu, Y . Y uan, H. Qiu, et al. AGF-PPIS: A protein-protein interaction s ite predictor based on an attention mechanism and graph con volutional networks. Methods , 222:142–151, 2024. [29] C. Xiao, Z. Zhou, J. She, et al. PEL-PVP: Application of plant vacuolar protein discriminator based on PEFT ESM-2 and bilayer LSTM in an unbalanced dataset. International Journal of Biolo gical Macr omolecules , 277:134317, 2024. [30] ¸ S. K. Çorbacıo ˘ glu and G. Aksel. Receiver operating characteristic curve analysis in diagnostic accuracy studies: A guide to interpreting the area under the curve v alue. T urkish J ournal of Emerg ency Medicine , 23(4):195–198, 2023. 11
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment