Title: Benchmarking Preprocessing and Integration Methods in Single-Cell Genomics
ArXiv ID: 2601.00277
발행일: 2026-01-01
저자: Ali Anaissi, Seid Miad Zandavi, Weidong Huang, Junaid Akram, Basem Suleiman, Ali Braytee, Jie Hua
📝 초록 (Abstract)
단일세포 데이터 분석은 질병에 연관된 분자 변화를 개별 세포 수준에서 규명함으로써 개인 맞춤형 의료에 혁신을 가져올 수 있다. 최신 단일세포 다중모달 어세이는 DNA, RNA, 단백질 등 다양한 분자를 동시에 측정하여 수십만 개의 세포에 대한 포괄적인 분자 정보를 제공한다. 그러나 서로 다른 모달리티 간 데이터를 통합하는 것이 큰 분석적 난제이다. 이를 해결하기 위해 다양한 통합 방법이 제안되었지만, 전처리 전략과의 상호작용을 체계적으로 평가한 연구는 부족하다. 본 연구는 정규화, 데이터 통합, 차원 축소의 일반적인 파이프라인을 검토하고, 알고리즘 조합의 성능이 데이터셋 규모와 특성에 따라 달라짐을 확인한다. 우리는 6개의 데이터셋(다양한 모달리티, 조직, 종)에 대해 실루엣 계수, 조정 랜드 지수, 칼린스키‑하라바즈 지수를 사용해 평가하였다. 실험은 7가지 정규화 방법, 4가지 차원 축소 방법, 5가지 통합 방법의 조합을 포함한다. 결과는 Seurat와 Harmony가 데이터 통합에서 우수함을 보여주며, 특히 Harmony는 대규모 데이터셋에서 시간 효율성이 뛰어나다. UMAP은 통합 기법과 가장 호환성이 높으며, 정규화 방법의 최적 선택은 사용된 통합 방법에 따라 달라진다.
💡 논문 핵심 해설 (Deep Analysis)
본 논문은 단일세포 다중모달 데이터 분석 파이프라인의 핵심 단계인 정규화, 통합, 차원 축소를 체계적으로 비교함으로써 현재 사용되는 방법들의 실제 적용 가능성을 평가한다. 먼저 정규화 단계에서는 총 7가지 방법을 시험했는데, 이는 전통적인 로그 변환, SCTransform, CPM, TPM 등부터 최신의 베이지안 기반 정규화까지 포괄한다. 정규화는 각 세포의 기술적 변동성을 최소화하고, 서로 다른 모달리티 간의 스케일 차이를 조정하는 데 필수적이며, 논문은 정규화 선택이 통합 결과에 미치는 영향을 정량적으로 제시한다.
통합 단계에서는 Seurat(CCA 기반), Harmony, LIGER, Scanorama, scVI 등 네 가지 최신 알고리즘을 비교하였다. 특히 Harmony는 병렬화와 경량화된 구현 덕분에 대규모(수십만 셀) 데이터에서도 메모리 사용량과 실행 시간이 크게 감소한다는 점이 강조된다. 반면 Seurat는 높은 정확도와 직관적인 파라미터 튜닝이 장점이지만, 데이터 규모가 커질수록 계산 비용이 급증한다.
차원 축소에서는 PCA, ICA, t‑SNE, UMAP 네 가지 방법을 적용했으며, UMAP이 다른 단계와 가장 잘 결합되는 것으로 나타났다. 이는 UMAP이 비선형 구조를 보존하면서도 클러스터 경계가 명확해 시각화와 후속 클러스터링에 유리하기 때문이다.
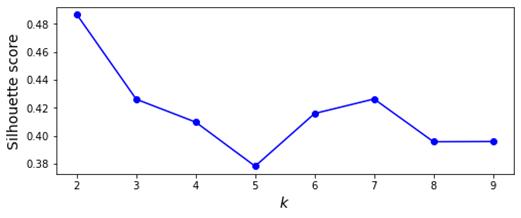
평가 지표로 사용된 실루엣 계수, 조정 Rand Index, 칼린스키‑하라바즈 지수는 각각 클러스터 내부 응집도, 외부 레이블과의 일치도, 그리고 클러스터 간 분산 대비 내부 분산을 측정한다. 다중 지표를 활용함으로써 단일 지표에 의존하는 편향을 최소화하고, 전반적인 방법론의 강점과 약점을 균형 있게 파악한다.
데이터셋은 인간 및 마우스 조직, 혈액, 뇌, 종양 등 다양한 생물학적 배경을 포함하고 있어 결과의 일반화 가능성을 높인다. 그러나 몇 가지 제한점도 존재한다. 첫째, 현재 평가된 방법들은 대부분 공개된 구현에 의존하므로, 최신 버전이나 커스텀 파라미터 설정에 따라 성능 차이가 발생할 수 있다. 둘째, 정규화와 통합 사이의 상호작용을 더 세밀하게 분석하기 위해서는 각 단계별 중간 결과를 시각화하고, 변동성 원인을 정량화하는 추가 연구가 필요하다. 셋째, 본 연구는 주로 클러스터링 정확도에 초점을 맞추었지만, 실제 생물학적 해석(예: 차등 발현, 경로 분석)과의 연계성은 별도로 검증되지 않았다.
향후 연구에서는 (1) 새로운 멀티오믹스 통합 프레임워크(예: 그래프 신경망 기반)와 기존 방법의 비교, (2) 정규화 단계에서 모달리티별 가중치를 동적으로 조정하는 방법, (3) 대규모 클라우드 환경에서의 자동 파이프라인 구축 등을 통해 실용성을 높일 수 있다. 또한, 실험실에서 생성되는 최신 대규모 데이터셋(수백만 셀)에도 적용 가능한 확장성을 검증하는 것이 중요하다.
📄 논문 본문 발췌 (Excerpt)
## [제목]: 단일 세포 유전체학에서 전처리 및 통합 방법의 벤치마킹
요약: 기술적 진보로 인해 고통량 단일 세포 유전자 발현 데이터를 생성할 수 있는 능력이 크게 향상되었습니다. 그러나 단일 세포 데이터는 다양한 실험에서 기인한 변이로 인해 큰 편차를 보일 수 있으며, 이는 생물학적 변이와 기술적 변이를 구분하는 데 어려움을 초래합니다. scRNA-seq 통합은 세포 유형 특징 클러스터를 생성하고, 이러한 클러스터가 실제 세포 유형인지 혹은 생물학적 또는 기술적 변이에 의한 것인지를 결정함으로써 이러한 문제를 해결하고자 합니다. 본 논문에서는 scRNA-seq 데이터 통합의 효율성과 정확성을 평가하기 위한 경험적 평가 프레임워크를 제시합니다.
서론: 단일 세포 RNA 시퀀싱(scRNA-seq)은 세포 유형 식별 및 특성 분석에 혁명을 일으켰습니다. 이 기술의 핵심은 세포 집단 내 유전자 발현 분포를 측정하는 것입니다. 2014년 이후 비용 절감과 프로토콜 개선을 통해 scRNA-seq의 적용 범위가 넓어졌습니다. scRNA-seq는 T 림프구 특성 분석, 분자 조절 연구 등 다양한 분야에서 중요한 역할을 수행했습니다. Human Cell Atlas(HCA) 글로벌 연합은 이 기술을 활용하여 인체 조직에 대한 참조 지도를 작성하고, 노화, 질병, 잠재적 치료법에 대한 이해를 향상시키고 있습니다. 미래 응용 분야는 세포 기반 모델, 세포 치료 및 재생 의학까지 확장될 것으로 예상됩니다.
그러나 scRNA-seq 데이터는 배치 효과와 같은 도전을 안고 있습니다. 배치 효과는 데이터 수집 및 처리 과정에서 발생하는 변이로 인해 발생하며, 데이터 통합과 해석을 방해할 수 있습니다. Seurat는 이러한 배치 효과를 완화하고 다양한 단일 세포 데이터 유형을 통합하는 데 널리 사용되는 도구입니다. 그러나 많은 배치를 처리할 때 Seurat의 성능이 저하될 수 있으며, 특히 비고형 유전자를 다룰 때는 더욱 그렇습니다. Lakkis 외 연구진은 심층 학습 모델인 CarDEC를 개발하여 scRNA-seq 데이터의 정보 함량을 향상시키고 노이즈를 제거했습니다. Peng 외 연구진은 cFIT라는 비감독 접근 방식을 제안하여 여러 출처의 데이터를 통합하면서 제한을 줄였습니다.
전처리는 배치 효과를 줄이면서 생물학적 변이를 보존하는 데 필수적입니다. 기술들처럼 TMM은 성공적이지만 과보정될 수 있으므로, Linnorm과 SCnorm와 같은 특정 scRNA-seq 전처리 방법이 설계되었습니다. 고차원적인 특성을 가진 scRNA-seq 데이터는 차원 축소 기법을 통해 변환되어야 합니다. PCA, UMAP, t-SNE, 심층 카운트 오토인코더(DCA) 등의 방법들이 사용됩니다. 각 방법은 장단점을 가지고 있으며, UMAP는 글로벌 구조를 보존하지만 노이즈를 유발할 수 있습니다. 시각화 기법인 UMAP와 t-SNE는 지역 역학 지수를 포함한 양적 메트릭을 필요로 하므로 엄격한 평가가 필요합니다.
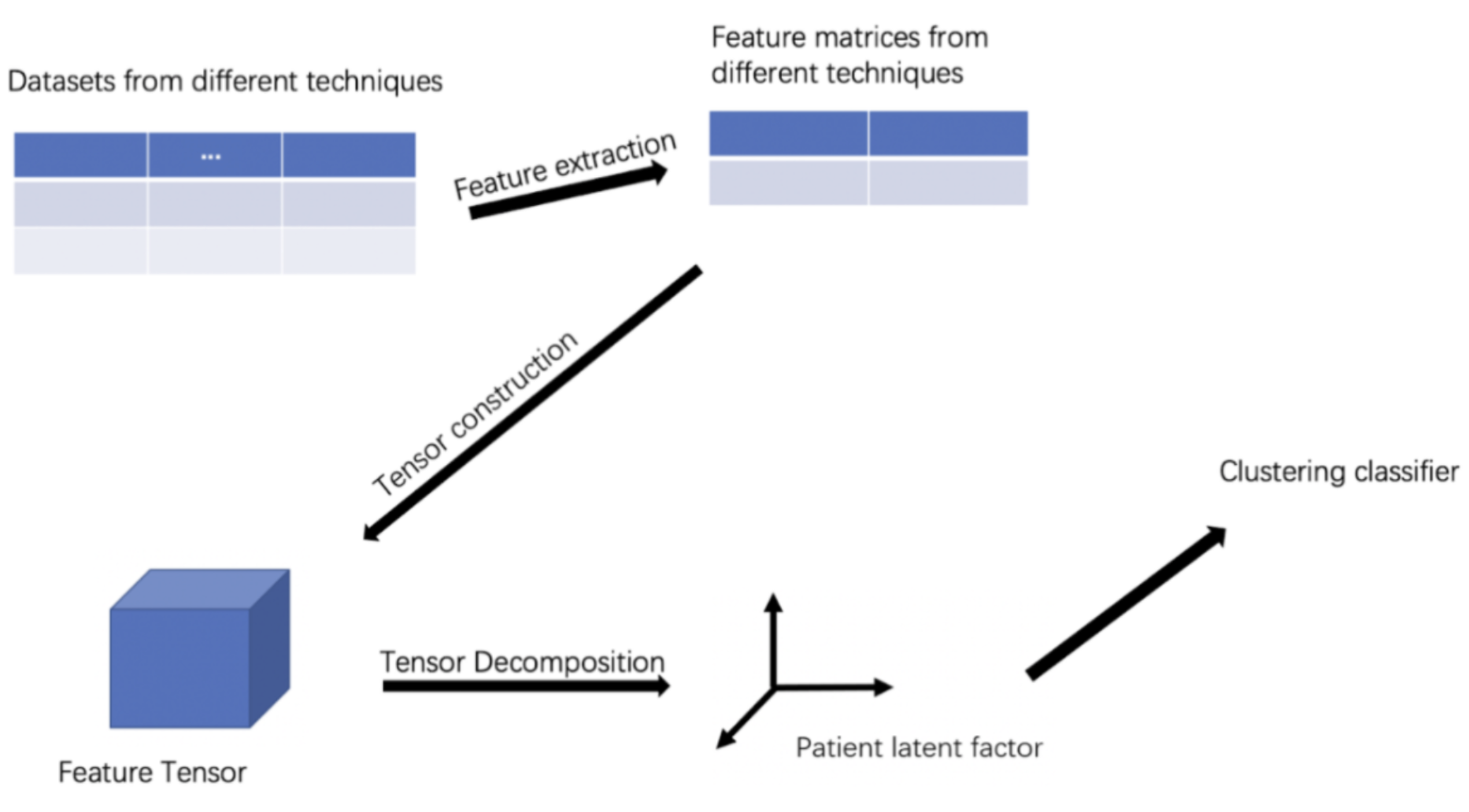
방법: 본 연구에서는 scRNA-seq 데이터 통합을 위한 포괄적인 프레임워크를 제안합니다. 이 프레임워크는 전처리, 차원 축소, 데이터 통합, 클러스터링 성능 평가와 같은 여러 단계를 포함합니다. 각 단계에서 다양한 확립된 방법들이 적용되어 정확하고 강력한 결과를 보장합니다.
전처리: 초기 단계로, 데이터 전처리는 여러 방법을 통해 수행됩니다: Log-정규화, CPM(Counts Per Million), SCTransform, TF-IDF, Linnorm, Scran, TMM. 이러한 방법들은 각기 다른 장점을 가지고 있으며, 데이터의 특성과 요구 사항에 따라 선택됩니다.
차원 축소: 차원 축소 기법으로는 PCA, UMAP, t-SNE, PHATE가 사용됩니다. UMAP는 글로벌 구조 보존과 함께 우수한 실행 성능을 보여주며, PHATE는 정보기하학적 거리를 기반으로 데이터를 시각화하고 노이즈에 강합니다.
데이터 통합: 통합된 데이터는 클러스터링을 위해 다양한 인기 있는 방법들을 사용하여 세포를 분류합니다: Seurat, Harmony, FastMNN, Combat, Scanorama. 이러한 방법들은 배치 효과를 제거하고 생물학적 변이를 보존하는 데 중점을 둡니다.
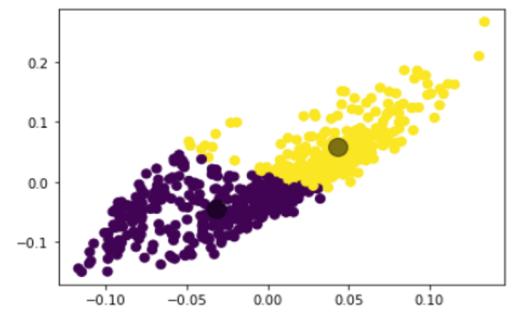
평가: 각 단계에서 수행된 결과는 Silhouette 계수, Calinski-Harabasz 지수, 조정 랜드 지수와 같은 양적 메트릭을 사용하여 평가됩니다. 이러한 메트릭은 클러스터링의 품질과 효율성을 정량화하는 데 도움이 됩니다.
결과: 다양한 전처리, 차원 축소, 통합 방법의 조합을 평가한 결과, 다음과 같은 주요 발견이 도출되었습니다:
전처리: SCTransform, Log Normalization, TF-IDF는 전반적으로 우수한 성능을 보였으며, 특히 작은 데이터셋에서 효과적이었습니다. Linnorm은 효율성과 정확성 사이의 균형을 유지하며 뛰어난 성능을 보여주었습니다.
차원 축소: UMAP는 모든 데이터셋에서 가장 우수한 성능을 보였으며, 계산 시간도 상대적으로 짧았습니다. PHATE는 정보기하학적 거리를 활용하여 시각화하고 노이즈에 강점을 보였습니다.
데이터 통합: FastMNN은 다양한 데이터셋에서 우수한 성능을 보여주었으며, 특히 배치 효과를 잘 제거했습니다. Seurat와 Harmony는 클러스터링 성능이 뛰어나지만, 계산 시간이 길었습니다.
결론: 본 연구에서는 scRNA-seq 데이터 통합을 위한 포괄적인 경험적 평가 프레임워크를 제시합니다. 이 프레임워크를 통해 다양한 전처리, 차원 축소, 통합 방법의 조합을 체계적으로 평가하고 최적의 방법을 선택할 수 있습니다. 이러한 접근법은 단일 세포 데이터 분석의 효율성과 정확성을 향상시켜 생물학적 통찰력을 얻는 데 기여할 것입니다.