Sparse autoencoders (SAEs) are a mechanistic interpretability technique that have been used to provide insight into learned concepts within large protein language models. Here, we employ TopK and Ordered SAEs to investigate an autoregressive antibody language model, p-IgGen, and steer its generation. We show that TopK SAEs can reveal biologically meaningful latent features, but high feature concept correlation does not guarantee causal control over generation. In contrast, Ordered SAEs impose an hierarchical structure that reliably identifies steerable features, but at the expense of more complex and less interpretable activation patterns. These findings advance the mechanistic interpretability of domain-specific protein language models and suggest that, while TopK SAEs are sufficient for mapping latent features to concepts, Ordered SAEs are preferable when precise generative steering is required.
Antibodies are a key part of the body's adaptive immune response. They are characterised by their ability to bind to a specific antigen and subsequently neutralise it or initiate an immune response. Their extensive sequence-and therefore structural-diversity enables binding to virtually any target antigen (Chiu et al., 2019).
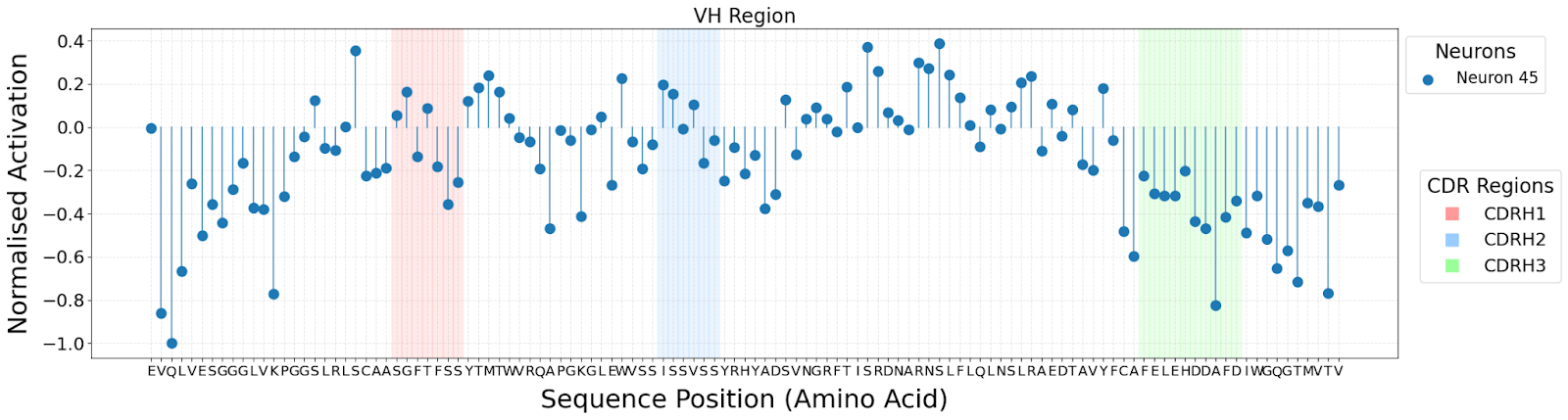
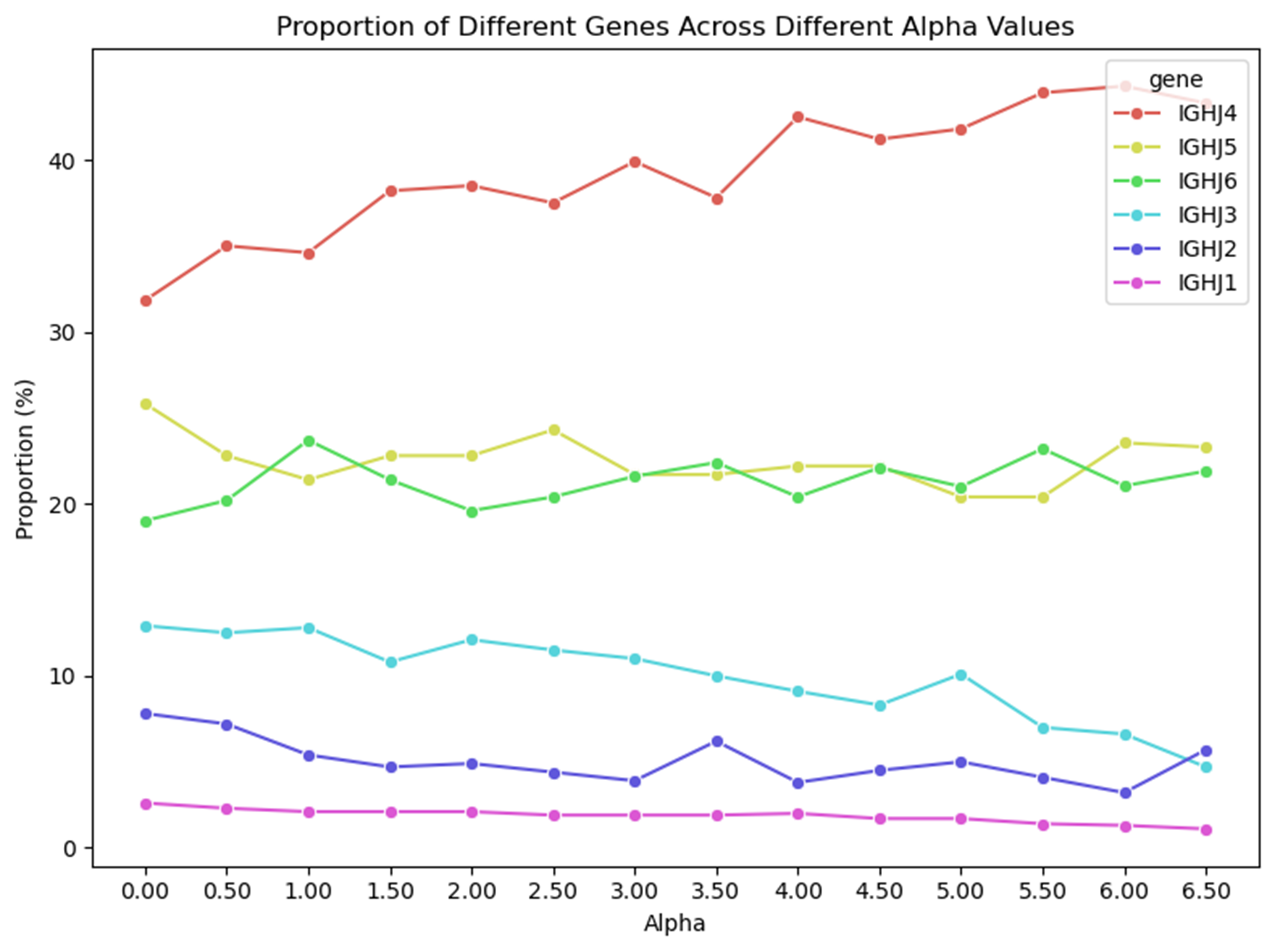
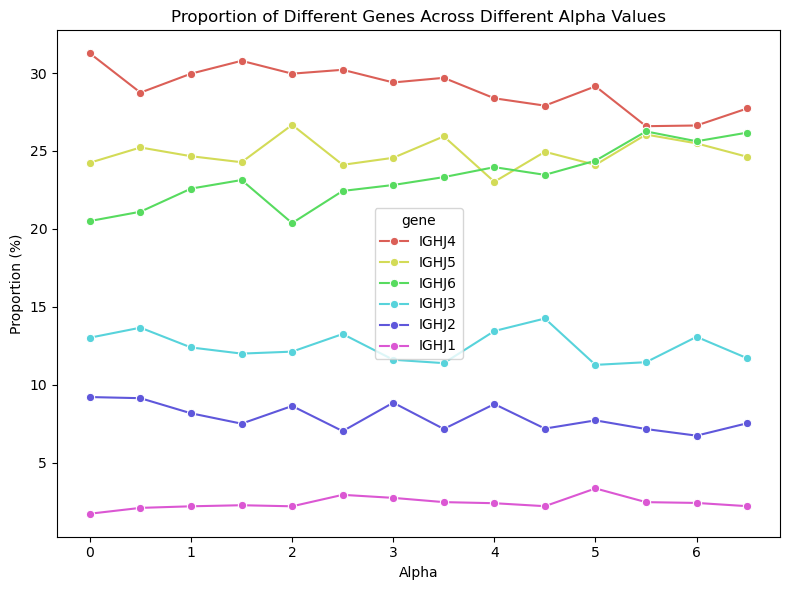
The antigen-binding domain of antibodies is made up of variable heavy (VH) and variable light (VL) chains. Antibody-antigen binding specificity and affinity are largely determined by structural units known as complementaritydetermining regions (CDRs), with each VH and VL chain having 3 CDRs, making a total of 6. Within our genome, there are numerous V, D, and J gene segments which together code for the VH and VL chains.
The combinatorial assembly of the discrete gene segments-V (variable), D (diversity), and J (joining) for the VH (heavy) chain, and V and J for the VL (light) chain-give rise to the final sequence diversity within the VH/VL chains. Somatic hypermutations, characterised by random nucleotide substitutions occurring at rates considerably higher than the genomic background, in the joint V(D)J segment further increases sequence diversity (Andreano & Rappuoli, 2021).
The ability to bind any target antigen with high specificity and affinity makes antibodies ideal candidates for drug discovery. As a result, antibody drugs hold a major and growing share of the total pharmaceutical market (Crescioli et al., 2025). Antibody drug development pipelines need to identify candidates which bind specifically and with high affinity to the target antigen, while also being ‘developable’ (Jarasch et al., 2015). ‘Developability’ refers to properties required for a successful drug such as immunogenicity, solubility, specificity, stability, manufacturability, and storability (Raybould & Deane, 2022).
Antibody language models have been used to optimise multiple steps of antibody-drug development pipelines from library generation (Turnbull et al., 2024) to humanisation during lead optimisation (Chinery et al., 2024). p-IgGen is a GPT-like decoder-only model trained on antibody-sequence data, consisting of 17M parameters (Turnbull et al., 2024). The authors released a paired model, as well as a finetuned version capable of generating diverse antibody libraries with developable properties.
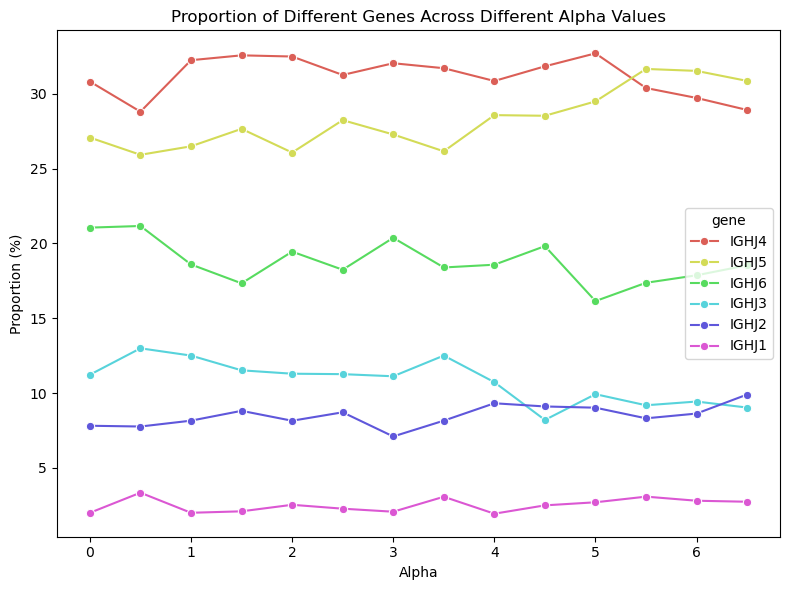
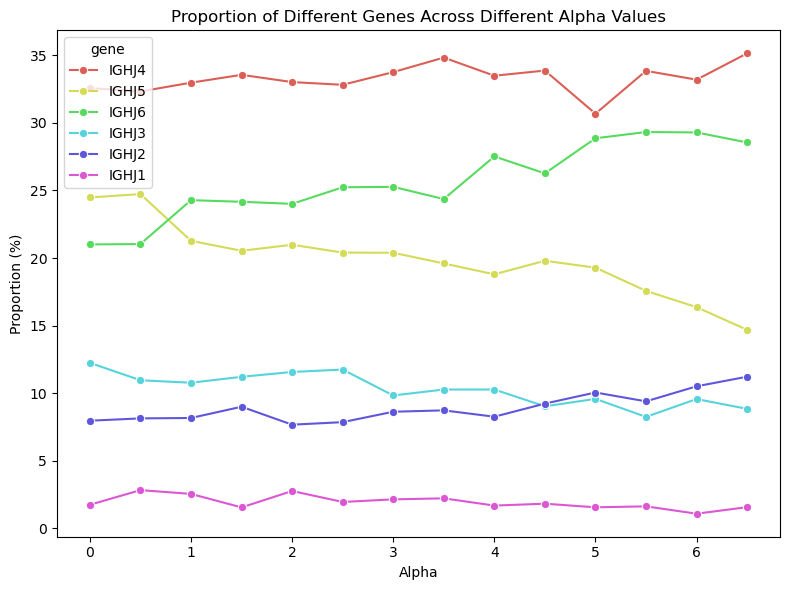
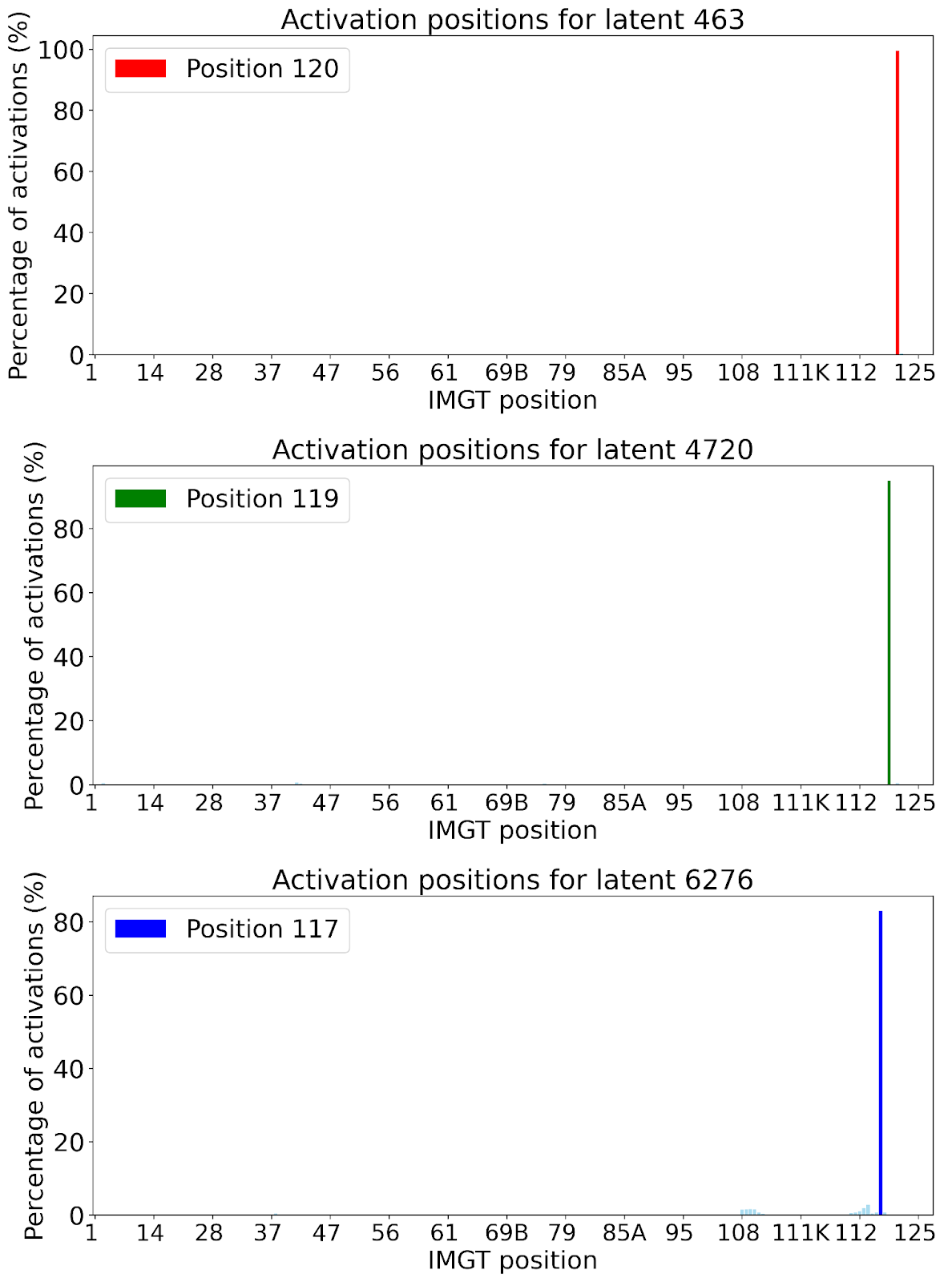
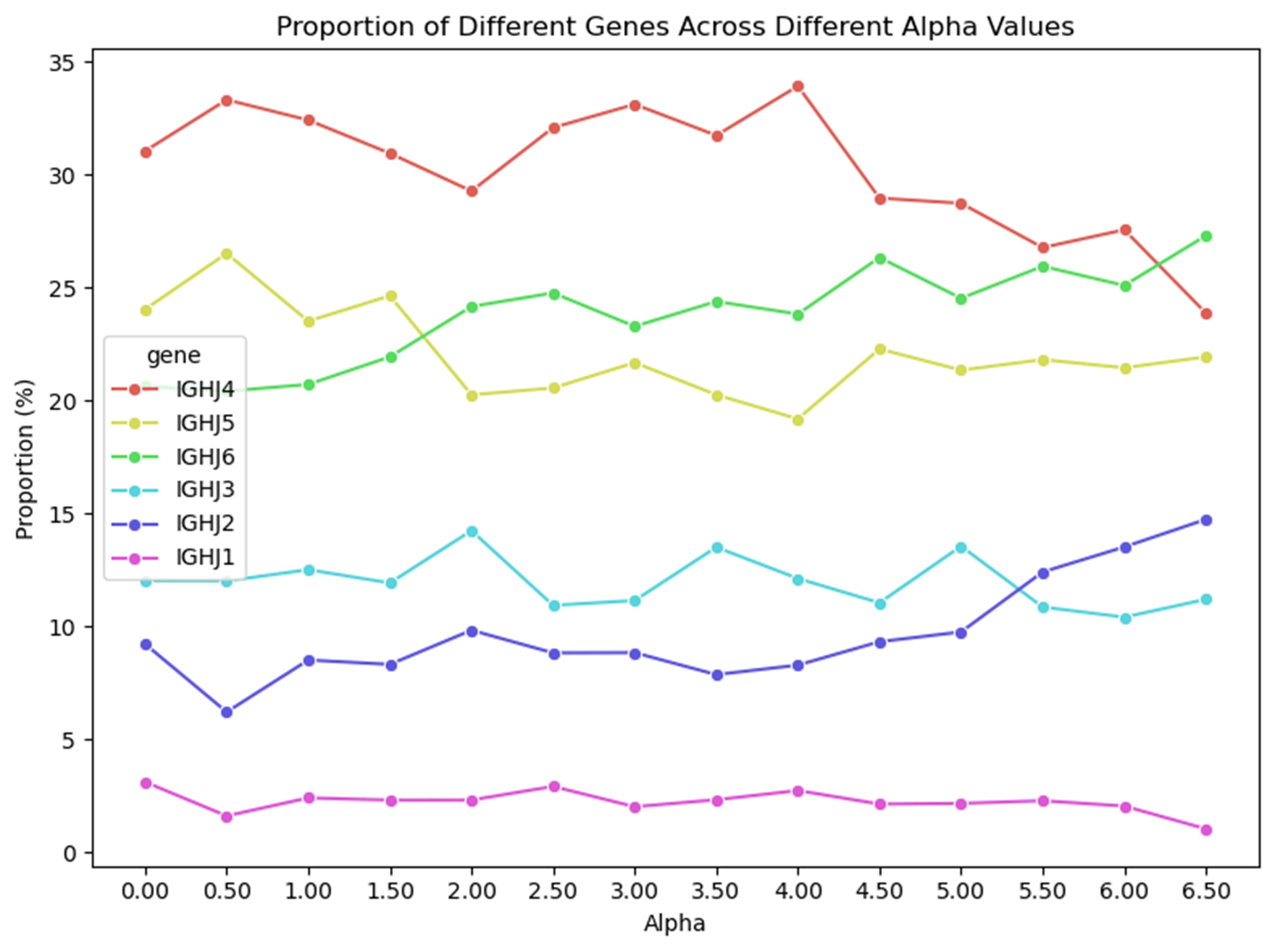
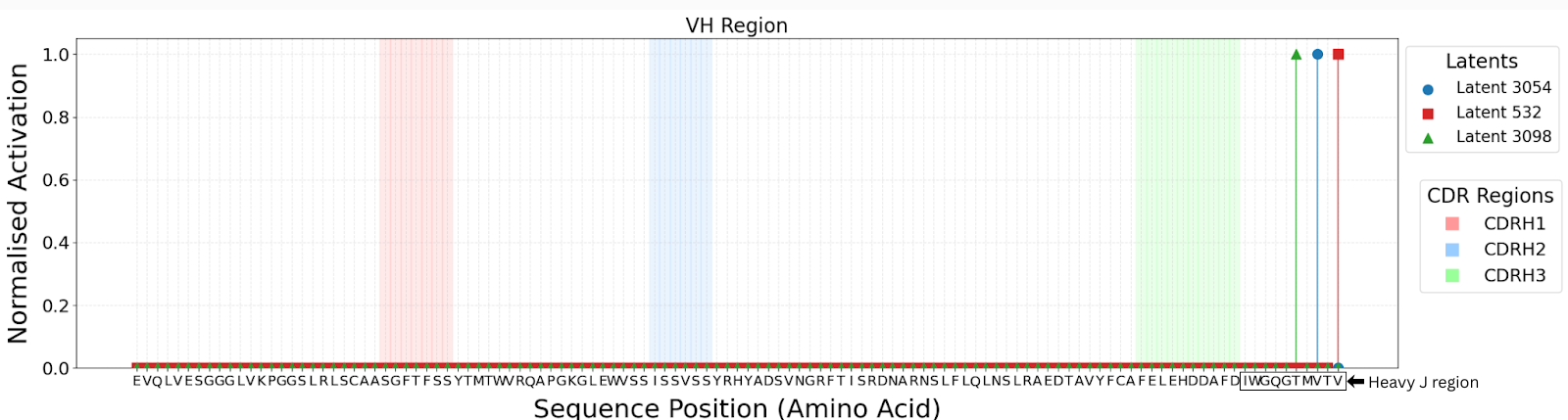
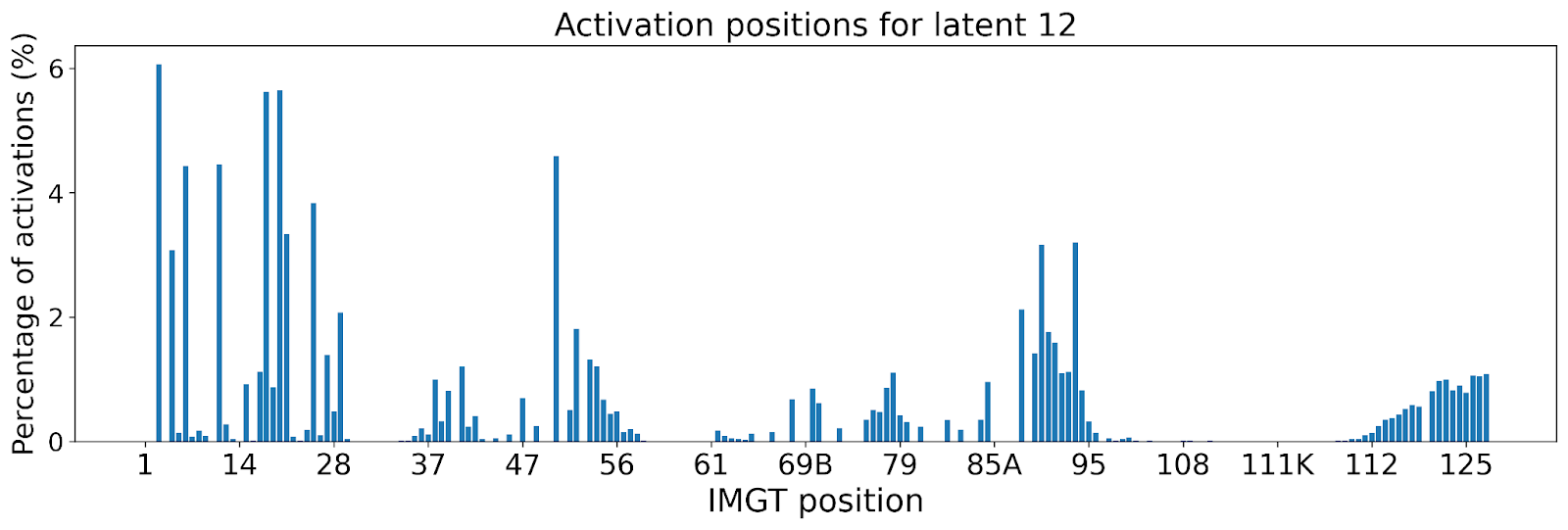
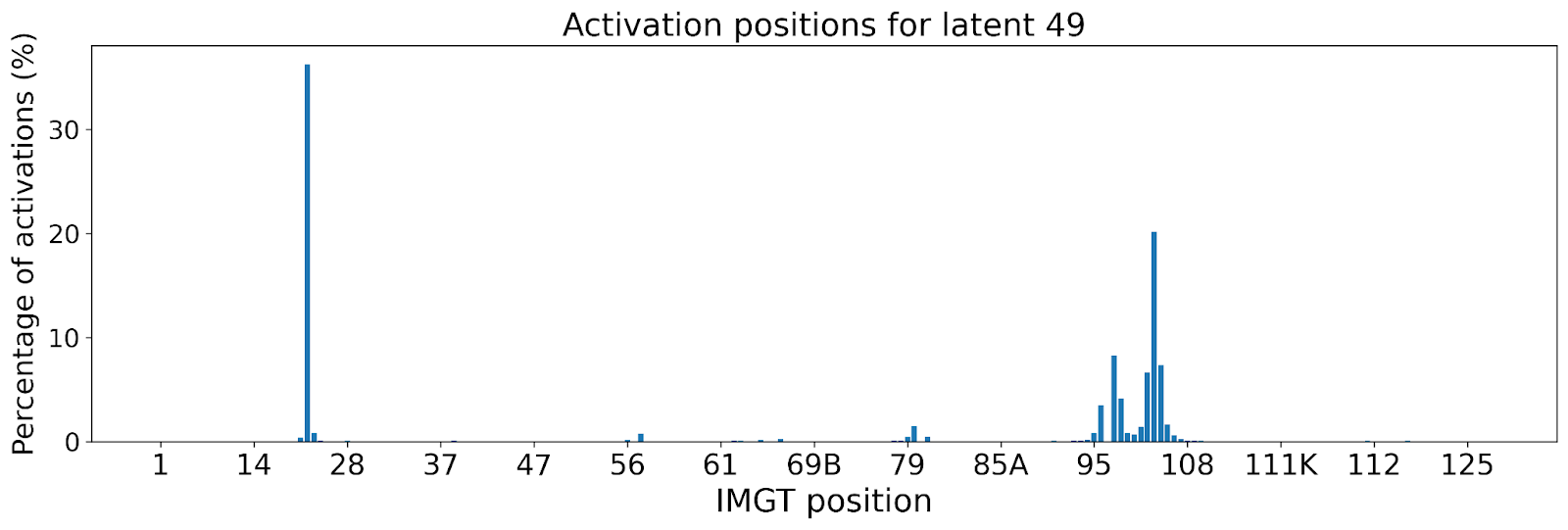
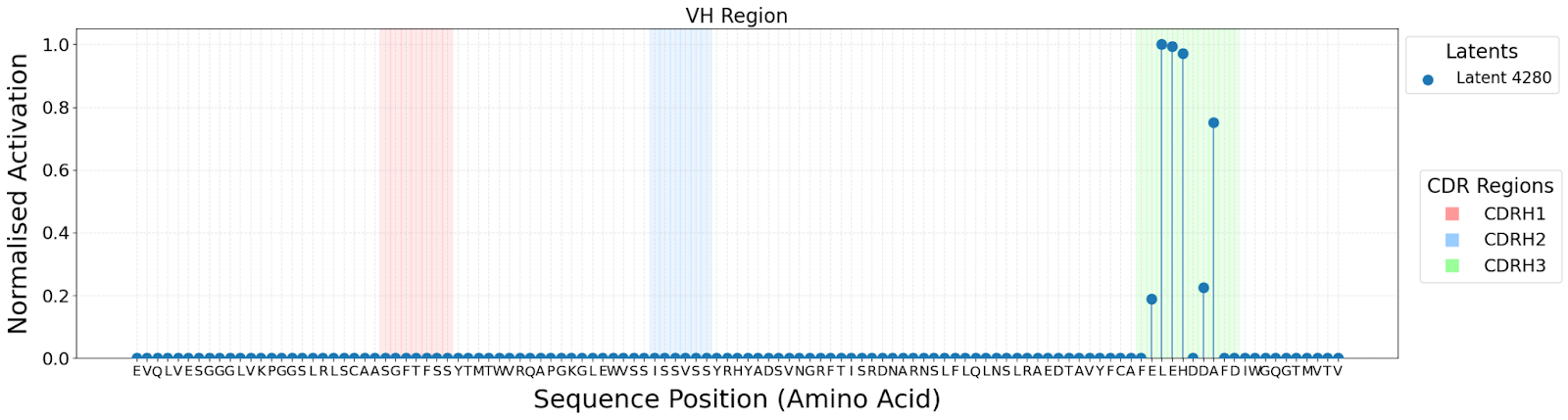
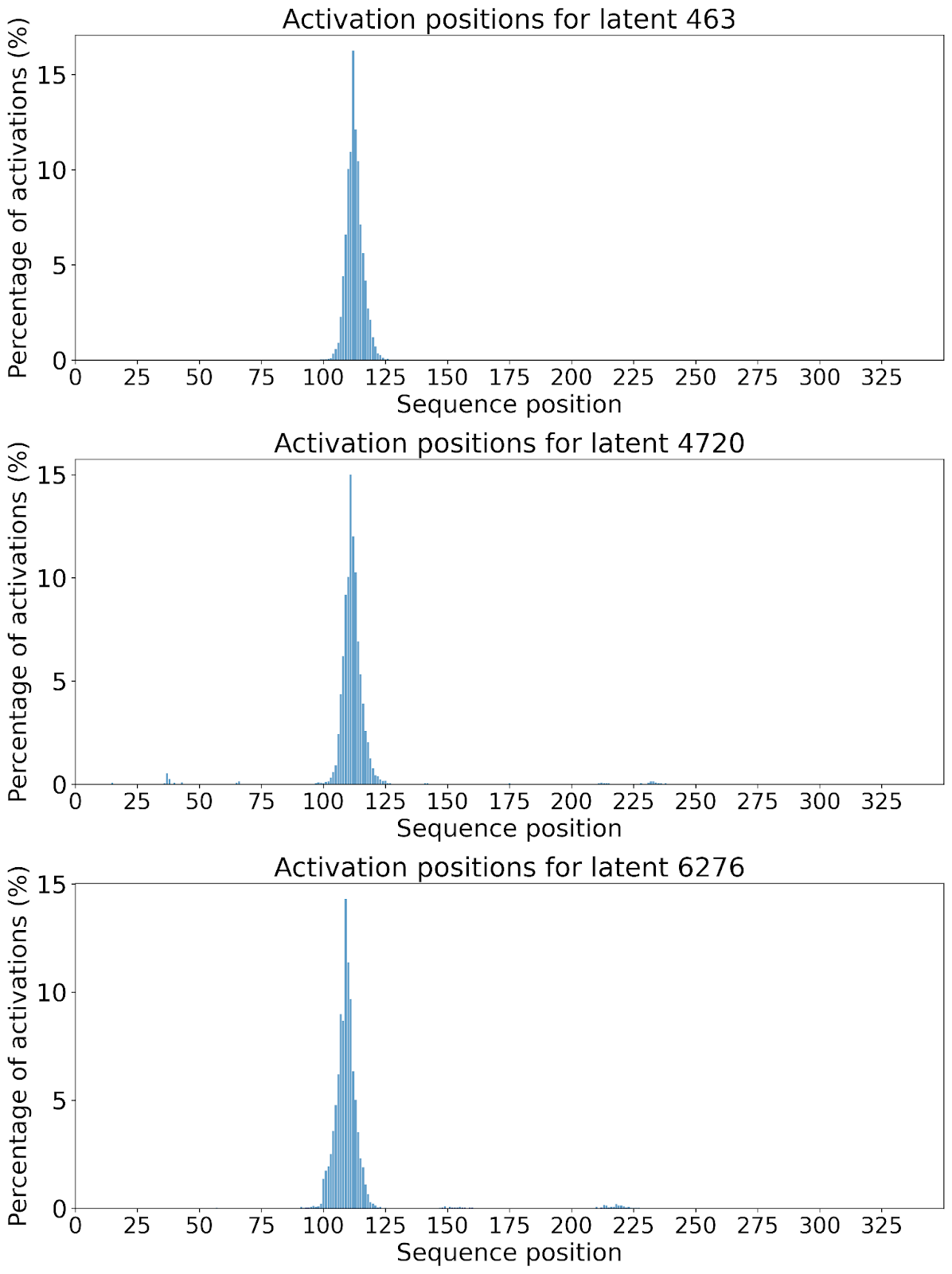
The lack of interpretability of machine learning models contributes to a lack of trust in model predictions, difficulty determining whether biologically relevant features are being used to make predictions and difficulty detecting overfitting. Collectively, these pose a barrier when employing language models for drug discovery (Chen et al., 2023). SAEs offer a promising approach to identify human-interpretable concepts learned by models and steer their generation (Chen et al., 2025;Templeton et al., 2024). Prior works have used SAEs to understand the inner mechanisms of PLMs (Adams et al., 2025;Parsan et al., 2025b;Simon & Zou, 2024), and steer model output. However, to date, SAEs have not been used to interrogate autoregressive protein or antibody-specific language models. This work advances the interpretability of antibody language models, using SAEs to identify biologically relevant features of interest learned by p-IgGen, and predictably steer its generation. We identify antibody-specific features, such as the complementarity-determining region (CDR) identity and germline gene identity, and use them to steer p-IgGen generation for specific germline gene identities. Overall, this work shows the applicability of SAEs for incorporating rational design principles to antibody library generation. We show that TopK SAEs can accurately identify interpretable latents underpinning model generation, whereas Ordered SAEs can identify steerable features capable of tuning model generation.
Mechanistic interpretability refers to the approach of explaining complex machine learning systems through the behaviour of their functional units (Kästner & Crook, 2024) by decomposing or reverse-engineering systems into their more elementary computations (Rai et al., 2025). The eventual goal is to discover causal relationships between model inputs and corresponding outputs.
Within the context of transformer-based language models, there are three main ideas relevant for mechanistic interpretability research: features, circuits and universality (Rai et al., 2025). Features refer to human-interpretable properties that are encoded by model activations (Templeton et al., 2024). Circuits inform how these features are extracted from model inputs and processed to influence model outputs (Olah et al., 2020b). Finally, universality determines whether features and circuits identified for a specific model and task exist in other models and tasks (Olah et al., 2020a). This paper specifically focuses on the identification of features from language models and using these features to steer model generation.
Sparse Autoencoders (SAEs) have spec
This content is AI-processed based on open access ArXiv data.