JEPA-DNA: Grounding Genomic Foundation Models through Joint-Embedding Predictive Architectures
Genomic Foundation Models (GFMs) have largely relied on Masked Language Modeling (MLM) or Next Token Prediction (NTP) to learn the language of life. While these paradigms excel at capturing local genomic syntax and fine-grained motif patterns, they o…
Authors: Ariel Larey, Elay Dahan, Amit Bleiweiss
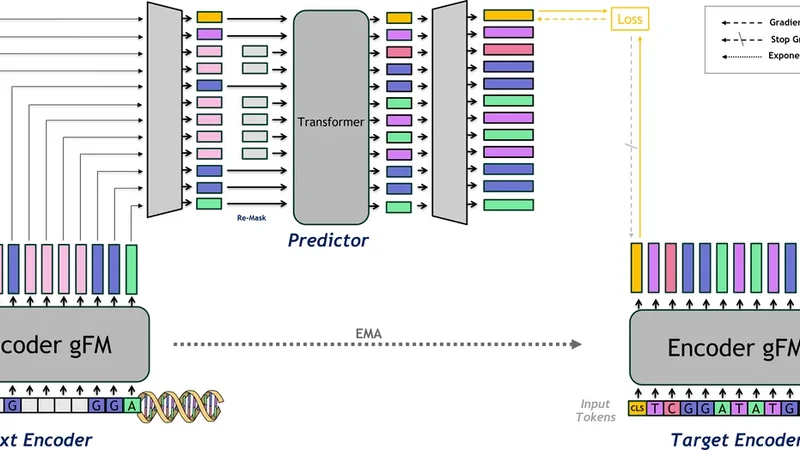
JEP A-DN A: Gr ounding Genomic F oundation Models thr ough Joint-Embedding Pr edicti ve Ar chitectur es Ariel Larey 1 Elay Dahan 2 Amit Bleiweiss 3 Raizy Kellerman 4 Guy Leib 4 Omri Nayshool 4 Dan Ofer 4 T al Zinger 4 Dan Dominissini 4 Gideon Rechavi 4 Nicole Bussola 5 Simon Lee 5 Shane O’Connell 5 Dung Hoang 5 Marissa Wirth 5 Alexander W . Charney 5 Nati Daniel 1 , ∗ Y oli Shavit 1 , ∗ Abstract Genomic Foundation Models (GFMs) have largely relied on Masked Language Modeling (MLM) or Next T oken Prediction (NTP) to learn the language of life. While these paradigms e xcel at capturing local genomic syntax and fine-grained motif patterns, they often fail to capture the broader functional context, resulting in representations that lack a global biological perspectiv e. W e introduce JEP A-DNA, a novel pre-training framework that integrates the Joint-Embedding Predictiv e Architecture (JEP A) with traditional generativ e objectiv es. JEP A-DN A introduces latent grounding by coupling token-le vel recov ery with a predictive objecti ve in the latent space by supervising a CLS token. This forces the model to predict the high-level functional embeddings of masked genomic se gments rather than focusing solely on individual nucleotides. JEP A-DN A extends both NTP and MLM paradigms and can be deployed either as a standalone from-scratch objecti v e or as a continual pre-training enhancement for existing GFMs. Our ev aluations across a div erse suite of genomic benchmarks demonstrate that JEP A-DNA consis- tently yields superior performance in supervised and zero-shot tasks compared to generativ e-only baselines. By pro viding a more robust and biologically grounded representation, JEP A-DN A of fers a scalable path to ward foundation models that understand not only the genomic alphabet, but also the underlying functional logic of the sequence. 1 Introduction Genomic Foundation Models (GFMs) ha ve the potential to significantly adv ance our understanding of genomes and ho w DN A elements at v arious scales interact to gi ve rise to comple x functions [ 1 ]. These models, including DNABER T -2 [ 2 ], Nucleotide Transformer [ 3 ], HyenaDN A [ 4 ], and Evo [ 5 ], are Large Language Models (LLMs) adapted for DN A sequences, operating with conte xt windo ws ranging from a few thousand base pairs to megabase-scale inputs. T ypically , these architectures ∗ Co-corresponding authors: ndaniel@nvidia.com , yolis@nvidia.com . 1 Applied AI Architecture, NVIDIA, Israel. 2 W orldwide Field Ops, NVIDIA, Israel. 3 Dev eloper Programs, NVIDIA, Israel. 4 Cancer Research Center and W ohl Institute of T ranslational Medicine, Sheba Medical Center , T el Hashomer, Israel. 5 W indreich Department of AI and Human Health, Icahn School of Medicine at Mount Sinai, Ne w Y ork, USA. Preprint. rely on self-supervised token prediction objecti ves, such as Masked Language Modeling (MLM) or autoregressi ve Ne xt T oken Prediction (NTP), to learn genomic representations. While effecti ve for identifying local motifs and sequence patterns [ 2 , 3 ], these methods face a fundamental limitation in capturing the broader, functional logic of the genome [ 1 , 6 ]. W e term this limitation the "granularity trap" . In the MLM/NTP paradigms, the model is tasked with reconstructing individual masked tokens (e.g., A, C, T , G ). While this encourages a high-fidelity understanding of local syntax, it does not inherently require the model to internalize the high-le vel biological consequences of a sequence. Consequently , these models may ov er-allocate capacity to high-frequency "noise," such as non-coding repetitive elements or neutral polymorphisms, while failing to ground representations in global functional contexts, such as long-range enhancer-promoter interactions. T o bridge the gap between genomic syntax and biological semantics, we introduce JEP A-DNA , a novel frame work that incorporates the Joint-Embedding Predictiv e Architecture (JEP A) [ 7 ] into genomic pre-training. Unlike generativ e objectives that operate in the raw token space, the JEP A paradigm predicts the latent representations of mask ed segments. While Joint-Embedding architec- tures have seen preliminary success in transcriptomics for modeling gene expression vectors [ 8 ], JEP A-DN A represents the first adoption of this paradigm to the high-resolution, multiscale domain of genomic sequences. By coupling token-le vel recovery with a predicti ve objectiv e in the latent space, supervised via a [ C LS ] token, JEP A-DNA encourages the learning of abstract, functional features that are in variant to lo w-lev el sequence noise. Our approach is uniquely versatile: JEP A-DN A can be deplo yed as a standalone pre-training objecti ve or as a continuous pre-training phase to "ground" existing GFMs, extending both NTP and MLM paradigms and different architectures. This "latent grounding" serves as a correctiv e layer for pre- trained models, anchoring their token-le vel kno wledge to a more stable, semantic world model of genomic function. W e ev aluate JEP A-DNA across a suite of genomic benchmarks, specifically focusing on linear probing and zero-shot protocols to isolate the quality of the learned representations. Our empirical results demonstrate that latent grounding consistently ele vates performance across functional tasks compared to generati ve-only baselines. Ultimately , JEP A-DN A demonstrates that moving be yond local nucleotide reconstruction is essential for developing foundation models that internalize the high-lev el regulatory mechanisms governing the genome. In summary , our contributions are as follo ws: • W e introduce a nov el application of Joint-Embedding Predictive Architectures to the genomic domain, shifting the pre-training focus from literal token reconstruction to latent feature prediction. • W e empirically demonstrate that by operating in the embedding space, our model captures higher-order functional semantics that standard MLM and NTP objecti ves may ignore. • Our proposed method can be used for training models from scratch or as a refinement phase for GFMs and is compatible across architectures, pro viding a consistent means to learn functional sequence features. • Through linear probing and zero-shot experiments, we establish that JEP A-DN A learns more linearly-separable and biologically-relev ant features than generativ e baselines that use standard LLM objectiv es. 2 Related W ork 2.1 Pre-training Paradigms of Genomic Foundation Models (GFMs) The de velopment of GFMs has been largely inspired by the success of Large Language Models (LLMs) in Natural Language Processing. Early iterations, such as DNABER T [ 9 ], adapted the BER T architecture [ 10 ] replacing its original W ordPiece subword tokenizer with k-mer tokenization to capture bidirectional context within the genome. More recently , DN ABER T -2 [ 2 ] and the Nucleotide T ransformer [ 3 ] expanded this scale by training on diverse multi-species datasets, demonstrating that larger conte xt windo ws and higher parameter counts can improv e "out-of-the-box" performance on downstream tasks lik e promoter prediction and v ariant effect prediction. 2 Beyond T ransformer-based architectures, recent advancements ha ve focused on overcoming the quadratic scaling of self-attention to model longer genomic dependencies. HyenaDN A [ 4 ] utilizes the Hyena operator to process sequences at single-nucleotide resolution across long contexts. Similarly , Evo2 [ 5 ] le verages a StripedHyena backbone to push the conte xt size to me gabase scales. Despite these structural innov ations, nearly all current GFMs rely on tok en-lev el reconstruction objecti ves, namely MLM and NTP . While these methods are ef fective for learning the "syntax" of DN A, they often fail to capture the structural "logic" or functional state of the genome [ 6 ] because the loss function is applied solely in the potentially "noisy" token space. Alternativ e self-supervised strate gies hav e sought to move beyond literal token reconstruction by incorporating ev olutionary or contrastiv e constraints. Models such as GPN-MSA [ 11 ] lev erage multi- sequence alignment (MSA) to learn from cross-species conservation patterns, identifying functional constraints through substitution probabilities. In parallel, contrasti ve learning framew orks like DN ASimCLR [ 12 ] employ data augmentations to learn representations by minimizing the distance between similar sequence "views" in latent space. Ho wever , these paradigms in volv e significant trade-offs: MSA-based methods require computationally expensiv e preprocessing and are limited by the av ailability of high-quality alignments, while contrasti ve methods often necessitate non-tri vial augmentations or costly negativ e mining. In contrast, JEP A-DN A captures functional in variants directly from the raw sequence. By utilizing a predictive latent objective, our approach bypasses the need for negati ve samples or complex alignment pipelines, of fering a more scalable path to ward grounding genomic foundation models in biological semantics. 2.2 Joint-Embedding Predictiv e Architectur e (JEP A) In computer vision, the Joint-Embedding Predictiv e Architecture (JEP A) has emerged as a po werful alternati ve to generati ve modeling. In this paradigm, a model is task ed with predicting the latent repre- sentation of a masked segment rather than its literal pixels [ 13 ]. This shift from signal reconstruction to representation prediction allows the model to ignore unpredictable, lo w-level details and focus on semantically rich features. This concept was recently extended to the language domain with LLM- JEP A [ 14 ], which proposes a first step tow ards coupling token-lev el recovery with sequence-le vel latent grounding for natural language tasks. W ithin the biological domain, the JEP A framework w as recently introduced by GeneJEP A for single- cell transcriptomics [ 8 ]. Ho we ver , GeneJEP A operates on gene-expression vectors, design to learn and reason ov er gene-gene relationships within a cell. In contrast, JEP A-DNA focuses on the primary genomic sequence: the "blueprint" itself. Unlike transcriptomic models that process tabular gene sets, our framew ork must handle the multiscale nature of DNA, where functional meaning is encoded through sequential motifs and long-range dependencies. By applying the JEP A paradigm directly to raw sequences, we enable the learning of "world models" [7] for genomic function. 3 Method The JEP A-DN A framew ork treats the genome not merely as a sequence of tokens, but as a structured signal with both local syntax and global semantics. W e achiev e this by augmenting a standard backbone E with a Joint-Embedding Predicti ve Architecture (JEP A) branch, enabling a dual-objecti ve learning process. 3.1 Architectur e Components The JEP A-DN A architecture is illustrated in Fig. 1. It consists of three primary modules: • Context Encoder ( E θ ): A sequence backbone (e.g., T ransformer, State-Space Model) that processes the input sequence x . W e prepend a learnable [ C LS ] token to the input, such that h cls ∈ R d captures a global summary of the visible context. • T arget Encoder ( E ¯ θ ) : A structural duplicate of the context encoder , whose weights ¯ θ are updated via an Exponential Moving A verage (EMA) of θ . This encoder processes the unmasked tar get sequence to provide stable latent tar gets. • Predictor Head ( P ϕ ): A network designed to map context representations into the tar get latent space. While the predictor is conditioned on the entire encoded sequence from the 3 Figure 1: The JEP A-DN A Architecture. context encoder le veraging both spatial token information and the global [ C LS ] embedding, its objectiv e is specifically to predict the [ C LS ] latent representation of the target sequence. This allo ws the predictor to utilize the full richness of the context to estimate a high-le vel summary of the target. 3.2 Masking and Re-masking Strategy The JEP A-DN A framew ork relies on a dual-masking process, to ensure the predictor does not see the masked content. Initial Masking. Giv en an input sequence, a subset of tokens is replaced by a special [ M AS K ] token. This masked sequence is subsequently processed by the context encoder E θ to yield latent representations. In contrast to standard Masked Language Modeling (MLM) protocols that typically mask random independent tokens (approximately 15%), our approach employs a span-based masking strategy . W e sample multiple contiguous target regions rather than individual tokens, resulting in a higher aggregate masking ratio (typically exceeding 20%), in alignment with the JEP A context encoder masking protocol in vision [ 13 ]. In particular , this masking configuration is propagated to both the JEP A loss and the standard LLM loss, thereby enforcing a uniformly more challenging reconstruction objectiv e. Re-masking Strategy . T o prevent the predictor from having a tri vial mapping to the targets, we introduce a re-masking step, where the outputs of E θ at the masked positions are replaced by the [ M AS K ] . Note that the predictor head recei ves the full sequence of conte xt encodings, including the global [ C LS ] representation. By conditioning the predictor on the entire encoded sequence rather than a single vector , the model can lev erage both localized spatial information and the global sequence summary to accurately reconstruct the target [ C LS ] latent. 3.3 Multi-Objective Pre-training The model is trained by minimizing a composite loss function that balances language modeling, latent prediction, and embedding div ersity . 4 3.3.1 LLM Loss ( L llm ) T o maintain nucleotide-level precision, we retain the standard LLM objecti ve (MLM or NTP). For a masked sequence with indices M , the loss is: L llm = − X i ∈M log P ( x i | h i ) (1) where h i denotes the hidden state of the genomic token, and the set of masked indices M is deriv ed from the masking strategy . 3.4 Latent Predicti ve Loss ( L j epa ) The JEP A objecti ve grounds the model by forcing the [ C LS ] token to capture functional semantics. The predictor head attempts to match the directional orientation of the embedding produced by the target encoder in the latent space. W e define this loss using cosine similarity: L j epa = 1 − P ϕ ( h cls ) · z targ et ||P ϕ ( h cls ) || 2 · || z targ et || 2 (2) where z targ et = E ¯ θ ( x ) cls . Minimizing this objecti ve encourages the predicted representation of the context encoder to align with the functional embedding of the tar get encoder . 3.4.1 V ariance and Covariance Regularization T o prevent the "collapse" problem common in non-contrastiv e methods (where the model outputs a constant vector), we utilize variance and cov ariance constraints on the latent vectors Z ∈ R B × d across a batch of size B , following the VICRe g framework [15]. V ariance Loss ( L v ar ). The v ariance loss ensures that each embedding dimension maintains suf fi- cient variance across the batch, pre venting informational collapse: L v ar = 1 d d X j =1 max(0 , γ − σ ( z : ,j )) (3) where σ ( z : ,j ) is the standard deviation of the j -th dimension across the batch, and γ is a constant threshold (typically γ = 1 ). Covariance Loss ( L cov ). The cov ariance loss encourages decorrelation between different embed- ding dimensions, prev enting redundant representations: L cov = 1 d X i = j C 2 ij (4) where C ∈ R d × d is the cov ariance matrix of the centered embeddings: C = 1 B − 1 ( Z − ¯ Z ) ⊤ ( Z − ¯ Z ) (5) and ¯ Z denotes the batch mean. By penalizing the squared of f-diagonal elements of the co variance matrix, this loss encourages each dimension to encode independent information. 3.4.2 T otal Objective The final optimization problem is defined as: min θ,ϕ L total = λ 1 L llm + λ 2 L j epa + λ 3 L v ar + λ 4 L cov (6) where λ 1 , 2 , 3 , 4 are hyper-parameters that weigh the contrib ution of each objective. 5 3.5 Compatibility across Model Architectur es and Generative Objectives The JEP A-DN A frame work is model-agnostic and compatible with MLM and NTP objecti ves. Its primary requirement is an aggregation operator that compresses a sequence into a latent representation. While we utilize the [ C LS ] token for this purpose, it can be seamlessly replaced by learned global pooling or alternativ e aggregation methods. While the [ C LS ] token is "native" to the Transformer Encoder architecture which is trained with MLM objectiv es [ 10 ], where the token is preprended to sequence, JEP A-DNA also extends naturally to T ransformer Decoders, State Space Models (SSMs) and Long-Con volution backbones (e.g., HyenaDN A [ 4 ]) which are trained with NTP objectiv es. In these architectures, we append an [ E O S ] token at the end of the sequence instead of using the last token. By supervising this appended special token with the JEP A loss, regardless of the generati ve objectiv e or specific encoder architecture, we enforce a global pooling mechanism, ef fecti vely compensating for the otherwise local or recurrent nature of these operators. 4 Experimental Results In this section, we e valuate the impact of the JEP A-DNA framework on genomic representation learning. W e compare our approach against a GFM baseline across a suite of supervised and zero-shot genomic benchmarks to demonstrate the robustness and adaptability of the learned features. 4.1 Experimental Setup Models. W e utilize DNABER T -2 as the backbone for our context and target encoders. This model is based on the BER T architecture with 12 T ransformer Encoder layers, a hidden dimension of 768, and 12 attention heads, totaling approximately 117M parameters. DN ABER T -2 employs Byte Pair Encoding (BPE) tokenization, enabling efficient handling of variable-length sequences up to 512 tokens. The model was originally pre-trained on approximately 35B base pairs from human and multi-species genomes using masked language modeling (MLM) [2]. The JEP A predictor is a lightweight 3-layer Transformer Encoder operating in a reduced latent space of 384 dimensions, with 3 attention heads. The predictor employs a pre-norm architecture with GELU activ ations and frozen sinusoidal positional embeddings. Input embeddings from the context encoder are first projected to the predictor dimension, with mask ed target positions replaced by learnable mask tokens before adding positional embeddings following the re-masking strategy described in Section 3.2. The predictor output for the [CLS] token is projected back to the original 768-dimensional space for computing the prediction loss against target encoder representation. Datasets. For pre-training, we utilize a subset of the training data used for DN ABER T -2. This comprises the human reference genome (GRCh38) [ 16 ], combined with genomic data from fi ve representativ e model organisms: mouse [ 17 ], zebrafish [ 18 ], fruit fly [ 19 ], nematode [ 20 ], and thale cress [ 21 ]. All genome sequences were obtained from the UCSC Genome Browser [ 22 ]. Sequences are filtered to include only valid nucleotides (A, T , C, G) and chunked into fixed-length segments with 50% ov erlap, yielding approximately 4.76M training sequences spanning approximately 7.6B base pairs. For do wnstream ev aluation, we test on a div erse suite of supervised and zero-shot tasks derived from established genomic benchmarks. Our supervised ev aluation includes three standard classification tasks from the GUE benchmark [ 2 ]: promoter prediction, transcription factor binding site prediction, and splice site prediction. Additionally , we utilize variant ef fect prediction tasks from V ariantBench- marks [ 23 ], cov ering coding and non-coding pathogenicity classification, common vs. rare variant identification, and quantitativ e trait loci (QTL) prediction for expression, methylation (meQTL), and splicing (sQTL) effects. W e also ev aluate on the causal eQTL task from the Long Range Benchmark (LRB) [24], which in volves high-conte xt inputs. For zero-shot e valuation, we assess performance on v ariant ef fect prediction without task-specific fine-tuning. This includes the BEND benchmark [ 25 ] for expression- and disease-associated variants, and T raitGym [ 26 ] for predicting Mendelian and comple x traits. Furthermore, we ev aluate clinical pathogenicity prediction using the Song-Lab ClinV ar dataset [ 27 ] and the non-coding pathogenic OMIM task from LRB [24]. 6 Evaluation Protocol. W e employ two primary e valuation strate gies to assess the quality of learned representations: • Linear Probing: T o isolate the quality of the pre-trained features, we keep the backbone frozen and train only a linear classifier on the e xtracted [CLS] token representations. Per- formance is measured using three complementary metrics: (1) Area Under the Receiver Operating haracteristic curve (A UR OC), which measures the model’ s ability to discriminate between classes across all classification thresholds; (2) Area Under the Precision-Recall Curve (A UPRC), which is particularly informative for imbalanced datasets common in genomics tasks; and (3) Matthe ws Correlation Coefficient (MCC), a balanced metric that accounts for all four confusion matrix categories and remains robust even when class distributions are highly sk ewed. • Zero-Shot Inference: W e ev aluate the model’ s out-of-the-box semantic understanding by computing the cosine similarity between embeddings of reference and variant sequences. For variant effect prediction tasks (e.g., ClinV ar or T raitGym tasks), we extract [CLS] representations for both the reference and mutant sequences and measure their embedding angular distance. W e assess the model’ s ability to rank functional v ariants without any task-specific training by computing A UR OC and A UPRC over the similarity scores. T raining & Implementation Details. All models were implemented in PyT orch with Flash Atten- tion for efficient self-attention computation and trained on two NVIDIA GPUs using DataParallel for multi-GPU support. The context and tar get encoders are both initialized from pre-trained DN ABER T -2 weights, while the predictor network is initialized from scratch using a truncated normal distrib ution with standard deviation 0.02 for all linear layer weights and zero initialization for biases. For masking strategy , we sample 1-to-3 contiguous target regions per sequence, cov ering 20-40% of the sequence length. The context encoder processes the masked sequence while the target encoder (updated via exponential mo ving av erage) processes the full unmasked sequence. T raining follo ws a multi-phase schedule optimized for continual training: • Phase 1 (Predictor W armup): The encoder is frozen for 1,000 steps while only the predictor is trained at a learning rate of 1 × 10 − 5 . This allo ws the predictor to learn meaningful initial representations before jointly training with the encoder . • Phase 2 (Full T raining): The encoder is unfrozen with a linear warmup of 500 steps from 3 × 10 − 6 to 5 × 10 − 6 peak learning rate, followed by cosine decay to 1 × 10 − 6 . W e use Stochastic Gradient Descent (SGD) [ 28 ] optimizer with momentum 0.9, batch size of 32 with gradient accumulation ov er 4 steps (ef fecti ve batch size of 128), and constant weight decay of 0.01. T raining proceeds for 5 epochs. The target encoder is updated via exponential mo ving av erage (EMA) with momentum scheduled from 0.996 to 1.0 ov er training. The training objectiv e combines multiple losses: • CLS-JEP A Loss: Cosine similarity loss between the predicted and target CLS representa- tions, encouraging the conte xt encoder to capture global sequence semantics from partial observations. • MLM Loss: Standard masked language modeling loss on the masked token positions, using the pre-trained MLM head. • V ariance Loss: Hinge-based variance loss with weight 25.0 to prevent representation collapse by ensuring v ariance abov e a threshold (1.0) across the batch dimension. The variance is computed on the [CLS] token embeddings from both the context encoder output and the predictor output. T o av oid artificial v ariance that does not reflect true representational di versity , we employ two strate gies: (1) v ariance calculations are performed via an additional forward pass through the conte xt encoder and predictor in ev aluation mode, eliminating stochastic effects from dropout and random masking; and (2) all sequences within each batch are truncated to a uniform length, prev enting spurious variance arising from heterogeneous padding patterns. 7 • Cov ariance Loss: W ith weight 0.5 to decorrelate embedding dimensions and encourage div erse feature learning. For downstream ev aluation, we encounter tasks where the default sequence length exceeds the 512-token context window of DNABER T -2. In these instances, inputs are truncated to retain the central region. W e utilize a linear probing approach where the encoder weights are frozen, and a linear classification head is trained on the [CLS] token representations. For standard single-sequence tasks, the projection layer operates directly on the [CLS] embedding. In variant effect prediction, the reference and variant sequences are processed independently , and their corresponding [CLS] embeddings are concatenated prior to classification. T o ensure rigorous comparison, all supervised tasks follow a unified training protocol: the classifier is trained for 3 epochs using the AdamW optimizer with a learning rate of 3 × 10 − 5 , a weight decay of 0.01, and a batch size of 32. 4.2 Downstr eam T asks Linear Evaluation. Extending a DNABER T -2 baseline with JEP A-DN A results in a consistent improv ement across various genomic tasks (T able 1). W e e valuate models via linear probing, keeping the encoder weights frozen while training a linear classifier on the extracted features. This approach ensures that the observed performance gains are a direct result of the superior feature representations acquired during JEP A-DN A pre-training, rather than task-specific fine-tuning. T able 1: Performance comparison across supervised tasks. W e ev aluate DNABER T -2 (baseline) and DN ABER T -2 with JEP A-DN A (ours), when fine-tuned (linear probing) on a suite of genomic tasks. Gain is calculated relative to the baseline A UROC; ’–’ denotes cases where both models exhibit near-random performance. T ask Seq. Len. DNABER T -2 JEP A-DNA (Ours) Gain MCC A UR OC A UPRC MCC A UR OC A UPRC (A UR OC) GUE TF Binding 100 0.432 0.783 0.765 0.459 0.808 0.795 +3.193% GUE Promoter 300 0.680 0.916 0.907 0.714 0.925 0.916 +0.983% GUE Splice Site 400 0.000 0.623 0.425 0.069 0.653 0.448 +4.815% VB Coding Pathogenicity 1024 -0.045 0.569 0.462 0.091 0.603 0.516 +5.975% VB Non-coding Pathogen. 1024 0.000 0.590 0.137 0.000 0.593 0.146 +0.508% VB Expression Effect 1024 0.192 0.627 0.600 0.217 0.633 0.595 +0.957% VB Common vs. Rare 1024 0.013 0.506 0.502 0.022 0.519 0.515 – VB meQTL 1024 0.020 0.563 0.495 -0.001 0.586 0.510 +4.085% VB sQTL 1024 0.098 0.567 0.567 0.095 0.564 0.565 -0.529% LRB Causal eQTL 12000 0.307 0.704 0.726 0.310 0.705 0.725 +0.142% Zero-Shot T asks. W e further ev aluate the quality of the learned representations through zero-shot inference, measuring the cosine similarity between embeddings of input sequences. As summarized in T able 2, the integration of JEP A-DNA yields consistent improvements o ver the DNABER T -2 baseline across a div erse range of sequence lengths and biological phenomena. Notably , we observe substantial gains in identifying e xpression effects (+6.939%) and Mendelian traits (+7.298%). The superior zero-shot performance indicates that forcing the model to predict global latent sequence representation, yields superior features for genomic tasks. 5 Limitations and Future W ork While our work provide a proof-of-concept for the introduction of JEP A objectiv e to GFM pre-training, we plan to extend it, considering se veral directions: • Pre-training Strategy: Evaluating the comparati ve eff ects of training models from scratch versus utilizing JEP A-DNA for continual pre-training on e xisting checkpoints. 8 T able 2: Performance comparison across zero-shot tasks. W e ev aluate DNABER T -2 (baseline) and DNABER T -2 with JEP A-DNA (ours), based on the cosine similarity of embeddings. Gain is calculated relativ e to the baseline A UROC; ’–’ denotes cases where both models exhibit near -random performance. T ask Seq. Len. DNABER T -2 JEP A-DNA (Ours) Gain A UR OC A UPRC A UROC A UPRC (A UR OC) BEND Expression Effect 512 0.490 0.072 0.524 0.080 +6.939% BEND Disease V ariant 512 0.498 0.072 0.512 0.075 – T raitGym Complex 4096 0.499 0.100 0.491 0.099 – T raitGym Mendelian 4096 0.507 0.107 0.544 0.126 +7.298% Songlab ClinV ar 5994 0.528 0.570 0.544 0.585 +3.030% LRB Pathogenic OMIM 12000 0.495 0.002 0.452 0.002 – • GFM Architectures: T esting the impact of JEP A-DNA across a wider v ariety of GFMs beyond DN ABER T -2, including SSMs and Long-Conv olution backbones. • Masking Strategies: Studying alternati ve strategies such as biologically-informed multi- block masking to capture higher -order genomic dependencies to ensure we keep global semantic intact. • Aggregation Strate gies: Exploring ho w local and global information are fused together to effecti vely support both global and local tasks. • Auxiliary T asks: In vestigating whether sequence representation learning can be further enhanced by incorporating additional tasks, such as next sequence prediction, inspired by the ’next-sentence-prediction’ of BER T architectures [10]. • JEP A-Centric Modeling: In vestigating the transition of JEP A from a supplementary objective to the primary pre-training task, potentially bypassing traditional generativ e objectiv es entirely . • Comprehensiv e Ablations: Conducting a deeper analysis of the predictor head architec- ture, the specific context provided to the predictor , loss hyper-parameters, and optimizer configurations to ensure stability and prev ent latent mode collapse. • Comparativ e Analysis of SSL paradigms: Comparing JEP A-DNA to generativ e and con- trastiv e baselines. • Significance Analysis: Reporting confidence interv als due to the mission-critical nature of biological and clinical tasks, and ev aluating the significance of our reported improvement. 6 Conclusion In this work, we introduced JEP A-DN A, a v ersatile frame work that adapts the Joint Embedding Predictiv e Architecture for genomic sequence modeling. By shifting the focus from tok en-lev el reconstruction to the prediction of latent representations, we provide a method that captures high- lev el biological context more effecti vely than traditional generati ve objectives alone. Our results demonstrate that JEP A-DN A consistently improves the performance of established backbones like DN ABER T -2 across both supervised downstream tasks and zero-shot e valuations. References [1] Gonzalo Benegas, Chengzhong Y e, Carlos Albors, Jianan Canal Li, and Y un S Song. Genomic language models: opportunities and challenges. T rends in Genetics , 2025. [2] Zhihan Zhou, Y anrong Ji, W eijian Li, Pratik Dutta, Ramana Davuluri, and Han Liu. Dnabert- 2: Efficient foundation model and benchmark for multi-species genome. arXiv pr eprint arXiv:2306.15006 , 2023. 9 [3] Lorenzo Dalla-T orre, Nicolás Bene gas, Daria Grechishnikov a, et al. The nucleotide transformer: Building and ev aluating robust foundation models for human genomics. Nature Methods , 2023. [4] Michael Poli, T ri Dao, et al. Hyenadna: Long-range genomic sequence modeling at single nucleotide resolution. arXiv preprint , 2023. [5] Eric Nguyen, Michael Poli, Matthew G Durrant, Brian Kang, Dhruva Katrekar , David B Li, Liam J Bartie, Armin W Thomas, Samuel H King, Garyk Brixi, et al. Sequence modeling and design from molecular to genome scale with ev o. Science , 386(6723):eado9336, 2024. [6] Gonzalo Benegas, Sanjit Singh Batra, and Y un S Song. Dna language models are powerful predictors of genome-wide v ariant ef fects. Pr oceedings of the National Academy of Sciences , 120(44):e2311219120, 2023. [7] Y ann LeCun. A path to wards autonomous machine intelligence. Open Review , 2022. [8] Elon Litman, T yler Myers, V inayak Agarwal, Ekansh Mittal, Orion Li, Ashwin Gopinath, and T imothy Kassis. Genejepa: A predicti ve world model of the transcriptome. bioRxiv , 2025. [9] Y u Ji, Zhiqiang Zhou, Han Liu, and Ramana V Davuluri. Dnabert: a comprehensiv e predictor for dna sequences based on deep transfer learning. Bioinformatics , 37(24):4776–4783, 2021. [10] Jacob De vlin, Ming-W ei Chang, Kenton Lee, and Kristina T outanov a. BER T: Pre-training of deep bidirectional transformers for language understanding. In Jill Burstein, Christy Doran, and Thamar Solorio, editors, Pr oceedings of the 2019 Confer ence of the North American Chapter of the Association for Computational Linguistics: Human Language T echnologies, V olume 1 (Long and Short P apers) , pages 4171–4186, Minneapolis, Minnesota, June 2019. Association for Computational Linguistics. [11] Gonzalo Benegas, Carlos Albors, Alan J A w , Chengzhong Y e, and Y un S Song. Gpn-msa: an alignment-based dna language model for genome-wide variant effect prediction. bioRxiv , pages 2023–10, 2024. [12] Minghao Y ang, Zehua W ang, Zizhuo Y an, W enxiang W ang, Qian Zhu, and Changlong Jin. Dnasimclr: a contrasti ve learning-based deep learning approach for gene sequence data classifi- cation. BMC bioinformatics , 25(1):328, 2024. [13] Mahmoud Assran, Quentin Duval, Ishan Misra, Piotr Bojanowski, Pascal V incent, Michael Rabbat, Y ann LeCun, and Nicolas Ballas. Self-supervised learning from images with a joint- embedding predictiv e architecture. In Pr oceedings of the IEEE/CVF Conference on Computer V ision and P attern Recognition (CVPR) , pages 15619–15629, 2023. [14] Hai Huang, Y ann LeCun, and Randall Balestriero. Llm-jepa: Large language models meet joint embedding predictiv e architectures. arXiv pr eprint arXiv:2509.14252 , 2025. [15] Adrien Bardes, Jean Ponce, and Y ann LeCun. V icreg: V ariance-in v ariance-cov ariance regular - ization for self-supervised learning. arXiv preprint , 2021. [16] V alerie A Schneider, Tina Graves-Lindsay , Kerstin Howe, Nathan Bouk, Hsiu-Chuan Chen, Paul A Kitts, T erence D Murphy , Kim D Pruitt, Françoise Thibaud-Nissen, Derek Albracht, et al. Evaluation of grch38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly . Genome resear ch , 27(5):849–864, 2017. [17] Deanna M Church, Leo Goodstadt, LaDeana W Hillier , Michael C Zody , Stev e Goldstein, Xingyi She, Carol J Bult, Richa Agarw ala, J Michael Cherry , Michael DiCuccio, et al. Modernizing reference genome assemblies. PLoS Biology , 9(7):e1001091, 2011. [18] K erstin Ho we, Matthe w D Clark, Carlos F T orroja, James T orrance, Camille Berthelot, Matthieu Muf fato, John E Collins, Sean Humphray , Karen McLaren, Lucy Matthews, et al. The zebrafish reference genome sequence and its relationship to the human genome. Natur e , 496(7446):498– 503, 2013. 10 [19] Roger A Hoskins, Joseph W Carlson, Kenneth H W an, Soo Park, Iv onne Mendez, Samuel E Galle, Benjamin W Booth, Barret D Pfeiff er , Reed A George, Robert Svirskas, et al. The release 6 reference sequence of the Drosophila melanogaster genome. Genome Researc h , 25(3):445–458, 2015. [20] The C. ele gans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for in vestigating biology . Science , 282(5396):2012–2018, 1998. [21] The Arabidopsis Genome Initiati ve. Analysis of the genome sequence of the flowering plant arabidopsis thaliana. Nature , 408(6814):796–815, 2000. [22] W James Kent, Charles W Sugnet, T errence S Furey , Krishna M Roskin, T om H Pringle, Alan M Zahler , and David Haussler . The human genome browser at UCSC. Genome Resear ch , 12(6):996–1006, 2002. [23] Aleksandr Medvede v , Karthik V iswanathan, Pra veenkumar Kanithi, Kirill V ishniakov , Prateek Munjal, Clement Christophe, Marco AF Pimentel, Ronnie Rajan, and Shadab Khan. Bioto- ken and biofm - biologically-informed tokenization enables accurate and ef ficient genomic foundation models. bioRxiv , 2025. [24] Evan T rop, Y air Schiff, Edgar Mariano Marroquin, Chia Hsiang Kao, Aaron Gokaslan, McKin- ley Polen, Mingyi Shao, A ymen Kallala, Bernardo P de Almeida, Thomas PIERRO T , Y ang I Li, and V olodymyr Kulesho v . The genomics long-range benchmark: Advancing DN A language models, 2025. [25] Frederikke I. Marin, Felix T eufel, et al. Bend: Benchmarking dna language models on biologi- cally meaningful tasks. arXiv preprint , 2024. [26] Gonzalo Benegas, Gökcen Eraslan, and Y un S Song. Benchmarking dna sequence models for causal regulatory v ariant prediction in human genetics. bioRxiv , pages 2025–02, 2025. [27] Gonzalo Benegas, Carlos Albors, Alan J. A w , Chengzhong Y e, and Y un S. Song. A dna language model based on multispecies alignment predicts the effects of genome-wide v ariants. Natur e Biotechnology , 43(12):1960–1965, 2025. [28] Léon Bottou. Large-scale machine learning with stochastic gradient descent. In Pr oceedings of COMPST AT’2010: 19th International Conference on Computational StatisticsP aris F rance, August 22-27, 2010 K eynote , In vited and Contributed P apers , pages 177–186. Springer , 2010. 11
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment