Scaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute
Protein interaction modeling is central to protein design, which has been transformed by machine learning with applications in drug discovery and beyond. In this landscape, structure-based de novo binder design is cast as either conditional generativ…
Authors: Kieran Didi, Zuobai Zhang, Guoqing Zhou
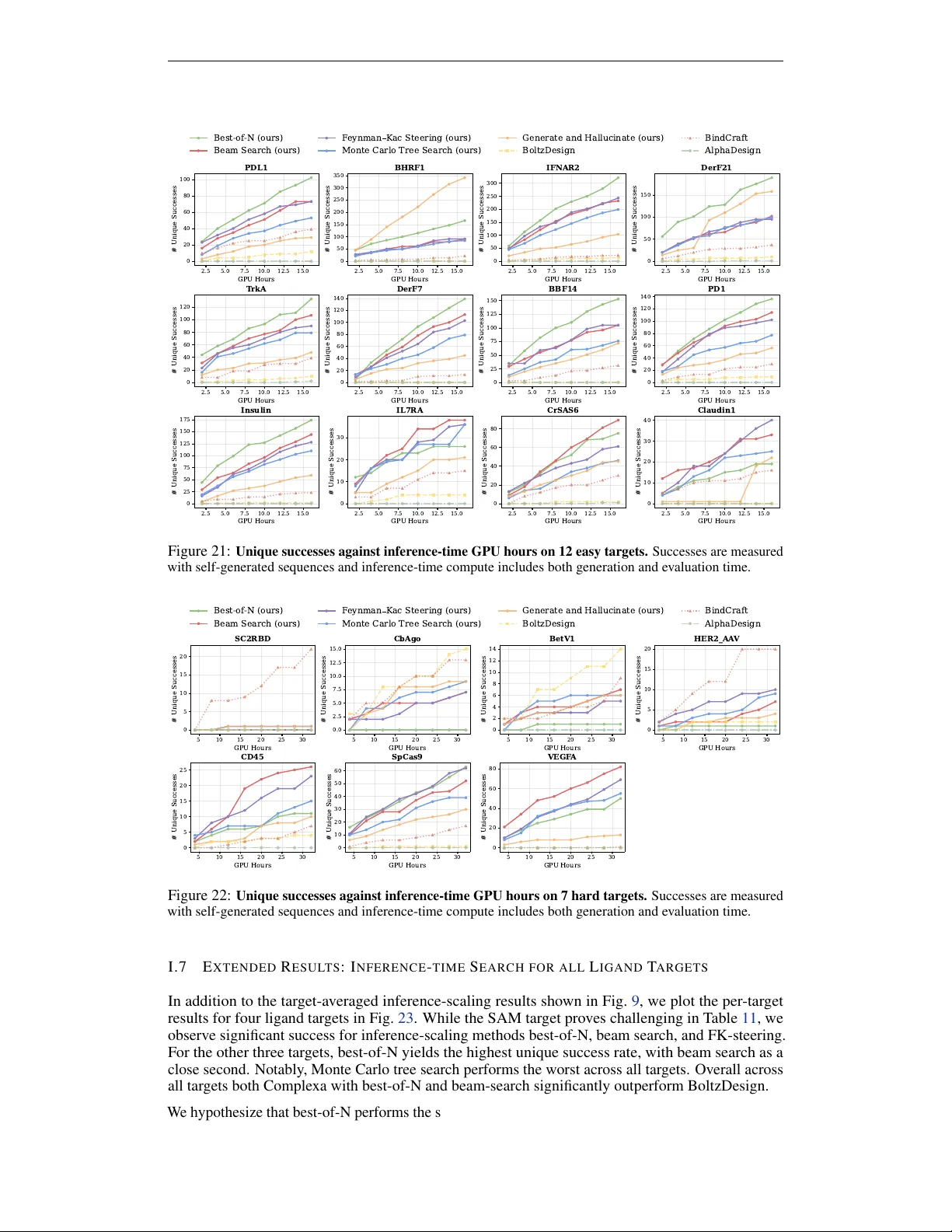
Published as a conference paper at ICLR 2026 S C A L I N G A T O M I S T I C P R OT E I N B I N D E R D E S I G N W I T H G E N E R A T I V E P R E T R A I N I N G A N D T E S T - T I M E C O M P U T E Kieran Didi 1,2,* Zuobai Zhang 1,3,4,* Guoqing Zhou 1,* Danny Reidenbach 1,* Zhonglin Cao 1,* Sooyoung Cha 8,9,* T omas Geffner 1 Christian Dallago 1 Jian T ang 3,5,6 Michael M. Bronstein 2,7 Martin Steinegger 8,9,10,11 Emine Kucukbenli 1, ♢ Arash V ahdat 1, ♢ Karsten Kreis 1,† 1 NVIDIA 2 Univ ersity of Oxford 3 Mila - Qu ´ ebec AI Institute 4 Univ ersit ´ e de Montr ´ eal 5 HEC Montr ´ eal 6 CIF AR AI Chair 7 AITHYRA 8 School of Biological Sciences, Seoul National Univ ersity 9 Interdisciplinary Program in Bioinformatics, Seoul National Univ ersity 10 Institute of Molecular Biology and Genetics, Seoul National Univ ersity 11 Artificial Intelligence Institute, Seoul National Univ ersity Pr oject page: https://research.nvidia.com/labs/genair/proteina- complexa/ A B S T R AC T Protein interaction modeling is central to protein design, which has been trans- formed by machine learning with applications in drug discov ery and beyond. In this landscape, structure-based de nov o binder design is cast as either conditional gener - ativ e modeling or sequence optimization via structure predictors (“hallucination”). W e argue that this is a false dichotomy and propose Pr ote ´ ına-Complexa , a novel fully atomistic binder generation method unifying both paradigms. W e extend recent flo w-based latent protein generation architectures and le verage the domain- domain interactions of monomeric computationally predicted protein structures to construct T eddymer , a ne w large-scale dataset of synthetic binder -target pairs for pretraining. Combined with high-quality e xperimental multimers, this enables train- ing a strong base model. W e then perform inference-time optimization with this gen- erativ e prior, unifying the strengths of previously distinct generati ve and hallucina- tion methods. Prote ´ ına-Complexa sets a new state of the art in computational binder design benchmarks: it deliv ers markedly higher in-silico success rates than existing generativ e approaches, and our novel test-time optimization strategies greatly out- perform pre vious hallucination methods under normalized compute b udgets. W e also demonstrate interface hydrogen bond optimization, fold class-guided binder generation, and extensions to small molecule targets and enzyme design tasks, again surpassing prior methods. Code, models and new data will be publicly released. 1 I N T RO D U C T I O N Designing binding proteins is a central challenge in computational biology . As protein interactions are mediated by structure, most binder design methods adopt a structure-centric vie w . Advances in ma- chine learning for protein structure prediction ( Jumper et al. , 2021 ; Abramson et al. , 2024 ) and struc- ture generation ( W atson et al. , 2023 ; Ingraham et al. , 2023 ) no w enable increasingly accurate de nov o binder design in silico ( Bennett et al. , 2023 ). Modern AI-based approaches fall into two classes: gener - ative methods , such as RFDif fusion ( W atson et al. , 2023 ), treat binder design as conditional generation, training on binder-tar get complex structures and producing ne w candidates for unseen targets; hal- lucination methods , exemplified by BindCraft ( Pacesa et al. , 2025 ), use the confidence and alignment scores of structure predictors to assess interfaces and optimize binder sequences via gradient feedback. This dichotomy contrasts with lar ge generation systems in language and image modeling, where a pre- trained base model is combined with adapti ve inference-time compute scaling and reasoning in a single framew ork ( W ei et al. , 2022 ; Snell et al. , 2025 ; Ma et al. , 2025 ). By analogy , current binder design methods resemble either pure training-time optimization (generativ e) or pure inference-time optimiza- tion without generati ve prior (hallucination). Inspired by inference-time scaling in language and vi- sion, we argue this split is false and introduce Pr ote ´ ına-Complexa (hereafter Complexa), to our kno wl- * Core contributor . ♢ Equal advising. † Project lead. 1 Published as a conference paper at ICLR 2026 Figure 1: (T op) Prote ´ ına-Complexa’ s target-conditioned generation process. (Bottom) Scaling test-time compute, we use Comple xa’ s generativ e prior for more ef ficient optimization than pre vious hallucination methods (Sec. 3.4 ). W e depict beam search, which steers stochastic generation to ward high-quality binders, guided by structure prediction models’ interface scores or hydrogen bond energies. Intermediate candidate states (blue) are scored via rollouts (blue, dotted), promising candidates are kept, and new trajectories are launched (orange). edge the first binder design frame work that unifies a strong flow-based base generati ve model with flexible inference-time optimization utilizing the generati ve prior , combining the strengths of both. T raining an expressi ve generator for atomistic binder design requires large binder -target datasets, yet experimentally resolv ed multimers in the Protein Data Bank (PDB) are limited. T o overcome this, we exploit domain-domain interactions from predicted monomer structures in the AlphaFold Database (AFDB). Using structural domain annotations from The Encyclopedia of Domains (TED) ( Lau et al. , 2024b ), we partition proteins into domains and assemble artificial protein dimers. After clustering and filtering, we obtain T eddymer , a ne w large-scale dataset of synthetic binder-tar get complexes. T o train on this data, Complexa builds on La-Prote ´ ına ( Geffner et al. , 2026 ), which combines a scalable partially latent protein representation with flow matching ( Lipman et al. , 2023 ) and efficient transformer neural networks for accurate fully atomistic protein generation. W e extend La-Prote ´ ına’ s architecture to binder design through a novel latent target conditioning mechanism. Inspired by pretraining and post-training alignment strategies from language and image generation, we adopt a staged training scheme on div erse monomers, T eddymer, and experimental multimer structures. Next, we enhance performance by scaling inference-time compute. W e adapt diffusion- and flow- based test-time scaling algorithms to binder design, including best-of-N sampling, beam search, Feynman–Kac steering, and Monte Carlo T ree Search ( Fernandes et al. , 2025 ; Ramesh & Mardani , 2025 ; Y oon et al. , 2025 ; Singhal et al. , 2025 ). Using interface confidence scores from structure predic- tors as rew ards, we steer the base model to high-quality in-silico binders (Fig. 1 ). This unifies halluci- nation and generati ve modeling by ef ficiently searching within the generativ e prior . W e also sho w that BindCraft-style hallucination can be accelerated by initializing from a generati ve model sample. In contrast to prior methods, Complexa does not require sequence re-design from backbone structures. W e comprehensiv ely ev aluate Complexa on binder design tasks for protein as well as small molecule targets. Our base model outperforms all prior generativ e models on established in-silico binding success metrics, and when compared to hallucination methods we achiev e higher success rates under normalized compute budgets, confirming Complexa’ s state-of-the-art performance. Since strong binding between proteins and tar gets is often facilitated through hydrogen bonds, we also analyze and show how we can explicitly optimize interface hydrogen bonding, proving the flexibility of our framework. Moreov er , we qualitativ ely demonstrate how fold class conditioning allows us to enhance binder div ersity in a controllable manner—pre vious methods often produce primarily alpha helical outputs. A key task in computational biology is enzyme design, and we also test Complexa on a recent enzyme design benchmark ( Ahern et al. , 2025 ), where we again outperform prior work by a large mar gin. Ablation studies on modeling decisions and the T eddymer data provide further insights. Modern generativ e AI systems scale both data and compute; the former during training, the latter dur- ing inference. T o our knowledge, Complexa is the first structure-based protein design method to follow this paradigm, bridging the false divide between generativ e vs. hallucination approaches. W e hope to enable efficient in-silico protein generation and design of binders for pre viously inaccessible targets. Key Contributions: (a) W e propose to combine previously distinct generativ e and hallucination methods—a nov elty in the field of protein design. (b) W e introduce T eddymer, a ne w large-scale syn- thetic dataset of protein dimers deriv ed from domain-domain interactions. (c) W e present Complexa, which extends La-Prote ´ ına to binder design, utilizes T eddymer , and implements efficient inference- 2 Published as a conference paper at ICLR 2026 Figure 2: Binders generated by Complexa, passing in-silico success criteria (more visualizations in Sec. K ). (a) TNF- α three-chain target. (b) Claudin-1 target, red interf ace hydrogen bonds. (c) OQO small molecule target. time optimization accelerated by the generative prior . (d) W e achie ve state-of-the-art in-silico success rates for both protein and small molecule targets as well as in an enzyme design benchmark without the necessity for sequence re-design. (e) W e further optimize and analyze interface hydrogen bonding, carry out ablation studies, and showcase fold class guidance for binder diversity . (f) W e will publicly release source code, model weights and the T eddymer dataset to benefit the community . 2 B A C K G RO U N D A N D R E L A T E D W O R K Flow Matching and La-Prote ´ ına. Complexa builds on top of La-Prote ´ ına ( Geffner et al. , 2026 ), whose core generation framew ork is flow-matching ( Lipman et al. , 2023 ; Albergo & V anden-Eijnden , 2023 ), which models a probability path p t ( x t ) transforming tractable noise p t =0 into data p t =1 via an ordinary differential equation (ODE) d x t = v θ ( x t , t ) dt defined by a learnable vector field v θ . T raining uses conditional flow matching (CFM), where conditional paths p t ( x t | x 1 ) make the target v ector field u t ( x t | x 1 ) tractable for simple p 0 . The network is trained by regressing v θ against u t ( x t | x 1 ) , yielding in expectation the same gradients as regression on the intractable mar ginal field u t ( x t ) . La-Prote ´ ına adopts the rectified flow formulation ( Liu et al. , 2023 ; Lipman et al. , 2023 ; Geffner et al. , 2025 ) with a linear interpolant x t = t x 1 + (1 − t ) x 0 and target x 1 − x 0 to model fully atomistic proteins using a partially latent flow matching framew ork. Specifically , it performs joint flo w matching ov er residues’ alpha carbon coordinates x C α and per-residue continuous latent variables z that encode amino acid identities s and the residues’ remaining atom coordinates x ¬ C α . This partially latent representation emerges from La-Prote ´ ına’ s variational autoencoder (V AE) framew ork with encoder E ( x C α , x ¬ C α , s ) and decoder D ( x C α , z ) , corresponding to approximate posterior and conditional likelihood in the V AE formalism, respectiv ely . La-Prote ´ ına’ s decoder outputs atom coordinates in an Atom37 protein representation. See La-Prote ´ ına details in Sec. B . Geffner et al. ( 2026 ) introduces partially latent flow matching, shows high-quality fully atomistic monomer generation, analyzes biophysical v alidity and performs motif scaffolding. In contrast, our work is entirely focused on protein binder design and is therefore fully orthogonal and complementary . Generation vs. Hallucination. Structure-based protein binder design with deep learning has tradition- ally follo wed two distinct routes: Generative methods train generati ve models, often flo w or diffusion models ( Ho et al. , 2020 ; Song et al. , 2021 ), on binder-tar get complex es and produce ne w binders condi- tioning on unseen tar gets. This w as first shown for protein tar gets by the seminal RFDiffusion ( W atson et al. , 2023 ). RFDiffusion-AllAtom ( Krishna et al. , 2024 ) extended this to di verse tar get modalities. While these models generate backbone structures only , Protpardelle ( Chu et al. , 2024 ; Lu et al. , 2025 ) and APM ( Chen et al. , 2025 ) enable fully atomistic binder generation. The hallucination approach to binder design corresponds to directly optimizing binder amino acid sequences towards high confidence and alignment scores under structure prediction models without training any generators. The term was coined by Anishchenk o et al. ( 2021 ), and recently BindCraft ( P acesa et al. , 2025 ), building on Gov erde et al. ( 2023 ), scaled the approach via gradient-based optimization, using AlphaFold2 ( Jumper et al. , 2021 ). BoltzDesign ( Cho et al. , 2025 ) uses Boltz-1 ( W ohlwend et al. , 2025 ) and extends to small molecules, DN A and other targets. These methods rely on complex and ad-hoc modifications and relaxations of the sequence representation to obtain gradients. AlphaDesign ( Jendrusch et al. , 2025 ) does not use gradients, lev eraging genetic algorithms instead. BA GEL ( L ´ ala et al. , 2025 ) focuses on applications such as peptide design and intrinsically disordered tar gets. Many approaches, both generativ e and hallucination-based, apply in verse-folding methods ProteinMPNN ( Dauparas et al. , 2022 ) or LigandMPNN ( Dauparas et al. , 2025 ) to generated binder backbones for sequence re-design. Other binder design methods include PXDesign ( T eam et al. , 2025c ), LatentX ( T eam et al. , 2025b ), Chai-2 ( T eam et al. , 2025a ) and AlphaProteo ( Zambaldi et al. , 2024 ); these are proprietary models without av ailable source code or model weights and in the last three cases lack methodological details. 3 Published as a conference paper at ICLR 2026 3 P RO T E ´ I N A - C O M P L E X A Overview . Our Complexa binder design framework consists of sev eral key components: (a) T o train Complexa’ s generative model to high performance, we first deri ve a new lar ge-scale dataset of synthetic protein dimers, T eddymer . This is described in Sec. 3.1 . (b) Complexa’ s base generativ e model builds on top of La-Prote ´ ına and conditions La-Prote ´ ına’ s partially latent flow matching com- ponent on the binder tar get without the need for adapting La-Prote ´ ına’ s autoencoder . This nov el latent target conditioning mechanism as well as adjusted training objecti ves and strate gies are presented in Sec. 3.2 . (c) W e discuss in-silico success metrics, including interface hydrogen bonds, in Sec. 3.3 . (d) T o enhance performance during inference and unify generation-based and optimization-based modeling, we adapt test-time compute scaling methods from the diffusion literature to Complexa—a first in the area of structure-based binder design to the best of our kno wledge (see Sec. 3.4 ). T o this end, rew ards to guide the generation are derived from the pre viously discussed success criteria. 3 . 1 T E D DY M E R : B I N D E R - T A R G E T D A TA F R O M I N T E R A C T I N G P RO T E I N D O M A I N S T raining an expressi ve base generati ve model for binder design requires paired binder -target multimer data; howe ver , such data is limited primarily to experimental structures in the protein data bank (PDB) ( Berman et al. , 2000 ). Meanwhile, the AlphaFold database (AFDB) ( V aradi et al. , 2021 ) provides a lar ge set of computationally predicted monomers, but no similar repository of synthetic Figure 3: T eddymer dimers resemble realis- tic binder-target structures, including interface hydrogen bonding (zoom-in). Also see Sec. C . multimers exists. Howe ver , most AFDB monomers are multi-domain proteins, and recently Lau et al. ( 2024b ) released The Encyclopedia of Domains (TED) of structural domain assignments for the AFDB ( Lau et al. , 2024a ). Inspired by prior work ( Sen & Madhusudhan , 2022 ), we argue that the biophysical interactions between structural domains of AFDB monomers are qualitati vely similar to the interactions between chains in multimeric structures (see Fig. 3 ). Hence, we propose to split the AFDB multi-domain monomers into their individual domains and treat the resulting multimer structures as binder-tar get training data for Complexa. W e start from AFDB50, a smaller , clustered version of the AFDB, and select the subset of structures with TED domain annotations, corresponding to 47M samples. W e then split these structures into multimers, treating each domain as a separate chain. Next, we extract dimers from those multimers, filtering for spatial proximity of the dimer chains, and we use only structures with complete CA T annotations ( Dawson et al. , 2016 ). This results in 10M dimers. Finally , we cluster the data to reduce redundancy , resulting in 3.5M clusters. W e name this database of TED-based dimers T eddymer . Please see Sec. C for data processing details and extended analyses comparing T eddymer interfaces with PDB multimers. Figure 4: Filtered training datasets used by Complexa. T eddymer is substantially larger than the PDB, which consists of ≈ 225k entries and requires further filtering to extract high-quality dimers suitable for training. As we show , our large-scale T eddymer pro vides a valuable additional resource for training at scale. In practice, we use four datasets in our experiments: ( a ) Foldseek AFDB monomer cluster representativ es ( van Kempen et al. , 2024 ; Barrio-Hernandez et al. , 2023 ), also used in pre vious work on protein generation ( Geffner et al. , 2025 ; Lin et al. , 2024 ). ( b ) T eddymer dimer cluster representatives, filtered with interface pLDDT > 70, ipAE < 10, interface length > 10 . ( c ) Protein multimers filtered from PDB. ( d ) Filtered PLINDER protein-ligand dataset ( Durairaj et al. , 2024 ). See Sec. D for data processing details and Fig. 4 for dataset sizes. 4 Published as a conference paper at ICLR 2026 3 . 2 C O M P L E X A ’ S B A S E G E N E R A T I V E M O D E L W e build on top of La-Prote ´ ına ( Geffner et al. , 2026 ) for tw o reasons: On the one hand, La-Prote ´ ına offers state-of-the-art accurate fully atomistic protein generation capabilities, necessary to produce precise atomistic binder-tar get interfaces. On the other hand, the framework is scalable and ef ficient, relying on fast transformer networks ( Geffner et al. , 2025 ) without slow triangular multiplicative or attention layers ( Jumper et al. , 2021 ). This is critical as binder design often inv olves in-silico gen- eration of many candidates, especially when scaling compute during inference. Complexa introduces a series of adaptations to extend the La-Prote ´ ına framew ork to binder design, described below . T arget- and Hotspot-Conditioning. W e modify La-Prote ´ ına to generate partially latent represen- tations of binder proteins only . Thus, only the partially latent flow matching model conditions on the target, while the autoencoder simply encodes and decodes monomeric binders. T o repre- sent the target, we use the Atom37 scheme: each residue is assigned up to 37 three-dimensional atomic coordinates, determined by its amino acid type. These residue-wise Atom37 features are combined with amino acid identity features and binary hotspot tokens that mark interface residues near which the binder should be generated. During training, we extract hotspots from interface residues of binder-tar get training pairs; during inference, hotspots are typically known (for all benchmarks and in most applications; otherwise, preprocessing could include hotspot identification). Figure 5: Complexa’ s latent target conditioning . When train- ing the conditional denoiser , the encoder and decoder are frozen. The resulting target-conditioning features, denoted as c target , are linearly embedded and concatenated in the token dimension to La-Prote ´ ına’ s se- quence of alpha carbon coordinates x C α and latent variables z that encode the binder . This extended sequence of noisy binder embeddings and clean target embeddings is then jointly processed by the transformer denoiser network of the partially latent flow model, which applies pair-biased attention ( Geffner et al. , 2025 ; Jumper et al. , 2021 ). Pair representations are formed jointly o ver the extended sequence representation of binder and target. Our novel latent target conditioning mechanism is illustrated in Fig. 5 (note that a related architecture, but without the pair rep- resentation, was used by Geffner et al. ( 2026 ) for motif scaffolding tasks). For small molecule tar gets, we featurize at the atomic lev el, using atom type, atom name, 3D atom coordinates, charge, and graph Laplacian positional encodings as sequence features. These are embedded and concatenated to the binder embeddings as before. Pair features are derived both from bond order and bond masks within the small molecule, and from distances between target atoms and binder residues’ backbone atoms. Further architectural details in Sec. G . Objective with T ranslation Noise. Using our latent target conditioning architecture, the partially latent flow model’ s training objectiv e of Complexa’ s extended La-Prote ´ ına component is min ϕ E t x ,t z , ( x , c target ) ∼ p data , x C α 0 ∼ p C α 0 , z 0 ∼ p z 0 , d ∼ p d 0 v ϕ z x C α t x , z t z , c target , t x , t z − ( E ( x ) − z 0 ) 2 + v ϕ x x C α t x , z t z , c target , t x , t z − x C α − h x C α 0 + d 1 N i 2 , (1) where x and c target denote binder and target drawn from the training data distrib ution p data , E ( x ) is the monomer encoder applied to the binder x , and t x and t z are the interpolation times of alpha carbon coordinates and latents, respectiv ely , drawn follo wing Geffner et al. ( 2026 ) (the model uses separate schedules for t x and t z both during training and inference). Moreover , we ha ve x C α t x = t x x C α + (1 − t x )( x C α 0 + d 1 N ) , z t z = t z E ( x ) + (1 − t z ) z 0 , p C α 0 = N ( x C α 0 | 0 , I ) , and p z 0 = N ( z 0 | 0 , I ) . In contrast 5 Published as a conference paper at ICLR 2026 to Geffner et al. ( 2026 ), we additionally perturb the binder’ s N alpha carbon coordinates with a ran- dom global translation d ∼ p d 0 = N ( d | 0 , c 2 d I xy z ) in x, y , z dimensions, broadcast over the N binder residues, when applying the interpolant ( c d = 0 . 2 ; units in nm ). This explicit translation noise forces the model to reason ov er global positioning of the binder . This is irrelev ant in monomer generation, but critical when the model needs to accurately position the binder at the interface. W e can also view this from a Fourier perspecti ve: Regular flo w and diffusion models generate the lo west data frequen- cies at the beginning of the generative process without further refinement ( Falck et al. , 2025 ). Global translation corresponds to the lo west frequency mode, and our translation noise forces the model to refine positioning throughout generation (a related technique was used by Ahern et al. ( 2025 )). Stagewise T raining. Inspired by the training strate gies of large-scale generativ e AI systems, we adopt a multi-stage training pipeline. First, the autoencoder of Complexa’ s La-Prote ´ ına component is trained on AFDB monomers and then fine-tuned on PDB structures, as synthetic AFDB structures alone are ov erly idealized, being generated by a folding model. Next, we also pretrain the partially latent flow- matching model on encoded AFDB Foldseek cluster representativ e monomers, enabling it to acquire general protein structure generation capabilities. Only afterwards we train on binder target pairs: for protein binders, using T eddymer and PDB multimers; for small molecule binders, using PLINDER and AFDB monomers via LoRA ( Hu et al. , 2022 ). In the latter case, we use LoRA to a void overfitting considering the small size of PLINDER. Full architecture, training, and sampling details in Sec. G . Complexa Design Considerations. A key advantage of our model design is that the same autoencoder can be used re gardless of the target type, as the autoencoder only needs to model monomeric chains— the de nov o binders in our case. In fact, we employ the same autoencoder in all models, simplifying the frame work. Our design parallels modern generation frame works in vision, which are typically latent dif fusion or flow models where only the latent generator , not the autoencoder , is conditioned on text or other signals ( Rombach et al. , 2022 ; Blattmann et al. , 2023 ; Esser et al. , 2024 ; Brooks et al. , 2024 ), and where inference-time optimization of latent generation can like wise be applied ( Fernandes et al. , 2025 ; Singhal et al. , 2025 ). Combined with our streamlined, fully transformer-based networks, this makes Complexa not only a modern, but also a fast and highly efficient binder design framew ork. 3 . 3 I N - S I L I C O S U C C E S S M E T R I C S A N D I N T E R FAC E H Y D RO G E N B O N D S Figure 6: Fold class-conditional binder generation. Samples pass success criteria. IFN AR2 target. Protein Structure Prediction Scores. Interf ace confidence and alignment scores from structure prediction models correlate with wet-lab success when e v aluated on designed binder–target pairs ( Overath et al. , 2025 ). Consequently , such scores hav e become standard in-silico metrics for assessing binder qual- ity . Pr otein tar gets: Following Zambaldi et al. ( 2024 ), we use AlphaFold2-Multimer ( Evans et al. , 2022 ), implemented through Co- labDesign ( Ovchinnikov et al. , 2025 ). A generated binder sequence s is considered successful if, together with the target sequence and structure (omitted), it satisfies f pLDDT ( s ) > 90 , f ipAE ( s ) < 7 . 0 , and f Binder-RMSD ( s ) < 1 . 5 ˚ A . Small molecule tar gets: Following Cho et al. ( 2025 ), we use an AlphaFold3-like model, specifically RosettaFold-3 (RF3) ( Corley et al. , 2025 ), and define success by f min-ipAE ( s ) < 2 , f Binder-RMSD ( s ) < 2 ˚ A and f Ligand-RMSD ( s ) < 5 ˚ A . W e generally directly e valuate Complexa’ s generated sequences (co-generated with atomistic binder structures), in contrast to prior works which usually re-design the sequence with ProteinMPNN or LigandMPNN from backbones—this is not necessary in Comple xa. Importantly , structure prediction model confidence and alignment scores can also be used as rewards when searching for strong binders during inference. In particular , we use f ipAE as reward (see Sec. 3.4 ). Interface Hydrogen Bonding is central in mediating strong protein-tar get interactions and its model- ing can play a key role in protein structure prediction and design ( O’Meara et al. , 2015 ; Herschlag & Pinney , 2018 ). Therefore, we also explore optimizing interface hydrogen bond energies f H-Bond ( s ) of re-folded structures of our generated binder sequences s (again, we also use the target to fold and e val- uate energies, b ut omit this for brevity). T o calculate hydrogen bond energies, we follo w Rosetta ( Al- ford et al. , 2017 ), implemented via Tmol ( Leav er-Fay et al. , 2025 ), and we use HBPlus ( McDonald & Thornton , 1994 ) to detect hydrogen bonds in generated structures. See Sec. F for details on metrics. 6 Published as a conference paper at ICLR 2026 3 . 4 I N F E R E N C E - T I M E O P T I M I Z A T I O N Prior hallucination methods can be seen as pure inference-time optimization without use of a generativ e model. T o combine Complexa’ s flow-based generativ e component with inference-time optimization, we adapt test-time scaling methods from the diffusion literature. W e generally sample our partially latent flow model stochastically with reduced noise injection (see Geffner et al. ( 2026 )). Best-of-N Sampling is the simplest version of test-time compute scaling: Giv en an increasing com- pute budget, we gro w the number of generated samples N and select all binders with f ipAE ( s ) < 7 . 0 . Beam Search (visualization in Fig. 1 ). W e maintain a set of N (beam width) denoising trajectories B t x ,t z = { ( x C α t x , z t z ) } i = N i =1 , the “beam”. From each beam element i , we initiate L (branching factor) ne w stochastic denoising trajectories that we run for K denoising steps to obtain a total of N × L ne w candidate states C t x +∆ t K x ,t z +∆ t K z = { ( x C α t x +∆ t K x , z t z +∆ t K z ) i } i = N L i =1 . W e now stochastically simulate all candidates i tow ards clean partially latent states, decode, fold the resulting sequences, and calculate the candidates’ re wards R (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) , for instance using f ipAE or f H-Bond . W e can now select the top- N candidates to form the updated beam after K steps, which can be formalized as B t x +∆ t K x ,t z +∆ t K z = arg max T ⊆C , |T | = N X i ∈T R (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) . (2) This procedure is continued until we obtain fully denoised samples. In contrast to prior work ( Fer - nandes et al. , 2025 ; Ramesh & Mardani , 2025 ), we do not use T weedie’ s formula to estimate the rew ard of average one-shot denoising predictions and instead iterati vely roll out all candidate states. While this leads to stochastic re wards, the resulting samples on which decoder and structure predictor operate are clean—this is necessary , because our structure prediction-based rew ards are only reliable on realistic sequences. Due to the ef ficiency of Complexa’ s generator, rolling out full generation trajectories is computationally inexpensi ve, and we perform this search only e very K steps. Feynman–Kac Steering (FKS) ( Singhal et al. , 2025 ) is related to beam search, but instead of top- N uses importance sampling to sample the tilted distrib ution p ϕ ( x C α , z ) exp { β R ( x C α , z ) } , where p ϕ de- notes the model distribution and β is an in verse temperature scaling (we omit indicating c target ). Specif- ically , we sub-sample N new states from the N × L candidates C t x +∆ t K x ,t z +∆ t K z with probability p (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) ∝ exp { β R (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) } . (3) Monte Carlo T ree Search (MCTS) ( Y oon et al. , 2025 ; Ramesh & Mardani , 2025 ) treats the iterativ e generation process of flo w and dif fusion models as a tree, where dif ferent paths in the tree correspond to different stochastic denoising trajectories, and MCTS then searches within that tree. The method explores many possible denoising paths and then chooses the best next state along the tree. It is critical to balance exploration and exploitation when tra versing the denoising tree (via parameter C in Eq. ( 4 )) and child states ( x C α t x +∆ t K x , z t z +∆ t K z ) i are chosen according to the index selection criterion i = arg max i R (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) V (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) | {z } exploitation + C v u u t ln ( V (( x C α t x , z t z ) i )) V (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) | {z } exploration , (4) where ( x C α t x , z t z ) i denotes the parent of ( x C α t x +∆ t K x , z t z +∆ t K z ) i and V ( · ) counts ho w often a (noisy) partially latent state has already been visited during previous searches. Further technical innov ations are necessary to meaningfully apply MCTS in our flow’ s continuous state/action space; see Sec. H . Generate and Hallucinate (G&H). The above approaches inte grate search directly into the model’ s generati ve denoising process. W e can also take a simpler approach to combine generati ve with halluci- nation methods: W e propose to initialize a binder candidate with our generati ve model, and then refine its sequence through an established hallucination method, for which we choose the BindCraft frame- work ( Pacesa et al. , 2025 ). BindCraft uses sev eral stages of optimization that partially rely on ad-hoc sequence relaxations to enable backpropagation through discrete sequences (in contrast to our Best-of- N, Beam Search, FKS, and MCTS variants, which are all principled and stable search algorithms). W e study different optimization stages when combining BindCraft with generati ve model initialization. 7 Published as a conference paper at ICLR 2026 T able 2: Complexa’s generativ e performance f or protein targets without optimization vs. baselines. Self de- notes model sequence e valuation, MPNN full backbone-based re-design, and MPNN-FI the same with fixed inter- face amino acids. Note that RFDiffusion and Protpardelle only generate backbones and not their own sequences. Complete results in Sec. I . If methods tie on unique and absolute successes, we do not count this, see Sec. F . Model # Unique Successes ↑ # T imes Best Method ↑ T ime [s] ↓ Nov elty ↓ Self MPNN-FI MPNN Self MPNN-FI MPNN RFDiffusion ( W atson et al. , 2023 ) – – 4.68 – – 3 70.8 0.87 Protpardelle-1c ( Lu et al. , 2025 ) – – 0.73 – – 0 8.13 0.77 APM ( Chen et al. , 2025 ) 0.31 1.52 3.15 1 0 1 73.1 0.86 Complexa (ours) 9.10 13.6 14.4 14 14 14 15.6 0.80 T o our knowledge, we are the first to systematically explore test-time scaling for protein design with generative models and search and optimization algorithms. Details & algorithms in Sec. H . T able 1: Complexa’ s generative perf ormance f or small molecule targets without optimization. RFDiffusion-AllAtom uses LigandMPNN, we e valuate sequences produced by Comple xa. Model # Unique Successes ↑ Times [s] ↓ Novelty ↓ SAM OQO F AD IAI RFDiffusion-AllAtom 2 3 5 8 87.4 0.72 Complexa (ours) 10 6 17 19 13.5 0.71 4 E X P E R I M E N T S W e train two Complexa generative base models, one for protein tar gets, one for small molecule targets, using the stagewise training protocol and latent target conditioning mechanism (Sec. 3.2 ). Training and model de- tails provided in Sec. G , selected test targets in Sec. E . Generated binders shown in Fig. 2 and Sec. K . 4 . 1 G E N E R A T I V E B A S E M O D E L B E N C H M A R K I N G Protein T argets. W e first ev aluate Complexa’ s generative model without test-time optimization and compare to publicly av ailable generativ e methods that similarly do not rely on hallucination: RFDiffusion, Protpardelle-1c, and APM. F or each method and target, we generate 200 binders from 40 to 250 residues. As prior approaches often collapse into producing the same binder repeatedly , we report the a verage number of unique successes, that is, we calculate success following Sec. 3.3 , cluster successful samples, and count the clusters. W e also report nov elty against PDB, per-sample generation time, and the frequency with which each method achieves the best score across targets. In addition, we e valuate both self-generated model sequences and backbone-based re-design with ProteinMPNN, with and without preserving interface residues. Results in T ab. 2 . Complexa significantly outperforms the baselines, producing more unique successful binders across all settings and winning on most targets. Its sampling time is substantially faster than RFDif fusion and APM, while also yielding more novel binders. Protpardelle is somewhat faster and fa vors nov elty , but its success rates are poor . Importantly , while MPNN-based re-design can improve outcomes, e ven Complexa’ s self-generated sequences out- perform all baselines, including those relying on re-design, making such additional steps unnecessary . Small Molecule T argets. The only publicly av ailable purely generative method for small molecule binder design is RFDiffusion-AllAtom ( Krishna et al. , 2024 ). W e ev aluate four molecules (SAM, OQO, F AD, IAI) following Cho et al. ( 2025 ), with results in T ab . 1 , based on Complexa’ s self- generated sequences (Sec. I for extended results). W e again significantly surpass the baseline, matching no velty and achie ving much faster sampling speed. Combined with the protein tar get bench- marks, these results underscore Complexa’ s versatility and establish the state-of-the-art performance of its generative base model—without sequence re-design. Generated binders sho wn in Figs. 2 and 31 . Diverse Binders via Fold Class Guidance. Previous protein generators often produce primarily alpha helical outputs. Recently , Geffner et al. ( 2025 ) used fold class guidance to enhance secondary structure control in monomer modeling. Here, we extend this conditioning to binder design and train a Complexa model conditioned on CA T labels ( Dawson et al. , 2016 ). Qualitative results in Figs. 6 and 32 show ho w we can explicitly control the model to output beta sheet or alpha helix binders. 4 . 2 I N F E R E N C E - T I M E C O M P U T E S C A L I N G A N D O P T I M I Z A T I O N Protein T argets. Next, we ev aluate Complexa’ s inference-time compute scaling techniques (we use only f ipAE as reward, details in Sec. H ) against hallucination baselines, which can be viewed as brute-force optimization. W e compare to the publicly av ailable BindCraft ( Pacesa et al. , 2025 ), BoltzDesign ( Cho et al. , 2025 ) and AlphaDesign ( Jendrusch et al. , 2025 ). As optimization can be run with varying compute and different targets imply differe nt computational demands, we 8 Published as a conference paper at ICLR 2026 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 20 40 60 80 100 120 140 # Unique Successes Easy T argets 5 10 15 20 25 30 GP U Hours 0 5 10 15 20 25 # Unique Successes Hard T argets Best-of -N Beam Search F eynman Kac Steering Monte Carlo Tree Search Generate and Hallucinate BoltzDesign BindCraft AlphaDesign Figure 7: Scaling analysis of inference-time optimization across different target diffi- culties. A verage unique success rate on 12 easy and 7 hard tar gets against test-time GPU hours. Fiv e inference-time optimization algorithms from Sec. 3.4 are ap- plied to Complexa and compared with hallucination baselines BoltzDesign and BindCraft. plot success as a function of runtime and group targets into easy and hard classes (see Sec. E ). As shown in Fig. 7 , for easy targets simple methods like Best-of-N already outperform baselines, while on hard tar gets advanced searches such as Beam Sear ch , FKS , and MCTS are required. This aligns with intuition: brute-force sampling suf fices for easy cases, but structured search is es- sential when sampling becomes inefficient. Across both target sets, BindCraft, BoltzDesign and AlphaDesign perform poorly under matched compute, while our approaches consistently lead (on some targets, BindCraft is ahead, but ov erall Complexa wins by a large margin, see Sec. I.6 ). 5 10 15 20 25 30 GP U Hours 0 20 40 60 80 # Unique Successes Best-of -N (ours) Beam Search (ours) F eynman Kac Steering (ours) Monte Carlo Tree Search (ours) Generate and Hallucinate (ours) BoltzDesign BindCraft AlphaDesign Figure 8: Inference-time scaling for VEGF A multi-chain target (hard). Initializing BindCraft from our samples ( G&H ) accelerates search on easy but not hard targets, which is expected as the initial sample will often be poor for hard targets. Fig. 8 presents a case study on the hard two-chain target VEGF A, highlighting the superior performance of our inference-time optimization methods. Note that our methods here generally use Complexa’ s self-generated sequences without requiring re-design, in contrast to BindCraft and BoltzDesign, which rely on ProteinMPNN (note that BindCraft and BoltzDesign both achieve 0.80 nov elty score, on-par with Complexa, T ab . 2 ). W e attribute Complexa’ s superior performance to our direct integration of a strong generative prior with search, in contrast to the hallucination baselines, which do naiv e optimization. Small Molecule T argets. W e compare Complexa to BoltzDesign, the only a vailable hallucination baseline for small molecules; see averaged results ov er the four molecule targets in Fig. 9 . Again, Complexa’ s scaling methods are far superior . Results for all indi vidual tar gets can be found in Sec. I.6 . 2.5 5.0 7.5 10.0 12.5 15.0 17.5 20.0 22.5 GP U Hours 0 20 40 60 80 100 # Unique Successes Best-of -N (ours) Beam Search (ours) F eynman Kac Steering (ours) Monte Carlo Tree Search (ours) BoltzDesign Figure 9: Inference-time optimization for small molecule tar gets. Interface Hydrogen Bond Optimization. In T ab . 3 , we show results of optimizing not only with f ipAE , but includ- ing an interface hydrogen bond energy re ward f H-Bond (cf. Sec. 3.3 ). Optimizing both f ipAE and f H-Bond can boost the av erage unique success rate. Importantly , including f H-Bond significantly enhances the number of interface hydrogen bonds (also Fig. 11 ). These results highlight the generality of Complexa’ s inference-time optimization framework and show that structure-based and physical energy-based re wards can be optimized, too—prior hallucination methods only considered folding model scores. This has promising future applications, for instance, when designing proteins with complex structural motif constraints. T able 3: Inference-time optimization using beam search with dif ferent combinations of f olding and hydr ogen bond rewards. Details in Sec. I.6 . Model # Unique # H-Bonds Successes ↑ (avg.) ↑ Complexa (no re ward) 77.00 5.271 Complexa w/ f ipAE 83.36 5.524 Complexa w/ f H-Bond 82.36 7.154 Complexa w/ f ipAE + f H-Bond 86.26 6.518 TNF- α , H1, IL17A are challenging multi-chain tar- gets ( T eam et al. , 2025c ), not part of the benchmark abov e. None of the publicly available baselines were able to achie ve an y successes on them within < 32 GPU hours of optimization. T o showcase Com- plexa’ s scalability , we extended our search horizon for these targets beyond > 100 GPU hours, allo wing us to find 15 unique successes for TNF- α , 7 for H1, and 1 for IL17A. This highlights Complexa’ s ability to find in-silico binder candidates ev en for very difficult targets as well as the flexibility of our test-time optimization framew ork. Sec. I.4 for experiment details. Successful binders shown in Figs. 2 and 30 . 9 Published as a conference paper at ICLR 2026 Figure 10: En- zyme Design Benchmark. Com- plexa significantly outperforms RFD- iffusion2 in 38/41 AME benchmark tasks, both with re-designed and self-generated se- quences. Extended results in Sec. I.9 . 4 . 3 E N Z Y M E D E S I G N A critical task in computational biology is enzyme design, where a protein is designed to catalyze a chemical reaction of a substrate target molecule. Recently , RFDiffusion2 ( Ahern et al. , 2025 ) introduced the Atomic Motif Enzyme (AME) benchmark, where an atomistic motif of the enzyme activ e site together with the substrate molecule are given, and a protein needs to be designed that reconstructs the motif and binds the substrate molecule. T o tackle this task, we extended our Complexa model for small molecule targets with atomistic motif reconstruction capabilities. The AME benchmark consists of 41 tasks with varying numbers of catalytic residues that need to be reconstructed, org anized in separated residue islands. A designed protein is considered successful, if the catalytic residues are reconstructed and there are no clashes with the ligand ( Ahern et al. , 2025 ). As pre viously , we only consider unique success. W e find that Complexa significantly outperforms RFDiffusion2 on almost all tasks, both with self-generated sequences and re-designed sequences using LigandMPNN (Fig. 10 ). Sec. I.9 for implementation, ev aluation details and extended results. 4 . 4 A B L A T I O N S T U D I E S In Sections I.1 and I.2 , we perform ablation studies ov er our novel T eddymer data used for training Complexa and the translation noise (cf. Sec. 3.2 ). Please see T ab. 7 in the Appendix. W e find that both are critical and h ypothesize that without translation noise, the model cannot reason well o ver the binder’ s positioning. In early experiments we observed poor binder placement without translation noise. W ithout T eddymer-based training data, performance plummets. The data is critical to learn div erse protein-protein interactions, and filtered PDB data alone is too small (cf. Fig. 4 ). Therefore, we attribute our strong generati ve performance reported in T ab . 2 in part to the T eddymer data. Please see our Appendix for extended results (Sec. I ), additional visualization of generated binders (Sec. K ), and complete training, model, architecture, algorithm, sampling, ev aluation and data details. 5 C O N C L U S I O N S Figure 11: (a) Interface hydrogen bond optimization can create binders with extended interacting regions, forming strong hydrogen bonding (red, zoom-in). (b) Regular binders, ev en though passing success criteria, can be smaller with less interactions. V isualization shows successful binders for T rkA target. W e hav e introduced Prote ´ ına-Complexa, a fully atomistic frame work for protein binder genera- tion that bridges large-scale generativ e modeling with test-time compute scaling. Pretrained on T eddymer, a new dataset of synthetic dimers from AFDB domain–domain interactions, Com- plexa achieves state-of-the-art de novo binder design without the need for sequence re-design. Adapting test-time scaling techniques from the diffusion literature, we outperform prior hallucination methods by directly unifying generation and optimization. W e also optimize interface hydrogen bonding, underscoring both the flexibility of our framew ork and opportuni- ties for integrating physics- and learning-based modeling. Future w ork could train a single unified model capable of tar geting and generating different molecular modalities, similar to recent work on peptide, small molecule and antibody design ( K ong et al. , 2025 ). Complexa pa ves the w ay to ward ef fi cient and scalable binder design, unlocking new and challenging targets, and moti vates systematic exploration of test-time scaling in AI for protein design. 10 Published as a conference paper at ICLR 2026 R E P RO D U C I B I L I T Y S T A T E M E N T T o guarantee the reproducibility of our work, we provide complete details with respect to our no vel methodology as well as training, e valuation and data processing. Moreover , we will release source code, models and the new dataset. In the following we describe each aspect in more detail. Methodological Details. W e ensure that all methodological innovations are e xplained appropriately to enable their reimplementation. All methods are explained on a high le vel in the main text (Sec. 3 ), and details as well as complete algorithms (in particular for all inference-time optimization methods) are provided in the Appendix in Sections G and H . Moreov er, background on La-Prote ´ ına is provided in Sec. B . T raining Details. Model training is discussed in Sec. 3.2 and complete training details and hyperpa- rameters are provided in Sec. G . Evaluation Details. Evaluation is discussed throughout the main paper on a high lev el, while e valuation metric details are pro vided in Sec. F . Furthermore, Sec. G covers ho w we sample from our base generativ e model and Sec. J explains how we sampled all baselines. Algorithms for generation with our inference-time optimization framework are provided in Sec. H . The selected protein and small molecule targets are described in Sec. E . Data Pr ocessing Details. Training data and data processing details are e xplained in the main paper in Sec. 3.1 and in detail in Sections C and D . In that context, please note that our nov el T eddymer data is based on the existing and publicly av ailable datasets AFDB ( V aradi et al. , 2021 ) and TED ( Lau et al. , 2024a ), and its careful processing is described in detail in Sec. C , making the process fully reproducible. Code, Model, and Data Release. Upon acceptance of the work we will publicly release source code, model weights, and the nov el T eddymer dataset, under permissiv e licensing. E T H I C S S TA T E M E N T Generativ e approaches to de nov o protein binder design promise to unlock broad advances across science, technology , and society . In medicine, they could speed the discovery of v accines, antibodies, and targeted therapeutics that address urgent health challenges such as infectious diseases and cancer . In biotechnology , they may enable more ef ficient and flexible enzyme engineering. Beyond applications, these models hav e the potential to transform basic science. For instance, by generating and testing large libraries of protein-target interactions, they of fer powerful tools to probe the fundamental principles of biophysical interactions, molecular recognition and protein folding. While these tools hold enormous promise, it is equally critical to ackno wledge that generative models for binder design could be misapplied in ways that pose risks. For this reason, their use demands prudent ov ersight and a strong emphasis on responsible deployment. F U N D I N G A C K N O W L E D G M E N T S M.B. is partially supported by the EPSRC T uring AI W orld-Leading Research Fellowship No. EP/X040062/1 and EPSRC AI Hub No. EP/Y028872/1. M.S is supported by the National Re- search Foundation of Korea grants (NRF) (2020M3-A9G7-103933, RS-2021- NR061659, RS- 2021- NR056571, RS-2024-00396026 and RS-2020-NR049543), the Novo Nordisk Foundation NNF24SA0092560 and the Creati ve-Pioneering Researchers Program. J.T . acknowledges funding from the Canada CIF AR AI Chair Program R E F E R E N C E S Josh Abramson, Jonas Adler , Jack Dunger , Richard Evans, Tim Green, Alexander Pritzel, Olaf Ronneberger , Lindsay W illmore, Andrew J. Ballard, Joshua Bambrick, Sebastian W . Bodenstein, David A. Evans, Chia-Chun Hung, Michael O’Neill, Da vid Reiman, Kathryn T unyasuvunakool, Zachary W u, Akvil ˙ e Zemgulyte, Eirini Arvaniti, Charles Beattie, Otta via Bertolli, Alex Bridgland, 11 Published as a conference paper at ICLR 2026 Alex ey Cherepanov , Miles Congre ve, Alexander I. Co wen-Riv ers, Andrew Co wie, Michael Fig- urnov , Fabian B. Fuchs, Hannah Gladman, Rishub Jain, Y ousuf A. Khan, Caroline M. R. Low , Kuba Perlin, Anna Potapenk o, Pascal Savy , Sukhdeep Singh, Adrian Stecula, Ashok Thillaisun- daram, Catherine T ong, Sergei Y akneen, Ellen D. Zhong, Michal Zielinski, Augustin Zidek, V ictor Bapst, Pushmeet K ohli, Max Jaderberg, Demis Hassabis, and John M. Jumper . Accurate structure prediction of biomolecular interactions with alphafold 3. Natur e , 630:493–500, 2024. 1 , 25 , 27 , 29 , 50 W oody Ahern, Jason Y im, Doug T ischer, Saman Salike, Seth M. W oodbury , Donghyo Kim, Indrek Kalvet, Y akov Kipnis, Brian Coventry , Han Raut Altae-Tran, Magnus Bauer , Regina Barzilay , T ommi S. Jaakkola, Rohith Krishna, and David Bak er . Atom level enzyme acti ve site scaf folding using rfdiffusion2. bioRxiv , 2025. doi: 10.1101/2025.04.09.648075. URL https://www. biorxiv.org/content/early/2025/04/10/2025.04.09.648075 . 2 , 6 , 10 , 27 , 44 , 46 Michael Samuel Albergo and Eric V anden-Eijnden. Building normalizing flows with stochastic interpolants. In The Eleventh International Confer ence on Learning Repr esentations (ICLR) , 2023. 3 Rebecca F . Alford, Andre w Leaver -Fay , Jeliazko R. Jeliazko v , Matthew J. O’Meara, Frank P . DiMaio, Hahnbeom Park, Maxim V . Shapo valov , P . Douglas Renfrew , V ikram K. Mulligan, Kalli Kappel, Jason W . Labonte, Michael S. P acella, Richard Bonneau, Philip Bradley , Roland L. Jr . Dunbrack, Rhiju Das, David Baker , Brian Kuhlman, T anja Kortemme, and Jeffre y J. Gray . The rosetta all-atom energy function for macromolecular modeling and design. Journal of Chemical Theory and Computation , 13(6):3031–3048, 2017. 6 , 25 , 50 Ivan Anishchenk o, Samuel J. Pellock, T amuka M. Chidyausiku, Theresa A. Ramelot, Sergey Ovchin- niko v , Jingzhou Hao, Khushboo Bafna, Christof fer Norn, Alex Kang, Asim K. Bera, Frank DiMaio, Lauren Carter, Cameron M. Chow , Gaetano T . Montelione, and David Baker . De novo protein design by deep network hallucination. Nature , 600:547–552, 2021. 3 Inigo Barrio-Hernandez, Jingi Y eo, J ¨ urgen J ¨ anes, Milot Mirdita, Cameron L. M. Gilchrist, T anita W ein, Mihaly V aradi, Sameer V elankar, Pedro Beltrao, and Martin Steinegger . Clustering predicted structures at the scale of the known protein uni verse. Nature , 622:637–645, 2023. 4 , 21 , 23 Nathaniel R. Bennett, Brian Cov entry , Inna Goreshnik, Buwei Huang, Aza Allen, Dionne V afeados, Y ing Po Peng, Justas Dauparas, Minkyung Baek, Lance Ste wart, Frank DiMaio, Steven De Munck, Savv as N. Sa vvides, and Da vid Baker . Improving de nov o protein binder design with deep learning. Natur e Communications , 14:2625, 2023. 1 , 24 Helen M. Berman, John D. W estbrook, Zukang Feng, Gary L Gilliland, T alapady N. Bhat, Helge W eissig, Ilya N. Shindyalov , and Philip E. Bourne. The protein data bank. Nucleic Acids Resear ch , 28(1):235–42, 2000. 4 Andreas Blattmann, Robin Rombach, Huan Ling, T im Dockhorn, Seung W ook Kim, Sanja Fidler , and Karsten Kreis. Align your Latents: High-Resolution V ideo Synthesis with Latent Dif fusion Models. In Pr oceedings of the IEEE/CVF Confer ence on Computer V ision and P attern Recognition (CVPR) , 2023. 6 T im Brooks, Bill Peebles, Connor Homes, W ill DePue, Y ufei Guo, Li Jing, Da vid Schnurr , Joe T aylor , T roy Luhman, Eric Luhman, Clarence Wing Y in Ng, Ricky W ang, and Aditya Ramesh. V ideo generation models as world simulators, 2024. URL https://openai.com/research/ video- generation- models- as- world- simulators . 6 Zhonglin Cao, Mario Geiger , Allan Dos Santos Costa, Danny Reidenbach, Karsten Kreis, T omas Geffner , Franco Pellegrini, Guoqing Zhou, and Emine Kucukbenli. Efficient molecular con- former generation with SO(3)-a veraged flo w matching and reflow . In F orty-second International Confer ence on Machine Learning (ICML) , 2025. 27 Ruizhe Chen, Dongyu Xue, Xiangxin Zhou, Zaixiang Zheng, Xiangxiang Zeng, and Quanquan Gu. An all-atom generati ve model for designing protein comple xes. In International Confer ence on Machine Learning , 2025. 3 , 8 , 46 12 Published as a conference paper at ICLR 2026 Y ehlin Cho, Martin P acesa, Zhidian Zhang, Bruno E. Correia, and Ser gey Ovchinnik ov . Boltzdesign1: In verting all-atom structure prediction model for generalized biomolecular binder design. bioRxiv , 2025. doi: 10.1101/2025.04.06.647261. URL https://www.biorxiv.org/content/ early/2025/04/06/2025.04.06.647261 . 3 , 6 , 8 , 24 , 43 Alexander E. Chu, Jinho Kim, Lucy Cheng, Gina El Nesr, Minkai Xu, Richard W . Shuai, and Po-Ssu Huang. An all-atom protein generativ e model. Proceedings of the National Academy of Sciences , 121(27):e2311500121, 2024. 3 Nathaniel Corley , Simon Mathis, Rohith Krishna, Magnus S. Bauer , T uscan R. Thompson, W oody Ahern, Maxwell W . Kazman, Rafael I. Brent, Kieran Didi, Andre w Kubaney , Lilian McHugh, Arnav Nagle, Andre w Fa vor , Meghana Kshirsagar , Pascal Sturmfels, Y anjing Li, Jasper Butcher , Bo Qiang, Lars L. Schaaf, Raktim Mitra, Katelyn Campbell, Odin Zhang, Roni W eissman, Ian R. Humphreys, Qian Cong, Jonathan Funk, Shreyash Sonthalia, Pietro Li ` o, David Bak er, and Frank DiMaio. Accelerating biomolecular modeling with atomworks and rf3. bioRxiv , 2025. doi: 10.1101/2025.08.14.670328. URL https://www.biorxiv.org/content/early/ 2025/08/14/2025.08.14.670328 . 6 , 24 , 29 , 35 , 46 R ´ emi Coulom. Efficient selectivity and backup operators in monte-carlo tree search. In International confer ence on computers and games , pp. 72–83. Springer , 2006. 31 Julian Cremer , T uan Le, Mohammad M. Ghahremanpour , Emilia Sługocka, Filipe Menezes, and Djork-Arn ´ e Clev ert. Flo wr .root: A flow matching based foundation model for joint multi-purpose structure-aware 3d ligand generation and affinity prediction. arXiv pr eprint arXiv:2510.02578 , 2025. 49 Justas Dauparas, Iv an Anishchenko, Nathaniel Bennett, Hua Bai, Robert J Ragotte, Lukas F Milles, Basile IM Wick y , Ale xis Courbet, Rob J de Haas, Ne ville Bethel, et al. Robust deep learning–based protein sequence design using proteinmpnn. Science , 378(6615):49–56, 2022. 3 , 24 Justas Dauparas, Gyu Rie Lee, Robert Pecoraro, Linna An, Ivan Anishchenko, Cameron Glasscock, and David Baker . Atomic context-conditioned protein sequence design using ligandmpnn. Nature Methods , 22:717–723, 2025. 3 , 24 , 46 , 50 Natalie L. Dawson, T ony E. Lewis, Sayoni Das, Jonathan G. Lees, David A. Lee, Paul Ashford, Christine A. Orengo, and Ian P . W . Sillitoe. Cath: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Resear ch , 45:D289 – D295, 2016. 4 , 8 , 53 Janani Durairaj, Y usuf Adeshina, Zhonglin Cao, Xuejin Zhang, Vladas Oleinikov as, Thomas Duignan, Zachary McClure, Xavier Robin, Danny Ko vtun, Emanuele Rossi, Guoqing Zhou, Srimukh V eccham, Clemens Isert, Y uxing Peng, Prabindh Sundareson, Mehmet Akdel, Gabriele Corso, Hannes St ¨ ark, Zachary Carpenter , Michael Bronstein, Emine Kucukbenli, T orsten Schwede, and Luca Naef. Plinder: The protein-ligand interactions dataset and e valuation resource. bioRxiv , 2024. doi: 10.1101/2024.07.17.603955. URL https://www.biorxiv.org/content/early/ 2024/07/17/2024.07.17.603955 . 4 , 23 Patrick Esser , Sumith Kulal, Andreas Blattmann, Rahim Entezari, Jonas M ¨ uller , Harry Saini, Y am Levi, Dominik Lorenz, Ax el Sauer, Frederic Boesel, Dustin Podell, T im Dockhorn, Zion English, Kyle Lace y , Alex Goodwin, Y annik Marek, and Robin Rombach. Scaling rectified flow transform- ers for high-resolution image synthesis. In International Confer ence on Machine Learning (ICML) , 2024. 6 Richard Evans, Michael O’Neill, Ale xander Pritzel, Natasha Antropova, Andre w Senior , Tim Green, Augustin ˇ Z ´ ıdek, Russ Bates, Sam Blackwell, Jason Y im, Olaf Ronneberger , Sebastian Bodenstein, Michal Zielinski, Alex Bridgland, Anna Potapenk o, Andrew Co wie, Kathryn T unyasuvunak ool, Rishub Jain, Ellen Clancy , Pushmeet K ohli, John Jumper , and Demis Hassabis. Protein complex pre- diction with alphafold-multimer . bioRxiv , 2022. doi: 10.1101/2021.10.04.463034. URL https: //www.biorxiv.org/content/early/2022/03/10/2021.10.04.463034 . 6 , 35 Fabian F alck, T eodora Pandev a, Kiarash Zahirnia, Rachel Lawrence, Richard T urner , Edward Meeds, Javier Zazo, and Sushrut Karmalkar . A fourier space perspectiv e on diffusion models. arXiv pr eprint arXiv:2505.11278 , 2025. 6 , 27 13 Published as a conference paper at ICLR 2026 Guilherme Fernandes, V asco Ramos, Rege v Cohen, Idan Szpektor , and Jo ˜ ao Magalh ˜ aes. Latent beam diffusion models for decoding image sequences. arXiv pr eprint arXiv:2503.20429 , 2025. 2 , 6 , 7 , 30 T omas Gef fner , Kieran Didi, Zuobai Zhang, Dann y Reidenbach, Zhonglin Cao, Jason Y im, Mario Geiger , Christian Dallago, Emine Kucukbenli, Arash V ahdat, and Karsten Kreis. Proteina: Scal- ing flow-based protein structure generativ e models. In International Confer ence on Learning Repr esentations (ICLR) , 2025. 3 , 4 , 5 , 8 , 26 , 27 , 30 , 53 T omas Gef fner , Kieran Didi, Zhonglin Cao, Dann y Reidenbach, Zuobai Zhang, Christian Dallago, Emine Kucukbenli, Karsten Kreis, and Arash V ahdat. La-proteina: Atomistic protein generation via partially latent flow matching. In The F ourteenth International Confer ence on Learning Repr esentations , 2026. 2 , 3 , 5 , 6 , 7 , 19 , 30 Casper A. Go verde, Benedict W olf, Hamed Khakzad, St ´ ephane Rosset, and Bruno E. Correia. De nov o protein design by in version of the alphafold structure prediction netw ork. Protein Science , 32(6):e4653, 2023. doi: https://doi.org/10.1002/pro.4653. URL https://onlinelibrary. wiley.com/doi/abs/10.1002/pro.4653 . 3 Scott R Granter , Andrew H Beck, and David J Papke Jr . Alphago, deep learning, and the future of the human microscopist. Ar chives of pathology & laboratory medicine , 141(5):619–621, 2017. 32 Daniel Herschlag and Margaux M. Pinney . Hydrogen bonds: Simple after all? Biochemistry , 57(24): 3338–3352, 2018. 6 Jonathan Ho, Ajay Jain, and Pieter Abbeel. Denoising diffusion probabilistic models. In Advances in Neural Information Pr ocessing Systems (NeurIPS) , 2020. 3 Edward J Hu, yelong shen, Phillip W allis, Zeyuan Allen-Zhu, Y uanzhi Li, Shean W ang, Lu W ang, and W eizhu Chen. LoRA: Low-rank adaptation of large language models. In International Confer ence on Learning Repr esentations (ICLR) , 2022. 6 , 28 John Ingraham, Max Baranov , Zak Costello, V incent Frappier, Ahmed Ismail, Shan T ie, W ujie W ang, V incent Xue, Fritz Obermeyer , Andrew Beam, and Ge vorg Grigoryan. Illuminating protein space with a programmable generativ e model. Nature , 623:1070–1078, 2023. 1 Michael A Jendrusch, Alessio L J Y ang, Elisabetta Cacace, Jacob Bobonis, Carlos G P V oogdt, Sarah Kaspar , Kristian Schweimer , Cecilia Perez-Borrajero, Karine Lapouge, Jacob Scheurich, Kim Remans, Janosch Hennig, Athanasios T ypas, Jan O Korbel, and S Kashif Sadiq. Alphadesign: a de nov o protein design framew ork based on alphafold. Molecular Systems Biology , 21(9):1166–1189, 2025. doi: https://doi.org/10.1038/s44320- 025- 00119- z. URL https://www.embopress. org/doi/abs/10.1038/s44320- 025- 00119- z . 3 , 8 John Jumper , Richard Evans, Alexander Pritzel, T im Green, Michael Figurno v , Olaf Ronneberger , Kathryn T unyasuvunakool, Russ Bates, Augustin Zidek, Anna Potapenko, Alex Bridgland, Clemens Meyer , Simon A. A. Kohl, Andre w J. Ballard, Andrew Cowie, Bernardino Romera- Paredes, Stanisla v Nikolov , Rishub Jain, Jonas Adler , Tre vor Back, Stig Petersen, Da vid Reiman, Ellen Clancy , Michal Zielinski, Martin Steinegger , Michalina Pacholska, T amas Berghammer , Sebastian Bodenstein, David Silv er, Oriol V inyals, Andre w W . Senior , K oray Kavukcuoglu, Push- meet K ohli, and Demis Hassabis. Highly accurate protein structure prediction with alphafold. Natur e , 596:583–589, 2021. 1 , 3 , 5 , 25 , 35 Felix Kallenborn, Alejandro Chacon, Christian Hundt, Hassan Sirelkhatim, Kieran Didi, Sooyoung Cha, Christian Dallago, Milot Mirdita, Bertil Schmidt, and Martin Steinegger . Gpu-accelerated homology search with mmseqs2. Natur e Methods , 22(10):2024–2027, 2025. 21 W oosub Kim, Milot Mirdita, Eli Levy Karin, Cameron LM Gilchrist, Hugo Schweke, Johannes S ¨ oding, Emmanuel D Le vy , and Martin Steinegger . Rapid and sensiti ve protein comple x alignment with foldseek-multimer . Natur e methods , 22(3):469–472, 2025. 21 Diederik P Kingma. Adam: A method for stochastic optimization. arXiv pr eprint arXiv:1412.6980 , 2014. 28 14 Published as a conference paper at ICLR 2026 Lev ente Kocsis and Csaba Szepesv ´ ari. Bandit based monte-carlo planning. In Eur opean confer ence on machine learning , pp. 282–293. Springer , 2006. 31 Xiangzhe K ong, Zishen Zhang, Ziting Zhang, Rui Jiao, Jianzhu Ma, W enbing Huang, Kai Liu, and Y ang Liu. Unimomo: Unified generativ e modeling of 3d molecules for de nov o binder design. In F orty-second International Conference on Mac hine Learning , 2025. 10 , 19 Rohith Krishna, Jue W ang, W oody Ahern, Pascal Sturmfels, Preetham V enkatesh, Indrek Kalvet, Gyu Rie Lee, Felix S. Morey-Burro ws, Ivan Anishchenko, Ian R. Humphreys, Ryan McHugh, Dionne V afeados, Xinting Li, George A. Sutherland, Andrew Hitchcock, C. Neil Hunter , Alex Kang, Evans Brack enbrough, Asim K. Bera, Minkyung Baek, Frank DiMaio, and David Baker . Generalized biomolecular modeling and design with rosettafold all-atom. Science , 384(6693): eadl2528, 2024. doi: 10.1126/science.adl2528. URL https://www.science.org/doi/ abs/10.1126/science.adl2528 . 3 , 8 , 24 Jakub L ´ ala, A yham Al-Saf far , and Stefano Angiolleti-Uberti. Bagel: Protein engineering via explo- ration of an energy landscape. bioRxiv , 2025. doi: 10.1101/2025.07.05.663138. URL https: //www.biorxiv.org/content/early/2025/07/08/2025.07.05.663138 . 3 A. M. Lau, N. Bordin, S. M. Kandathil, I. Sillitoe, V . P . W aman, J. W ells, C. A. Orengo, and D. T . Jones. The encyclopedia of domains (ted) structural domains assignments for alphafold database v4 [data set]. Zenodo , 2024a. 4 , 11 , 21 A. M. Lau, N. Bordin, S. M. Kandathil, I. Sillitoe, V . P . W aman, J. W ells, C. A. Orengo, and D. T . Jones. Exploring structural diversity across the protein uni verse with the encyclopedia of domains. bioRxiv , 2024b. 2 , 4 Andrew Lea ver-F ay , Jeff Flatten, Alex Ford, Joseph Kleinhenz, Henry Solberg, David Baker , An- drew M. W atkins, Brian Kuhlman, and Frank DiMaio. tmol: a gpu-accelarated, pytorch im- plementation of rosetta’ s relax protocol. https://github.com/uw- ipd/tmol , 2025. 6 , 25 Y eqing Lin, Minji Lee, Zhao Zhang, and Mohammed AlQuraishi. Out of many , one: Designing and scaffolding proteins at the scale of the structural univ erse with genie 2. arXiv pr eprint arXiv:2405.15489 , 2024. 4 , 25 Y aron Lipman, Ricky T . Q. Chen, Heli Ben-Hamu, Maximilian Nickel, and Matthe w Le. Flow matching for generati ve modeling. In The Eleventh International Confer ence on Learning Repre- sentations (ICLR) , 2023. 2 , 3 Xingchao Liu, Chengyue Gong, and qiang liu. Flo w straight and fast: Learning to generate and transfer data with rectified flo w . In The Eleventh International Confer ence on Learning Repr esentations (ICLR) , 2023. 3 T ianyu Lu, Richard Shuai, Petr Kouba, Zhaoyang Li, Y ilin Chen, Akio Shirali, Jinho Kim, and Po-Ssu Huang. Conditional protein structure generation with protpardelle-1c. bioRxiv , 2025. doi: 10.1101/2025.08.18.670959. URL https://www.biorxiv.org/content/early/ 2025/08/18/2025.08.18.670959 . 3 , 8 Nanye Ma, Mark Goldstein, Michael S. Albergo, Nicholas M. Boffi, Eric V anden-Eijnden, and Saining Xie. Sit: Exploring flow and dif fusion-based generativ e models with scalable interpolant transformers. arXiv pr eprint arXiv:2401.08740 , 2024. 20 Nanye Ma, Shangyuan T ong, Haolin Jia, Hexiang Hu, Y u-Chuan Su, Mingda Zhang, Xuan Y ang, Y andong Li, T ommi Jaakkola, Xuhui Jia, and Saining Xie. Inference-time scaling for diffusion models beyond scaling denoising steps. arXiv preprint , 2025. 1 Ian K McDonald and Janet M Thornton. Satisfying hydrogen bonding potential in proteins. Journal of molecular biology , 238(5):777–793, 1994. 6 , 25 Pierre Moral. F eynman-Kac formulae: genealogical and inter acting particle systems with applica- tions . Springer , 2004. 31 15 Published as a conference paper at ICLR 2026 Serge y Ovchinniko v , Stephen Rettie, Andrew Fav or , Hunar Batra, and Keaun Amani. sokryp- ton/colabdesign: v1.1.3. Zenodo , 2025. 6 , 35 Max D. Overath, Andreas S.H. Rygaard, Christian P . Jacobsen, V alentas Brasas, Oliver Morell, Pietro Sormanni, and Timothy P . Jenkins. Predicting experimental success in de nov o binder design: A meta-analysis of 3,766 experimentally characterised binders. bioRxiv , 2025. doi: 10. 1101/2025.08.14.670059. URL https://www.biorxiv.org/content/early/2025/ 09/17/2025.08.14.670059 . 6 Matthew J. O’Meara, Andrew Leav er-F ay , Michael D. T yka, Amelie Stein, K evin Houlihan, Frank DiMaio, Philip Bradley , T anja Kortemme, David Baker , Jack Snoeyink, and Brian Kuhlman. Combined cov alent-electrostatic model of hydrogen bonding improv es structure prediction with rosetta. Journal of Chemical Theory and Computation , 11(2):609–622, 2015. 6 Martin Pacesa, Lennart Nickel, Christian Schellhaas, Joseph Schmidt, Ekaterina Pyatov a, Lucas Kissling, Patrick Barendse, Jagrity Choudhury , Srajan Kapoor, Ana Alcaraz-Serna, Y ehlin Cho, K ourosh H. Ghamary , Laura V inu ´ e, Brahm J. Y achnin, Andrew M. W ollacott, Stephen Buckley , Adrie H. W estphal, Simon Lindhoud, Sandrine Geor geon, Casper A. Goverde, Geor gios N. Hat- zopoulos, Pierre G ¨ onczy , Y annick D. Muller , Gerald Schwank, Daan C. Sw arts, Alex J. V ecchio, Bernard L. Schneider , Sergey Ovchinnik ov , and Bruno E. Correia. One-shot design of functional protein binders with bindcraft. Natur e , 2025. doi: https://doi.org/10.1038/s41586- 025- 09429- 6. 1 , 3 , 7 , 8 , 23 , 33 Saro Passaro, Gabriele Corso, Jeremy W ohlwend, Mateo Rev eiz, Stephan Thaler, V ignesh Ram Somnath, Noah Getz, T ally Portnoi, Julien Ro y , Hannes Stark, et al. Boltz-2: T owards accurate and efficient binding af finity prediction. BioRxiv , pp. 2025–06, 2025. 22 , 35 , 49 W illiam Peebles and Saining Xie. Scalable diffusion models with transformers. In Pr oceedings of the IEEE/CVF International Confer ence on Computer V ision (ICCV) , 2023. 21 , 26 V ignav Ramesh and Morteza Mardani. T est-time scaling of diffusion models via noise trajectory search. arXiv pr eprint arXiv:2506.03164 , 2025. 2 , 7 , 30 , 32 Robin Rombach, Andreas Blattmann, Dominik Lorenz, Patrick Esser , and Bj ¨ orn Ommer . High- Resolution Image Synthesis with Latent Diffusion Models. In Pr oceedings of the IEEE/CVF Confer ence on Computer V ision and P attern Recognition (CVPR) , 2022. 6 Neeladri Sen and Mallur S. Madhusudhan. A structural database of chain–chain and domain–domain interfaces of proteins. Protein Science , 31(9):e4406, 2022. doi: https://doi.org/10.1002/pro.4406. URL https://onlinelibrary.wiley.com/doi/abs/10.1002/pro.4406 . 4 Raghav Singhal, Zachary Horvitz, Ryan T eehan, Mengye Ren, Zhou Y u, Kathleen McKeo wn, and Rajesh Ranganath. A general frame work for inference-time scaling and steering of diffusion models. In F orty-second International Confer ence on Machine Learning , 2025. 2 , 6 , 7 , 30 Charlie V ictor Snell, Jaehoon Lee, Kelvin Xu, and A viral Kumar . Scaling LLM test-time com- pute optimally can be more effecti ve than scaling parameters for reasoning. In The Thirteenth International Confer ence on Learning Representations , 2025. 1 Y ang Song, Jascha Sohl-Dickstein, Diederik P Kingma, Abhishek Kumar , Stefano Ermon, and Ben Poole. Score-Based Generative Modeling through Stochastic Differential Equations. In International Confer ence on Learning Representations (ICLR) , 2021. 3 Martin Steinegger and Johannes S ¨ oding. Mmseqs2 enables sensitiv e protein sequence searching for the analysis of massiv e data sets. Nature biotec hnology , 35(11):1026–1028, 2017. 35 Chai Discovery T eam, Jacques Boitreaud, Jack Dent, Danny Geisz, Matthe w McPartlon, Joshua Meier, Zhuoran Qiao, Alex Rogozhnik ov , Nathan Rollins, Paul W ollenhaupt, and K evin W u. Zero-shot antibody design in a 24-well plate. bioRxiv , 2025a. doi: 10.1101/2025.07.05.663018. URL https: //www.biorxiv.org/content/early/2025/07/06/2025.07.05.663018 . 3 16 Published as a conference paper at ICLR 2026 Latent Labs T eam, Alex Bridgland, Jonathan Crabb ´ e, Henry Kenlay , Daniella Pretorius, Sebastian M. Schmon, Agrin Hilmkil, Rebecca Bartke-Croughan, Robin Rombach, Michael Flashman, T omas Matteson, Simon Mathis, Alexander W . R. Nelson, Da vid Y uan, Annette Obika, and Simon A. A. K ohl. Latent-x: An atom-level frontier model for de nov o protein binder design. arXiv preprint arXiv:2507.19375 , 2025b. 3 Protenix T eam, Milong Ren, Jinyuan Sun, Jiaqi Guan, Cong Liu, Chengyue Gong, Y uzhe W ang, Lan W ang, Qixu Cai, Xinshi Chen, and W enzhi Xiao. Pxdesign: Fast, modular , and accurate de nov o design of protein binders. bioRxiv , 2025c. doi: 10.1101/2025.08.15.670450. URL https: //www.biorxiv.org/content/early/2025/08/16/2025.08.15.670450 . 3 , 9 Michel v an Kempen, Stephanie S. Kim, Charlotte T umescheit, Milot Mirdita, Jeongjae Lee, Cameron L. M. Gilchrist, Johannes S ¨ oding, and Martin Steine gger . Fast and accurate protein structure search with foldseek. Nat Biotechnol. , 42:243–246, 2024. 4 , 25 Mihaly V aradi, Stephen Anyango, Mandar Deshpande, Sreenath Nair, Cindy Natassia, Galabina Y ordanova, Da vid Y uan, Oana Stroe, Gemma W ood, Agata Laydon, Augustin ˇ Z ´ ıdek, T im Green, Kathryn T unyasuvunakool, Stig Petersen, John Jumper , Ellen Clancy , Richard Green, Ankur V ora, Mira Lutfi, and Sameer V elankar . Alphafold protein structure database: Massiv ely expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Researc h , 50:D439–D444, 2021. 4 , 11 , 35 Douglas V estal, Ren ´ e Carmona, and Jean-Pierre Fouque. Interacting particle systems for the compu- tation of CDO tranche spreads with rare defaults. preprint , 2008. 31 Y uyang W ang, Ahmed A Elhag, Navdeep Jaitly , Joshua M Susskind, and Miguel Angel Bautista. Swal- lowing the bitter pill: Simplified scalable conformer generation. arXiv pr eprint arXiv:2311.17932 , 2023. 27 Joseph L. W atson, David Juergens, Nathaniel R. Bennett, Brian L. Trippe, Jason Y im, Helen E. Eisenach, W oody Ahern, Andrew J. Borst, Robert J. Ragotte, Lukas F . Milles, Basile I. M. W icky , Nikita Hanikel, Samuel J. Pellock, Alexis Courbet, W illiam Sheffler , Jue W ang, Preetham V enkatesh, Isaac Sappington, Susana V ´ azquez T orres, Anna Lauko, V alentin De Bortoli, Emile Mathieu, Regina Barzilay , T ommi S. Jaakkola, Frank DiMaio, Mink yung Baek, and Da vid Baker . De nov o design of protein structure and function with rfdiffusion. Natur e , 620:1089–1100, 2023. 1 , 3 , 8 Jason W ei, Xuezhi W ang, Dale Schuurmans, Maarten Bosma, brian ichter , Fei Xia, Ed H. Chi, Quoc V Le, and Denny Zhou. Chain of thought prompting elicits reasoning in large language models. In Advances in Neural Information Pr ocessing Systems , 2022. 1 Jeremy W ohlwend, Gabriele Corso, Saro Passaro, Noah Getz, Mateo Reveiz, Ken Leidal, W ojtek Swiderski, Liam Atkinson, T ally Portnoi, Itamar Chinn, Jacob Silterra, T ommi Jaakkola, and Regina Barzilay . Boltz-1 democratizing biomolecular interaction modeling. bioRxiv , 2025. doi: 10.1101/2024.11.19.624167. URL https://www.biorxiv.org/content/early/ 2025/05/06/2024.11.19.624167 . 3 Jaesik Y oon, Hyeonseo Cho, Doojin Baek, Y oshua Bengio, and Sungjin Ahn. Monte carlo tree diffusion for system 2 planning. In F orty-second International Confer ence on Machine Learning (ICML) , 2025. 2 , 7 , 32 V inicius Zambaldi, David La, Alexander E. Chu, Harshnira P atani, Amy E. Danson, Tristan O. C. Kwan, Thomas Frerix, Rosalia G. Schneider , David Saxton, Ashok Thillaisundaram, Zachary W u, Isabel Moraes, Oskar Lange, Eliseo P apa, Gabriella Stanton, V ictor Martin, Sukhdeep Singh, Lai H. W ong, Russ Bates, Simon A. K ohl, Josh Abramson, Andre w W . Senior , Y ilmaz Alguel, Mary Y . W u, Irene M. Aspalter , Katie Bentley , David L. V . Bauer, Peter Cherepanov , Demis Hassabis, Pushmeet K ohli, Rob Fergus, and Jue W ang. De novo design of high-af finity protein binders with alphaproteo, 2024. 3 , 6 , 23 , 24 , 35 Y ang Zhang and Jeffre y Skolnick. Scoring function for automated assessment of protein structure template quality . Proteins: Structure , Function, and Bioinformatics , 57(4):702–710, 2004. 25 17 Published as a conference paper at ICLR 2026 A ppendix A Limitations and Future W ork 19 B La-Prote ´ ına Background 19 B.1 Overvie w . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19 B.2 Autoencoder . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20 B.3 Partially Latent Flo w Matching . . . . . . . . . . . . . . . . . . . . . . . . . . 20 B.4 Sampling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20 B.5 Architectures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21 C T eddymer Data 21 C.1 Comparison to Protein-Protein Interfaces from Protein Databank . . . . . . . . . 21 D Other Datasets 22 D.1 PDB Data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22 D.2 PLINDER . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 D.3 AFDB Monomer Pretraining Data . . . . . . . . . . . . . . . . . . . . . . . . . 23 E Protein and Small Molecule T argets 23 F Evaluation Metrics 24 G Architecture, Model, T raining and Sampling Details 25 G.1 Denoiser Architecture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 G.2 Features used as input to the Denoiser . . . . . . . . . . . . . . . . . . . . . . . 26 G.3 Training . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27 G.4 Sampling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 G.5 Cropping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 H Inference-time Optimization Method Details 29 H.1 Best-of-N Sampling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 H.2 Beam Search . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 H.3 Feynman–Kac Steering (FK Steering) . . . . . . . . . . . . . . . . . . . . . . . 30 H.4 Monte Carlo Tree Search (MCTS) . . . . . . . . . . . . . . . . . . . . . . . . . 31 H.5 Generate & Hallucinate (G&H) . . . . . . . . . . . . . . . . . . . . . . . . . . 33 I Ablation Studies and Additional Experiments 34 I.1 Ablation Experiment: T raining without T eddymer . . . . . . . . . . . . . . . . 34 I.2 Ablation Experiment: T raining without T ranslation Noise . . . . . . . . . . . . 36 I.3 Ablation Experiment: BindCraft Stages in Generate & Hallucinate . . . . . . . . 36 I.4 Inference-time Scaling for V ery Hard T argets . . . . . . . . . . . . . . . . . . . 39 I.5 Extended Results: Generativ e Model Benchmark with Sequence Re-Design . . . 39 I.6 Extended Results: Inference-time Search for all Protein T argets . . . . . . . . . 41 I.7 Extended Results: Inference-time Search for all Ligand T argets . . . . . . . . . 42 I.8 Extended Results: Interface Hydrogen Bond Optimization for All T argets . . . . 43 I.9 Extended Results: Enzyme Design . . . . . . . . . . . . . . . . . . . . . . . . 44 I.10 Case Study: Long Inference-time Search and Plateau Behavior . . . . . . . . . . 47 I.11 ipAE Scores and Hydrogen Bond Correlations . . . . . . . . . . . . . . . . . . 48 I.12 Binding Affinities for Small Molecule T argets . . . . . . . . . . . . . . . . . . 49 18 Published as a conference paper at ICLR 2026 I.13 Biophysical Interface Analyses for Protein T argets . . . . . . . . . . . . . . . . 49 J Baseline Evaluations 49 K Additional Visualizations 51 L Declaration on Usage of Large Language Models 51 A L I M I T A T I O N S A N D F U T U R E W O R K The focus of this work is to introduce and v alidate Complexa’ s novel atomistic binder design frame- work, which unifies state-of-the-art generati ve modeling with modern inference-time optimization, combining the strengths of previously separate generati ve and hallucination approaches. T o this end, we restrict our applications to designing binders and enzymes for protein and small molecule targets. A natural extension would be to apply Complexa to other molecular modalities, such as DN A and RN A. T o this end, it would be interesting to also train a single, unified model that can both condition on and flexibly generate different molecular modalities, including proteins, peptides, small molecules, nucleic acids, antibodies and more. Joint training on div erse molecules may also boost ov erall performance due to transfer learning across the modalities. This idea was recently explored by UniMoMo in the context of small molecule, peptide and antibody design ( K ong et al. , 2025 ). While our e v aluations are limited to in-silico success metrics, an important next step will be experimen- tal validation of generated binders in the wet lab . Future work could also target additional molecular properties—such as specificity or thermostability—by integrating suitable computational predictors into Complexa’ s inference-time search framework. W e leave these directions to future in vestigation. B L A - P RO T E ´ I N A B A C K G R O U N D Below , we describe La-Prote ´ ına ( Geffner et al. , 2026 ) on a high-lev el. This section is purely for reference purposes. W e adopt their notation, and emphasize that all content here is explained in more detail in Geffner et al. ( 2026 ). B . 1 O V E RV I E W La-Prote ´ ına is a generativ e model of atomistic proteins, producing both the amino acid sequence and fully atomistic three dimensional structures. It was dev eloped for unconditional monomer generation and atomistic motif scaf folding tasks. The model achieves state-of-the-art performance, generates structures with higher biophysical validity than baseline methods, and is designed to be ef ficient and scalable (by av oiding the use of computationally expensi ve triangular update layers). The core modeling choice in La-Prote ´ ına is its partially latent representation of proteins. In this scheme, the coordinates of the α -carbon atoms ( C α ) are modeled explicitly , while the sequence and all other atomistic details (non- C α atoms and side chains) are encoded into continuous, fixed-size, per-residue latent variables using an autoencoder . La-Prote ´ ına then trains a flow matching model in this continuous, fixed-size space to serve as its main generati ve component. This partially latent approach offers sev eral key advantages: (1) It transforms the complex generative problem from a mixed discrete/continuous space (sequence and coordinates) with v ariable dimensionality (different number of side chains atoms for different residue types) into a more manageable, fixed-size (per residue), and fully continuous space. La-Prote ´ ına then uses flow matching in this partially latent space as its generativ e model. (2) Separately modeling the α -carbon coordinates allows for the use of distinct generation schedules during inference. This enables a faster generation scheme for α -carbon backbone than the latent v ariables, which has been observed to be critical to achie ve high performance in the La-Prote ´ ına empirical e valuation. (3) This design enhances scalability . The per-residue latent v ariables function as additional channels on top of the α -carbon coordinates, which enables the use of powerful transformer architectures without increasing the model’ s sequence length representations. La-Prote ´ ına is composed of three neural networks: an encoder that maps fully atomistic proteins to their partially latent representation; a decoder that reconstructs fully atomistic proteins from these la- 19 Published as a conference paper at ICLR 2026 tent v ariables and the corresponding α -carbon coordinates; and a denoiser network that predicts clean samples from noisy latent variables and α -carbon coordinates as part of the flo w matching process. B . 2 A U T O E N C O D E R The autoencoder in La-Prote ´ ına is a V ariational Autoencoder (V AE), responsible for mapping between the full protein structure and its partially latent representation. Following the notation used in La- Prote ´ ına, we use L to denote number of residues in a protein, x C α ∈ R L × 3 for α -carbon coordinates, x ¬ C α ∈ R L × 36 × 3 for non- α -carbon atom coordinates (using the Atom37 representation without α -carbon atoms), s ∈ { 0 , ..., 19 } L for the protein sequence, and z ∈ R L × 8 for the latent variables. Decoder . The V AE decoder takes latent variables z and α -carbon coordinates x C α as input and outputs a distribution ov er sequences and coordinates of non- C α atoms. Formally , the decoder is defined by the conditionally independent distribution p ϕ ( x ¬ C α , s | x C α , z ) = p ϕ ( s | x C α , z ) p ϕ ( x ¬ C α | x C α , z ) , (5) where p ϕ ( s | x C α , z ) is modeled as a factorized categorical distrib ution for the sequence (the decoder network outputs the logits) and p ϕ ( x ¬ C α | x C α , z ) is a factorized Gaussian with unit v ariance for the atomic coordinates (the decoder network outputs the mean). Encoder . The encoder maps a fully atomistic protein to its corresponding latent representation. Formally , the encoder parameterizes q ψ ( z | x C α , x ¬ C α , s ) , a factorized Gaussian. The network inputs the complete protein and outputs the mean and log-scale parameters for this distribution. V AE training. The encoder and decoder are trained jointly by maximizing the β -ELBO max ϕ,ψ E p data ( x C α , x ¬ C α , s ) ,q ψ ( z | ... ) [log p ϕ ( x ¬ C α , s | x C α , z )] − β K L ( q ψ ( z | x C α , x ¬ C α , s ) || p ( z )) , (6) where the prior ov er latent variables p ( z ) is a standard Gaussian distribution. B . 3 P A RT I A L L Y L A T E N T F L OW M A T C H I N G W ith the trained autoencoder , the primary generativ e task is simplified to learning the joint distrib ution ov er α -carbon coordinates and latent variables, p ( x C α , z ) , defined in a continuous, per-residue, fixed- size space. La-Prote ´ ına employs a flo w matching model for this purpose, which learns to transport samples from a standard Gaussian distribution to the target data distribution. This is achie ved by training a denoiser network v θ minimizing the conditional flow matching objecti ve min θ E [ || v x θ ( x C α t x , z t z , t x , t z ) − ( x C α − x C α 0 ) || 2 + || v z θ ( x C α t x , z t z , t x , t z ) − ( z − z 0 ) || 2 ] , (7) where the expectation is taken over ( x C α , z ) ∼ p data , noise variables x C α 0 ∼ N (0 , I ) and z 0 ∼ N (0 , I ) , and interpolation time distributions p t x ( t x ) and p t z ( t z ) . For the latter La-Prote ´ ına uses p t x = 0 . 02 Unif (0 , 1) + 0 . 98 Beta(1 . 9 , 1) and p t z = 0 . 02 Unif (0 , 1) + 0 . 98 Beta(1 , 1 . 5) . (8) B . 4 S A M P L I N G La-Prote ´ ına generates ne w protein samples by numerically simulating a system of stochastic differ - ential equations (SDEs) from ( t x , t z ) = (0 , 0) to ( t x , t z ) = (1 , 1) . These equations use the score, denoted by ζ , which represents the gradient of the log-probability of the intermediate densities (this score can be computed directly from the learned v elocity field v θ ( Ma et al. , 2024 )). The SDEs are giv en by: d x C α t x = v x θ ( x C α t x , z t z , t x , t z ) d t x + β x ( t x ) ζ x ( x C α t x , z t z , t x , t z ) d t x + p 2 β x ( t x ) η x d W t x (9) d z t z = v z θ ( x C α t x , z t z , t x , t z ) d t z + β z ( t z ) ζ z ( x C α t x , z t z , t x , t z ) d t z + p 2 β z ( t z ) η z d W t z . La-Prote ´ ına uses scaling functions β x ( t x ) = 1 /t x and β z ( t z ) = π 2 tan( π 2 (1 − t z )) to modulate the Lange vin-like term in the SDEs. It also employs noise scaling parameters η x , η z < 1 (typically set to 0.1), which functions as a form of ”low temperature” sampling, which impro ves (co-)designability . 20 Published as a conference paper at ICLR 2026 La-Prote ´ ına discretizes the SDEs using the Euler -Maruyama scheme. A critical aspect of the sampling process is the use of different schedules for the α -carbons and latent variables z ; specifically , an exponential schedule is used for x C α and a quadratic schedule for z , which ef fectively means that the alpha carbon coordinates are denoised at a faster rate than the the latent v ariables. After the flo w matching model generates a pair of ( x C α , z ) , these are passed through the V AE decoder to produce the final, fully atomistic protein structure and sequence. While the decoder defines a distribution o ver sequence and coordinates, La-Prote ´ ına deterministically selects the final output by taking the mean of the Gaussian distrib ution for the non- C α -carbon coordinates and the ar gmax of the categorical distrib ution’ s logits for the amino acid sequence s . B . 5 A R C H I T E C T U R E S All three neural networks in La-Prote ´ ına (encoder , decoder , denoiser) are b uilt upon the architecture of Proteina, which relies on transformers with pair-biased attention mechanisms. The denoiser network additionally conditions on the interpolation times ( t x , t z ) by integrating adapti ve layer normalization and output scaling techniques directly into its transformer blocks ( Peebles & Xie , 2023 ). C T E D DY M E R D A TA For T eddymer, we first processed the entire AlphaFold database (AFDB) with TED annotations ( Lau et al. , 2024a ), treating each domain as a chain. Since the database contains about 203M structures, which is extremely large, we filtered the database to retain only the structures belonging to AFDB50, a clustered version of AFDB generated by MMseqs2 at 50% sequence identity ( Barrio-Hernandez et al. , 2023 ). W e then extracted dimers from the database and filtered those where both chains were annotated by CA TH at least up to the C.A.T . lev el. Finally , we clustered the database to reduce redundancy , resulting in 3,556,223 clusters. The detailed procedure is described as below . 1. T eddb can be downloaded from foldseek. This database contains 203,057,497 structures and a total of 364,806,077 chains. T o provide a smaller version, we generated teddb afdb50. This database contains 47,180,623 structures and a total of 77,743,546 chains. 2. Dimers in teddb afdb50 (123M entries): W e extracted 123,606,001 dimers from multimers in teddb afdb50. Here, a dimer is defined as a pair of chains in which each chain has at least 4 residues within 10 ˚ A of the other in terms of CA-CA distance. T o ensure reliable and precise structures, we used CA TH annotations. W e generated a subset of this dimer database in which both chains (originally domains) are annotated by CA TH at least up to the C.A.T . lev el. The final database contains 10,089,503 dimers. 3. Clustered dimerdb (3.5M clusters): W e clustered the dimer database with GPU-accelerated Foldseek-Multimer ( Kallenborn et al. , 2025 ; Kim et al. , 2025 ) using both chain-lev el structural similarity and interface similarity . This resulted in 3,556,223 clusters: 587,687 nonsingletons and 2,968,536 singletons. The clustering was performed with the following command: foldseek easy-multimercluster teddb afdb50 dimerdb cath teddb afdb50 dimerdb cath clustered tmp -c 0.6 –chain-tm-threshold 0.7 –interface-lddt-threshold 0.3 –cluster -mode 0 From this database, we generate the final T eddymer -based training dataset by filtering for interface length > 10, interface-pAE < 10 and interface-pLDDT > 70, leading to 510.454 cluster reps and 7.112.609 ov erall datapoints. W e only train on the cluster representativ es. C . 1 C O M PAR I S O N T O P RO T E I N - P R OT E I N I N T E R FA C E S F RO M P RO T E I N D A TA B A N K T o quantitati vely compare the synthetic protein-protein interfaces of Complexa with e xperimental PDB multimers, we compute 6 metrics on the complex structures. Those metrics include: number of hydrogen bonds across the interf ace, hydrophobicity of the binder interf ace, hydrophobicity of the binder surface, shape complementarity of the binder interface, delta Solvent Accessible Surface Area (dSASA) of the binder interf ace, and the number of binder residues on the interface. W e randomly selected 2,000 complexes from both PDB multimers and T eddymer and plot the distributions of the metrics in Fig. 12 . On some metrics, we notice slightly more div ersity of the PDB data, which is expected as T eddymer was created from the domain-domain interactions of synthetic AFDB 21 Published as a conference paper at ICLR 2026 0 20 40 60 80 # Interface H- Bonds 0 200 400 600 Count 0 25 50 75 100 Binder interface hydr ophobicity (%) 0 100 200 300 0.00 0.25 0.50 0.75 1.00 Binder surface hydr ophobicity (%) 0 100 200 300 0.0 0.2 0.4 0.6 0.8 Binder interface shape complementarity 0 100 200 Count 0 5000 10000 15000 B i n d e r i n t e r f a c e d S A S A ( Å 2 ) 0 200 400 0 50 100 150 200 # Binder interface r esidues 0 200 400 T eddymer PDB Multimer Figure 12: Experimental vs. synthetic interfaces. Comparison between T eddymer and PDB multimer on various protein-protein interf ace metrics. monomers. Importantly , we find that there is nonetheless significant overlap in the distributions, indicating that T eddymer can serv e as a source of data augmentation when training lar ge-scale protein binder generativ e models like Complexa. This is aligned with our numerical results and ablations studies (Sec. I.1 ), which demonstrate that including T eddymer during training dramatically boosts performance. D O T H E R D A TA S E T S W e also create multiple other datasets for training as outlined below; for PDB multimer and PLINDER data, we use cropping to crop and extract binder -target complex es as detailed in Sec. G.5 . D . 1 P D B D AT A W e utilized the PDB multimer dataset for training protein binder models, with particular emphasis on the multimer aspect to handle not just dimers but complex multi-chain targets. W e applied a rigorous three-stage filtering strategy to ensure the quality of PDB data. Firstly , the dataset was processed to only include binders with lengths between 50 and 250 residues and tar get chains with minimum length of 50 residues. Next, we filtered structures based on crystallographic resolution, retaining only entries with resolution better than 5 ˚ A to ensure high-quality structural data. Thirdly , we le veraged the power of folding models to filter the PDB multimer dataset based on predicted metrics. Specifically , we folded the remaining samples after the second stage of filtering using Boltz2 ( Passaro et al. , 2025 ) and computed the ipAE (inter-chain predicted Aligned Error), complex ipLDDT (complex predicted Local Distance Difference T est with interface residues being upweighted), and ipTM (inter-chain predicted T emplate Modeling score) of the folded comple x structures. W e then keep only the samples that satisfy { ipAE ≤ 10 ˚ A , complex ipLDDT ≥ 0 . 8 , ipTM ≥ 0 . 5 } and have ≥ 1 residue at the predicted interface giv en 8 ˚ A cutoff. This stage served as an alignment of the training data to the predictiv e power of the state-of-the-art folding models, because these metrics provide confidence estimates for the accuracy of predicted protein-protein interfaces. W e want to emphasize that we included the original PDB structures in the training data and only used the folding model as a filtering tool. The final size of PDB multimer dataset is 45,856 after all filtering stages. 22 Published as a conference paper at ICLR 2026 T able 4: Selected protein targets with their structural information, binding specifications, and data source. T arget Name PDB ID T arget Chain & Residues Hotspot Residues Binder Length Source BBF-14 9HAG A1-112 – 70-250 BindCraft Betv1 AF2 prediction A1-159 A24 70-185 BindCraft CbAgo (P AZ) AF2 prediction A180-276 A268 70-160 BindCraft CD45 AF2 prediction d1 domain A1 80-200 BindCraft Claudin1 AF2 prediction A1-188 A31, A46, A55, A152 80-175 BindCraft CrSAS6 AF2 prediction A1-145 – 90-200 BindCraft Derf21 AF2 prediction A1-118 A10 70-185 BindCraft Derf7 AF2 prediction A1-196 A115 70-185 BindCraft HER2-AA V 1N8Z C23-268 C165, C170 60-100 BindCraft IFNAR2 2LAG A1-103 A45, A73, A75, A77, A89, A91 60-175 BindCraft PD-1 AF2 prediction A1-115 A33, A95, A97, A102 80-150 BindCraft SpCas9 AF2 prediction A96-174, A306-446 A360 70-150 BindCraft BHRF1 2WH6 A2-158 A65, A74, A77, A82, A85, A93 80-120 AlphaProteo H1 5VLI A1-50, A76-80, A107-111, A258-322, B1-68, B80-170 B21, B45, B52 40-120 AlphaProteo IL7RA 3DI3 B17-209 B58, B80, B139 50-120 AlphaProteo IL17A 4HSA A17-29, A41-131, B19-127 A94, A116, B67 50-140 AlphaProteo Insulin 4ZXB E6-155 E64, E88, E96 40-120 AlphaProteo PD-L1 5O45 A17-132 A56, A115, A123 50-120 AlphaProteo SARS-CoV -2 RBD 6M0J E333-526 E485, E489, E494, E500, E505 80-120 AlphaProteo TNF- α 1TNF A12-157, B12-157, C12-157 A113, C73 50-120 AlphaProteo TrkA 1WWW X282-382 X294, X296, X333 50-120 AlphaProteo VEGF A 1BJ1 V14-107, W14-107 W81, W83, W91 50-140 AlphaProteo D . 2 P L I N D E R For training small molecule binder models, we employed the PLINDER (Protein-Ligand INteraction Dataset and Evaluation Resource) dataset ( Durairaj et al. , 2024 ). Starting with the full PLINDER dataset, we applied similar filtering procedures used by the PLINDER authors for DiffDock training. Follo wing the established PLINDER curation protocol, we used their pre-filtered data and applied additional filtering for sanitized SMILES strings and ligands processable by RDKit. The curation process in volved identifying small molecules that are not considered PLINDER ligands, including artifacts and single non-hydrogen atom or ion entities. Entities containing single atoms (excluding basic organic elements C, H, N, O, P , S) were retained as auxiliary entities referred to as “ions, ” regardless of oxidation state (e.g., Fe 2+ , Xe, Cl − ). T o enrich for biologically and therapeutically relev ant ligands, we excluded molecules with fe wer than 2 carbons or fe wer than 5 non-hydrogen atoms, those that are highly char ged (absolute charge > 2 ), contain long unbranched hydrocarbon linkers ( > 12 atoms), contain unspecified atoms, or are common experimental b uffer artifacts. Artifact identification was based on common definitions and included additional buf fer reagents while excluding biologically rele vant cofactors and sugars. Each detected ligand was assigned a unique canonical SMILES representation. For single residue ligands, we used the Chemical Component Dictionary (CCD) for SMILES identification. D . 3 A F D B M O N O M E R P R E T R A I N I N G D AT A W e utilized cluster representati ves from the AlphaFold Database (AFDB) ( Barrio-Hernandez et al. , 2023 ) for both autoencoder training and latent model pretraining. The AFDB monomer data was also incorporated in a mixed manner when training small molecule binders. This dataset was filtered for a minimum av erage pLDDT of 80 and lengths between 32 and 256. E P RO T E I N A N D S M A L L M O L E C U L E T A R G E T S For protein targets, we aggregated the 10 protein targets from the main results of AlphaProteo ( Zambaldi et al. , 2024 ), including BHRF1, SC2RBD, IL-7RA, PD-L1, TrkA, IL-17A, VEGF-A, insulin, H1 and TNF- α . W e then added 12 of the 14 protein targets from BindCraft ( Pacesa et al. , 2025 ), including BBF-14, Betv1, CbAgo(P AZ), Claudin1, CrSAS6, Derf21, Derf7, IFN AR2, PD-1 and SpCas9) to create a total set of 22 targets (we excluded the tw o PD-L1 targets since PD-L1 was already represented in the AlphaProteo tar gets). Details below in T able 4 , for each tar get we chose the corresponding hotspots and croppings from the rele v ant papers; if there were multiple hotspot specifications av ailable we chose one of them as listed below in the table. 23 Published as a conference paper at ICLR 2026 T able 5: Selected small molecule targets with their structural information, binding specifications, and data source. T arget Name PDB ID T arget Chain Binder Length SAM A A 100 F AD B A 100 IAI C A 100 OQO D A 100 From the original publications we exclude H1, IL17A and TNF- α from AlphaProteo from our main benchmarks since none of the methods achiev e success in 20h runtime as indicated in the target table. This way , we end up with 19 targets in our final benchmarks. For CD45 from BindCraft, the intended hotspots are dif ferent domains instead of specific residues, which does not align with the rest of our benchmark; we therefore choose the d1 domain as the specified tar get and residue A1 as hotspot as indicated in the table. For inference time scaling experiments, we consider tar gets either as easy or hard targets depending on the performance of all generati ve methods in the fixed sample budget benchmark. Based on the results of the sample budget benchmark (Fig. 20 ), we choose the follo wing categories: • Easy: IFNAR2, BHRF1, BBF14, DerF21, TrkA, PD1, Insulin, DerF7, PDL1, IL7RA, CrSAS6, Claudin1 • Hard: VEGF A, SpCas9, SC2RBD, CbAgo, CD45, BetV1, HER2-AA V For small molecule tar gets, we adapt the same targets as chosen in RFDif fusion-AllAtom ( Krishna et al. , 2024 ) and BoltzDesign1 ( Cho et al. , 2025 ) as described in the table below and adopt the benchmark setup of generating 200 structures of length 100 per target. F E V A L U A T I O N M E T R I C S Protein target e valuation pipeline: W e generate 200 samples for ev ery target. For each sample, we generate 8 re-designed binder sequences using ProteinMPNN ( Dauparas et al. , 2022 ), and call those them MPNN sequences. For models that generate all-atom binders, we additionally generate another 8 re-designed binder sequences with the interface residue identity fixed ( MPNN-FI sequences). The re-designed sequences, plus the self-generated sequence by model ( Self sequences, both MPNN-FI and Self sequences are exclusi vely to all-atoms models) are then folded by structure prediction model. W e used ColabDesign implementation of AF2 for protein-protein complex folding. For this we enable target templating as well as the initial guess option in line with pre vious work ( Bennett et al. , 2023 ) that initialises the AF2 pair representation with the pairwise distances of the designed binder . Inter-chain predicted alignment error (ipAE, ↓ ), complex predicted local distance dif ference test (complex pLDDT , ↑ ), and self-consistent RMSD of the binder (binder scRMSD, ↓ ) metrics are then calculated for each folded structure. Next, we apply AlphaProteo ( Zambaldi et al. , 2024 ) filters on the best metric values of all sequences in each one of the MPNN/MPNN-FI/Self categories. A sample is considered to be successful in a category if it satisfies all three requirements: 1. ipAE < 7 ˚ A 2. complex pLDDT > 0 . 9 3. binder scRMSD < 1 . 5 ˚ A Ligand target ev aluation pipeline: W e generate 200 samples for every tar get. For each sample we generate a single re-designed binder sequence with LigandMPNN ( Dauparas et al. , 2025 ) ( MPNN sequence) as well as a re-designed binder sequences with the interface (within 6 ˚ A) residue identity fixed ( MPNN-FI sequence). The MPNN , the MPNN-FI , and self-generated sequences by model ( MPNN-FI and Self sequences are exclusiv ely to all-atoms models), are then folded by RF3 ( Corley et al. , 2025 ) for ligand-protein complex co-folding. A protein-ligand sample is considered to be successful in a category if it satisfies all three requirements: 1. min ipAE < 2 2. binder C α scRMSD < 2 ˚ A 24 Published as a conference paper at ICLR 2026 3. binder aligned ligand scRMSD < 5 ˚ A where min ipAE calculates the minimum entry of the cross-chain elements in the pAE matrix predicted by RF3. The ligand scRMSD is calculated after the complex has been aligned to the binder structure to make sure that the ligand pose/pocket is not altered significantly . The successes are clustered by Foldseek ( van K empen et al. , 2024 ) based on the binder structures, and the number of clusters are reported as the unique successes. The novelty metrics is based on the TM-Score ( Zhang & Skolnick , 2004 ), which measures the structural similarity between generated successes and a reference set. TM-Score ranges from 0 to 1 and lo wer score indicates lo wer structural similarity hence higher nov elty . In this work, we use the PDB database from F oldseek as the reference set. The novelty v alues reported in T ab . 2 are the averaged TM-Score of all successes for each target. Sampling time: For each diffusion-based models, we set the sampling batch size as 1 and recorded the sampling time of each sample. For hallucination-based model such as BindCraft and BoltzDesign1, we recorded the sampling time as the time the algorithm took to generate 1 final optimized design. The sampling time of each target was the a veraged sampling time of all 200 samples of various length. W e set a time limit of 4 hours on 1 NVIDIA A100 GPU for each sample. The sampling time of AlphaDesign sometimes exceeded the time limit for large target comple xes. Therefore, the av eraged sampling time of AlphaDesign is the av eraged sampling time of all finished samples for each target. For e valuation of hydrogen bonds, we utilise HBPlus ( McDonald & Thornton , 1994 ) during ev al- uation and search. For search we also tested tmol ( Leaver -Fay et al. , 2025 ), an open-source GPU implementation of the Rosetta energy function ( Alford et al. , 2017 ) and calculate the hydrogen bond energy at the interf ace of binder and target chains via this. G A R C H I T E C T U R E , M O D E L , T R A I N I N G A N D S A M P L I N G D E T A I L S In this work we introduce Complexa, an extension of the La-Prote ´ ına framew ork to the task of de nov o binder design. Complexa operates in a conditional setting: it receiv es a target protein structure as input and generates a no vel, fully atomistic protein binder predicted to interact with that target. Our methodology closely follo ws La-Prote ´ ına. W e lev erage exactly the same V AE architecture, trained on monomeric proteins, to learn a partially latent representation of proteins. The core modification lies in the partially latent flow matching model, which is now trained to generate a binder protein conditioned on a given target. W e emphasize that our approach does not generate the full tar get-binder complex; the target protein serves e xclusively as conditioning information. While our neural network architecture processes the binder and tar get jointly to learn their structural relationship, the generati ve output of our flow matching model is solely the binder . G . 1 D E N O I S E R A R C H I T E C T U R E Follo wing La-Prote ´ ına, our model’ s denoiser is built upon a transformer architecture that uses pair-biased attention mechanisms. The denoiser network is designed to process a target protein (coordinates and sequence) alongside a potentially noisy , partially latent representation of a binder ( α -carbon coordinates and latent variables). These inputs are jointly featurized into two tensors: a sequence representation with shape [ L binder + L target , c seq ] and a pair representation with shape [ L binder + L target , L binder + L target , c pair ] . Intuitively , the sequence representation encodes residue-level features, while the pair representation captures features between pairs of residues (e.g., inter-atomic distances). The transformer blocks iteratively update the sequence representation, with the pair representation used to bias the attention logits through linear layers ( Jumper et al. , 2021 ). Follo wing La-Prote ´ ına, the pair representation remains static throughout the network. While prior work in structural biology has used triangular update layers to refine the pair representation ( Lin et al. , 2024 ; Abramson et al. , 2024 ), these layers are computationally and memory-expensi ve, which sev erely limits architectural scalability . By omitting them, our model is highly efficient, a property that is particularly advantageous for applying inference-time scaling techniques. W e use c seq = 768 , c pair = 256 , and 14 transformer layers. This leads to a denoiser architecture of ∼ 160 M parameters. Although the denoiser network processes a joint representation of the target and binder , its generativ e output is the velocity field for the binder . This architectural choice mirrors the approach used for unindex ed motif scaffolding in La-Prote ´ ına. In that context, features corresponding to the structural 25 Published as a conference paper at ICLR 2026 motif were concatenated to the input representation of the protein being generated, yet the model’ s output was similarly restricted to the generated scaf fold, not the motif itself. Conditioning on diffusion times and CA T embeddings. The denoiser is conditioned on sev eral inputs be yond the protein structures themselves. First, it recei ves the diffusion times, t x and t z , which correspond to the noise lev els of the binder’ s α -carbon coordinates and latent variables, respecti vely . Follo wing La-Prote ´ ına, these time v alues are integrated into the network’ s transformer blocks using adaptiv e layer normalization and output scaling techniques ( Peebles & Xie , 2023 ). Additionally , we condition the model on the structural class of the tar get and binder proteins using their CA T labels. Each CA T label is mapped to a learnable embedding, which is then incorporated into the network using the same adaptiv e normalization and output scaling layers, as done by Geffner et al. ( 2025 ). G . 2 F E A T U R E S U S E D A S I N P U T T O T H E D E N O I S E R G . 2 . 1 I N I T I A L S E Q U E N C E R E P R E S E N TA T I O N . The initial sequence representation is a concatenation of two independently constructed tensors: one for the binder , with shape [ L binder , c seq ] , and one for the tar get, with shape [ L target , c seq ] . For the binder , the per-residue feature vector is derived from three sources: (i) the raw coordinates of the noisy α -carbons, (ii) the ra w v alues of the noisy latent variables, and (iii) a Fourier embedding of the residue index. These features are concatenated and projected through a linear layer to produce the initial binder feature tensor . For the target, the feature vector is constructed from fi ve sources: (i) a one-hot representation of the amino acid sequence, (ii) the full-atom structure (Atom37 representation), (iii) a binary indicator for hotspot residues, (iv) backbone torsion angles, binned into 20 bins between − π and π , and (v) side chain angles, also binned into 20 bins between − π and π . These are similarly concatenated and passed through a linear layer to yield the target’ s feature tensor . Finally , the binder and target sequence representations are concatenated along the length dimension to form a single input tensor of shape [ L binder + L target , c seq ] . G . 2 . 2 I N I T I A L P A I R R E P R E S E N TA T I O N . A similar modular construction is used for the initial pair representation, which has a final shape of [ L binder + L target , L binder + L target , c pair ] . This tensor is assembled from three distinct sub-tensors: one describing interactions within the binder , one for interactions within the target, and one for the cross-interactions between the binder and target. The binder -binder pair representation , with shape [ L binder , L binder , c pair ] , is constructed follo wing La-Proteina. Its features include (i) the relativ e sequence separation, one-hot encoded and limited to ± 64 residues, and (ii) the pairwise distances between the noisy α -carbon coordinates, b ucketized into 1 ˚ A bins between 1 ˚ A and 30 ˚ A. These features are concatenated and projected through a linear layer . The target-target pair representation , with shape [ L target , L target , c pair ] , is deriv ed from features that describe intra-target residue pairs. These include: (i) the relativ e sequence separation, one-hot encoded and limited to ± 64 residues, (ii) the pairwise distances between backbone atoms, b ucketized into 1 ˚ A bins between 1 ˚ A and 30 ˚ A, (iii) a binary chain index (0 if residues are from the same chain, 1 if different), and (i v) a hotspot pair variable (0 if neither residue is a hotspot, 1 if one is, and 2 if both are). These features are concatenated and fed into a linear layer for the target pair representation. The cross-pair r epresentation , describing binder -target interactions with shape [ L binder , L target , c pair ] , is b uilt from features capturing inter-protein residue relationships. (Note: these features are analogous to the target-tar get features but are computed between binder-tar get residue pairs). They include: (i) the pairwise distances between the binder’ s noisy α -carbon coordinates and the target’ s backbone atoms, buck etized into 1 ˚ A bins between 1 ˚ A and 30 ˚ A, (ii) a binary chain index, and (iii) a hotspot pair v ariable (note that there are no hotspots in the binder , therefore in this case this variable can only be 0 or 1). These features are concatenated and fed into a linear layer for the cross-pair representation. Finally , these three tensors are assembled into the full initial pair representation that is passed to the network. W e note that this feature construction scheme is inherently compatible with multi-chain targets; such targets are simply treated as a single entity , where the chain index feature naturally distinguishes between residues belonging to different chains. 26 Published as a conference paper at ICLR 2026 G . 2 . 3 S M A L L M O L E C U L E S A S C O N D I T I O N I N G I N F O R M A T I O N Small molecule conditioning largely follo ws that of protein targets, with the ke y difference being that we directly featurize the fully atomistic structure, as small molecules do not hav e a natural sequence representation ( Abramson et al. , 2024 ). The sequence length of our protein-ligand complexes (binder -ligand pairs) is L binder + N target , where N target is the number of ligand heavy atoms. The protein, or binder, components of both the sequence and pair representations are largely similar to those described in the prior section, with a minor modification: we replace residue indices with binary chain break features. The key differences from our protein-target approach lies in how we enable fully atomistic target sequence conditioning and binder-tar get pair conditioning. Specifically , we focus on featurizing the target and the interactions between the binder and tar get. The target feature v ector is constructed from fiv e sources: (i) a one-hot representation of the hea vy atom element type, (ii) the full-atom structure ( N target × 3 ), (iii) heavy atom charges, (iv) graph Laplacian positional encoding ( Cao et al. , 2025 ; W ang et al. , 2023 ), and (v) one-hot representation of the atom name ( Abramson et al. , 2024 ). These features are concatenated and passed through a linear layer to yield the tar get’ s feature tensor of shape [ N target , c seq ] , which is then concatenated with that of the binder along the sequence dimension. Similar to protein tar gets, the pair tensor is assembled from interactions within the binder , interactions within the target, and cross-interactions between the binder and tar get. The target-target pair repr esentation , with shape [ N target , N target , c pair ] , is deriv ed from features that describe intra-target atom pairs, including (i) pairwise distances between all atoms b ucketized into 1 ˚ A bins between 1 ˚ A and 30 ˚ A , (ii) the bond adjacency matrix, and (iii) the bond order matrix to indicate how atoms are bonded, if at all. These features are concatenated and fed through a linear layer to obtain the target pair representation. The cross-pair r epresentation , describing binder -target interactions with shape [ L binder , N target , c pair ] , is built from features capturing inter-chain residue relationships, specifically the protein binder backbone (N-CA-C-CB)-ligand tar get pairwise distances. These features are concatenated and fed into a linear layer for the cross-pair representation. The binder-binder , binder-target, and tar get-target pair representations are then combined to obtain a final shape of [ L binder + N target , L binder + N target , c pair ] . G . 3 T R A I N I N G Time sampling distrib utions. W e follow the time sampling distrib ution from Eq. ( 8 ) in La-Prote ´ ına. Data centering / translation noise. During training, we center the complex so that the target lies at the origin. Howe ver , if we directly add noise to the binder using linear interpolation, x C α t x = t x x C α + (1 − t x ) x C α 0 with random Gaussian noise x C α 0 , then the center of mass becomes C ( x C α t x ) = t x C ( x C α ) + (1 − t x ) C ( x C α 0 ) = t x C ( x C α ) , where C ( · ) denotes the center of mass. In this setting, the center of the clean binder can be trivially recovered as C ( x C α ) = C ( x C α t x ) /t x . This shortcut allo ws the model to bypass learning ho w to position the binder relative to the target in a generalizable way . Such an issue does not arise in monomer generation, where the model only needs to learn protein structure without reasoning about global placement ( Geffner et al. , 2025 ). T o mitigate this problem, we perturb the binder’ s C α coordinates with a random global translation d ∼ p d 0 = N ( d | 0 , c 2 d ) during interpolation (we set c d = 0 . 2 nm). The interpolated states then become x C α t x = t x x C α + (1 − t x )( x C α 0 + d 1 ) , z t z = t z E ( x ) + (1 − t z ) z 0 , with priors p C α 0 = N ( x C α 0 | 0 , I ) and p z 0 = N ( z 0 | 0 , I ) . During training, the model predicts velocities relative to this globally perturbed noise. This explicit translation noise breaks the aforementioned shortcut and forces the model to reason about the binder’ s global positioning. Conceptually , this can be viewed as applying an additional dif fusion or flo w-matching process ov er the binder’ s center of mass, gradually moving it tow ard the correct global position. From a Fourier perspecti ve, standard flow and diffusion models generate lo w-frequency components first ( F alck et al. , 2025 ), and global translation corresponds to the lo west-frequency mode. By injecting translation noise, we force the model to refine the global position throughout generation, a technique related to prior work ( Ahern et al. , 2025 ). 27 Published as a conference paper at ICLR 2026 G . 3 . 1 S TAG E W I S E T R A I N I N G This section describes the main choices regarding model training. Full training hyperparameters details can be found in T ab . 14 . All models are trained using Adam ( Kingma , 2014 ). T raining V AE. W e train the V AE in tw o stages. First, on the AFDB monomer pretraining dataset (Sec. D.3 ) for 500k steps on 16 NVIDIA A100-80GB GPUs. Besides pretraining on synthetic monomer data, in a second stage we also finetune it on a realistic PDB dataset. The dataset is constructed by extracting single chains from monomers and multimers with max. 10 oligomeric states from PDB. W e filter out chains shorter than 50, longer than 256, or resolution worse than 5.0 ˚ A, with over 50% loops or with radius of gyration over 3.0 ˚ A. This results in a dataset with 110,976 chains. W e fine-tune the V AE on this dataset for 40k steps on 16 GPUs with batch size 16 per GPU. T raining Latent Diffusion for Protein T argets. W e also use a two-stage approach to train the partially latent flo w matching model. In the first stage we train on the AFDB monomer pretraining dataset for 540K steps on 32 NVIDIA A100-80GB GPUs (in this stage there is no target). On the second stage we further fine-tune the model checkpoint for 290K steps using 96 NVIDIA A100-80GB GPUs on the mixture of T eddymer and PDB dataset, filtered as detailed in App. C and App. D.1 . The mixture coefficient of T eddymer and PDB is 8:2. W e also fine-tune the same model checkpoint from the first stage on T eddymer-only data with additional CA T label conditioning. The CA T -conditioning model is fine-tuned on T eddymer dataset for 200K steps with 96 NVIDIA A100-80GB GPUs. Stagewise T raining for Small Molecule T argets. In contrast to protein targets, we employ a 3-stage training protocol for ligand conditioning. In the first stage, we pretrain our V AE on AFDB monomers with lengths in the range [32 , 512] for 140K steps on 32 NVIDIA A100-80GB GPUs. W e increase the maximum length from the protein target variant due to our filtered PLINDER subset having an a verage system size of 400. In the second stage, we train the partially latent flow matching model on the same AFDB subset for 270K steps on 48 NVIDIA A100-80GB GPUs. In the third and final stage, we le verage LoRA ( Hu et al. , 2022 ) fine-tuning on a mixture of AFDB monomers and PLINDER protein-ligand comple xes cropped to a length of 512 for 60K steps on 96 NVIDIA A100-80GB GPUs. Additionally , we mask out all ligand target features 50% of the time to pre vent ov erfitting to the ligand due to the low ligand di versity of the training data. G . 4 S A M P L I N G For all protein and ligand targets we follow the same sampling algorithm used by La-Prote ´ ına (see Sec. B.4 ), using 400 steps to simulate the corresponding SDEs, the same Langevin scaling terms β x ( t x ) and β z ( t z ) , noise scaling factors η x = 0 . 1 and η z = 0 . 1 , and the exponential schedule for the binder’ s alpha carbon coordinates and the quadratic schedule for its latent v ariables. T o align with the translation noise perturbing scheme in training, during inference we also sample a global translation noise d ∼ p d 0 = N ( d | 0 , c 2 d ) with c d = 0 . 2 nm, and add it to the initial noisy structure. CA T -Conditioned Sampling. W e follow Prote ´ ına to provide CA T labels during inference to support CA T conditioning. In this work, we only consider C-le vel conditioning for generation, i.e. , Mainly Alpha, Mainly Beta and Mixed Alpha Beta. V isualizations are shown in Fig. 6 and Fig. 32 . G . 5 C R O P P I N G Protein samples were cropped using a hierarchical procedure designed to preserve binding interfaces while limiting the number of residues for model input. The process operated as follows: For binder chain selection, residues at intermolecular interfaces were identified based on backbone C α distances, using an 8.0 ˚ A cutoff to define interface contacts. One interface residue was then selected at random as a binder seed , and the chain containing this residue was designated the binder chain. All other chains were considered target chains. After that, a contiguous subsequence of the binder chain was extracted. The subsequence length was randomly chosen between 1 and 250 residues, and the binder seed residue was required to lie within this subsequence. No additional flanking residues were enforced on either side of the seed residue. 28 Published as a conference paper at ICLR 2026 Algorithm 1 Protein Binder Best-of-N Sampling 1: Input: Model p ϕ , success criterion function S ( · ) , number of samples N 2: Output: Set of successful designs S 3: Initialize success set S ← ∅ 4: for i = 1 to N do 5: Generate candidate ( x C α , z ) i ∼ p ϕ ( x , z ) 6: Decode ( x C α , z ) i into structure-sequence tuple ( x i , s i ) 7: F old and score to compute metrics for ( x i , s i ) 8: if S ( x i , s i ) = T rue then 9: Add ( x i , s i ) to S 10: end if 11: end for 12: Return: S Then, cropping of the target chains w as performed in two sequential stages: • Spatial cropping: Candidate target residues were identified as those lying within 15 ˚ A of any residue in the cropped binder subsequence. • Contiguous cropping: From these candidates, contiguous stretches of residues were selected to fit into the remaining budget. At least 50 target residues were retained, and when more residues were av ailable than required, those closest to the binder subsequence were prioritized. Globally , the combined binder and target were restricted to a maximum of 500 residues in total. The number of target chains w as not limited, and the cropped structure retained atomic coordinates. This procedure ensured that cropped protein samples emphasized interfacial regions while maintaining a controlled and computationally tractable input size for model training. In the case of small molecules, we perform a spatial cropping similar to AF3 ( Abramson et al. , 2024 ) and use a modified implementation of the atomworks transform ( Corley et al. , 2025 ). First, we perform the standard spatial crop until a system size of 512 (binder residues + ligand hea vy atoms). W e then identify all connected components in the binder and discard any fragment with length < 20 that has no atom in that fragment within 15 ˚ A of the ligand. H I N F E R E N C E - T I M E O P T I M I Z A T I O N M E T H O D D E TA I L S In the follo wing, we explain our inference-time optimization approaches in detail, pro vide algorithms, and describe how the methods are adapted for protein binder generation in Complexa and differ from their typical implementations in the literature. H . 1 B E S T - O F - N S A M P L I N G Best-of-N sampling is the simplest form of inference-time scaling. W e generate N independent samples from the generative model, ev aluate each using a folding model, and return the subset of designs that meet pre-defined success criteria. In practice, we gradually increase the total number of generated samples N up to a maximum of 51,200. Generation is performed in batch mode, while e valuation (folding and scoring) is run in single-sample inference mode follo wing the implementation of ColabDesign and RF3. Sampling is stopped early if the wall-clock time limit is reached. Pseudo- code of the algorithm is shown in Algorithm 1 . H . 2 B E A M S E A R C H Beam search is a structured inference-time search strategy that balances exploration and exploitation by maintaining a fixed-size set of the top N partial trajectories, referred to as the beam . Starting from N independent noise samples at t x = 0 , t z = 0 , the algorithm proceeds iteratively by advancing each trajectory by K denoising steps. At each search step, ev ery trajectory in the beam is branched L times, yielding N × L intermediate candidates. These candidates form the set C t x +∆ t K x ,t z +∆ t K z = 29 Published as a conference paper at ICLR 2026 Algorithm 2 Protein Binder Beam Search with Rew ard Approximation 1: Input: Generativ e model p ϕ , success criterion function S ( · ) , rew ard function R ( · ) , beam width N , branch factor L , denoising step length K 2: Output: Set of successful designs S 3: Initialize success set S ← ∅ 4: Initialize beam B ← { ( x C α t x =0 , z t z =0 ) i } N i =1 // N initial noise samples 5: while t x < 1 or t z < 1 do 6: Initialize candidate set C ← ∅ 7: f or each state ( x C α t x , z t z ) in beam B do 8: f or j = 1 to L do 9: Run denoising for K steps with p ϕ to obtain intermediate state ( x C α t x +∆ t K x , z t z +∆ t K z ) 10: Roll out from ( x C α t x +∆ t K x , z t z +∆ t K z ) to ( t x = 1 , t z = 1) to get clean sample ( x , s ) 11: Compute rew ard r = R ( x , s ) 12: Add ( x C α t x +∆ t K x , z t z +∆ t K z , r ) to candidate set C 13: if S ( x , s ) = T rue then 14: Add ( x , s ) to success set S 15: end if 16: end f or 17: end f or 18: Update beam: B ← T op N ( C ) by rew ard 19: Update time inde x t x ← t x + ∆ t K x , t z ← t z + ∆ t K z 20: end while 21: // Evaluate final beam 22: for each state ( x C α , z ) in beam B do 23: Decode ( x C α , z ) into structure–sequence pair ( x , s ) 24: Compute re ward r = R ( x , s ) 25: if S ( x , s ) = T rue then 26: Add ( x , s ) to success set S 27: end if 28: end for 29: Return: S ( x C α t x +∆ t K x , z t z +∆ t K z ) i N L i =1 , where ∆ t K x and ∆ t K z correspond to a block of K discretized denoising steps for both backbones and latent states. W e then approximate the rewards of these candidates, for example using f ipAE or f H-Bond , and select the top N candidates to form the updated beam. This procedure is repeated until fully denoised samples are obtained. In contrast to prior work ( Fernandes et al. , 2025 ; Ramesh & Mardani , 2025 ), we do not use T weedie’ s formula to estimate rewards from one-shot denoising predictions, as intermediate states are too noisy for direct ev aluation. Instead, we roll out all candidates to fully denoised samples using low-temperature sampling ( Gef fner et al. , 2025 ; 2026 ). Different from standard beam search, instead of discarding these clean samples simulated during rew ard estimation, we add them into the success set if they meet the criteria. This significantly improves the total number of successes. In practice, we set N = 4 , L = 4 , and use 400 discretized denoising steps follo wing the LaProteina schedule, with K = 100 steps per beam search update. T o further scale inference-time compute, the entire beam search procedure is repeated multiple times until the wall-clock time limit is reached. Pseudo-code of the algorithm is shown in Algorithm 2 . H . 3 F E Y N M A N – K AC S T E E R I N G ( F K S T E E R I N G ) Feynman–Kac Steering (FK Steering) ( Singhal et al. , 2025 ) is a flexible framework for guiding diffusion-based generativ e models with arbitrary rew ard functions. The ke y idea is to sample from a tilted distribution that up-weights high-rew ard samples and down-weights low-re ward samples: ˜ p ( x , s ) ∝ p ϕ ( x , s ) exp { β R ( x , s ) } , where R ( x , s ) is the reward and β is an inv erse temperature controlling the bias toward high-re ward trajectories. Instead of relying on fine-tuning or gradient- 30 Published as a conference paper at ICLR 2026 Algorithm 3 Protein Binder Feynman–Kac Steering with Re ward Approximation 1: Input: Generati ve model p ϕ , success criterion S ( · ) , re ward function R ( · ) 2: Input: Beam width N , branch factor L , denoising step length K , inv erse temperature β 3: Output: Set of successful designs S 4: Initialize success set S ← ∅ 5: Initialize beam B ← { ( x C α t x =0 , z t z =0 ) i } N i =1 // N initial noise samples 6: while t x < 1 or t z < 1 do 7: Initialize candidate set C ← ∅ 8: f or each state ( x C α t x , z t z ) in beam B do 9: f or j = 1 to L do 10: Run denoising for K steps with p ϕ to obtain intermediate state ( x C α t x +∆ t K x , z t z +∆ t K z ) 11: Roll out from ( x C α t x +∆ t K x , z t z +∆ t K z ) to ( t x = 1 , t z = 1) to get clean sample ( x , s ) 12: Compute rew ard r = R ( x , s ) 13: Add ( x C α t x +∆ t K x , z t z +∆ t K z , r ) to candidate set C 14: if S ( x , s ) = T rue then 15: Add ( x , s ) to success set S 16: end if 17: end f or 18: end f or 19: Compute sampling probabilities for candidates c ∈ C : p ( c ) = exp { β R ( c ) } P c ′ ∈C exp { β R ( c ′ ) } 20: Sample N candidates from C according to p ( c ) ; update beam B ← sampled candidates 21: Update time inde x t x ← t x + ∆ t K x , t z ← t z + ∆ t K z 22: end while 23: // Evaluate final beam 24: for each state ( x C α , z ) in beam B do 25: Decode ( x C α , z ) into structure–sequence pair ( x , s ) 26: Compute re ward r = R ( x , s ) 27: if S ( x , s ) = T rue then 28: Add ( x , s ) to success set S 29: end if 30: end for 31: Return: S based guidance, FK Steering uses a particle-based inference-time procedure inspired by Feynman– Kac interacting particle systems (FK-IPS) ( Moral , 2004 ; V estal et al. , 2008 ). Multiple stochastic dif fusion trajectories ( particles ) are simulated in parallel, scored using intermediate re ward potentials, and resampled according to their potentials at intermediate steps. This resampling eliminates low- re ward particles and preferentially propagates high-re ward trajectories, making it ef fectiv e e ven when high-rew ard samples are rare under the original generative model. In practice, FK Steering can be implemented similarly to beam search, but instead of selecting the top- N candidates deterministically , N particles are resampled according to probabilities computed from their rewards. Here we use the same hyperparameters as our beam search ( N = 4 , L = 4 , K = 100 ), and set the in verse temperature to β = 10 to strongly fav or high-reward trajectories. Pseudo-code of the algorithm is shown in Algorithm 3 . H . 4 M O N T E C A R L O T R E E S E A R C H ( M C T S ) Monte Carlo T ree Search (MCTS) ( Coulom , 2006 ) is an algorithm that incrementally builds a search tree by simulating possible future outcomes. Each iteration consists of four steps: selection , where the tree is trav ersed with a strategy to balance exploration and exploitation ( Kocsis & Szepesv ´ ari , 2006 ); expansion , where a new node for an unexplored action is added; simulation , where a rollout estimates the expected re ward from that state; and backpr opagation , where the results are propagated 31 Published as a conference paper at ICLR 2026 Algorithm 4 Protein Binder MCTS with Stochastic Expansion 1: Input: Generativ e model p ϕ , success criterion S ( · ) , re ward R ( · ) , number of simulations N sim , roll-out length K , exploration probability ϵ 2: Output: Set of successful designs S 3: Initialize success set S ← ∅ 4: Initialize root node v cur ← ( x C α t x =0 , z t z =0 ) 5: Initialize global time indices t x , t z ← 0 6: while t x < 1 or t z < 1 do 7: f or simulation s = 1 to N sim do 8: Set node v ← v cur 9: Initialize local simulation time t sim x , t sim z ← t x , t z 10: while t sim x < 1 or t sim z < 1 do 11: if W ith probability ϵ or if v has no children / near t = 1 then 12: Expand new child by running K denoising steps as v new with our model p ϕ : 13: ( x C α t sim x , z t sim z ) → ( x C α t sim x +∆ t K x , z t sim z +∆ t K z ) 14: Add v new to the children set of node v . 15: else 16: Select child of v with highest UCB score as v new 17: end if 18: v ← v new 19: Update local simulation time t sim x ← t sim x + ∆ t K x , t sim z ← t sim z + ∆ t K z 20: end while 21: Decode fully denoised state ( x C α t sim x , z t sim z ) → ( x , s ) 22: Compute re ward r = R ( x , s ) 23: if S ( x , s ) = T rue then 24: Add ( x , s ) to success set S 25: end if 26: Backpropagate re ward r to update statistics of ancestor nodes 27: end f or 28: Mo ve current node v cur to child with highest expected re ward 29: Update global time indices t x ← t x + ∆ t K x , t z ← t z + ∆ t K z 30: end while 31: Return: S back up the tree to update visit counts and reward estimates. Repeating this process focuses the search on promising actions, making MCTS effecti ve for comple x tasks ( Granter et al. , 2017 ). Monte Carlo T ree Diffusion ( Y oon et al. , 2025 ; Ramesh & Mardani , 2025 ) treats the denoising process as a tree, where each node corresponds to a partial latent state ( x C α t x , z t z ) . Starting from the root node at t x = t z = 0 , the algorithm iterativ ely selects, expands, and evaluates trajectories while balancing exploration and exploitation (via a e xploration constant C ) with a modified Upper Confidence Bound (UCB) score: UCB score (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) = R (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) V (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) | {z } exploitation + C v u u t ln ( V (( x C α t x , z t z ) i )) V (( x C α t x +∆ t K x , z t z +∆ t K z ) i ) | {z } exploration , where ( x C α t x , z t z ) i denotes the parent state of ( x C α t x +∆ t K x , z t z +∆ t K z ) i and V ( · ) counts how often a (noisy) partially latent state has already been visited during pre vious searches. Empirically , each node maintains a set of children representing possible next states. When expanding a leaf node, sev eral denoising steps are performed to generate a new child, which is then added to the node’ s children set. For ev ery node, we record both the visit count and the av erage reward obtained after visiting that node. Once a trajectory is fully simulated to completion, its final rew ard is backpropagated to all ancestor nodes, updating their visit counts and av erage rewards to guide future search decisions. In classical MCTS with a finite action space, each node has a fixed number of children (branching factor L ), and the search iterati vely explores these discrete options. Howe ver , in flo w matching, the 32 Published as a conference paper at ICLR 2026 state space is ef fectively continuous and unbounded, making traditional MCTS with a fix ed branching factor inef ficient. T o address this, our implementation mer ges selection and expansion into a single stochastic decision at each node. At each step, with probability ϵ , a new child is e xpanded by running K denoising steps from the current node; with probability 1 − ϵ , the child with the highest expected rew ard is selected. If the current node has no children or the next step reaches t x = t z = 1 , a new child is always expanded. Once the current node reaches ( t x , t z ) = 1 , it will be decoded into a structure–sequence pair ( x , s ) , and the re ward is computed. This rew ard is then backpropagated to all ancestor nodes to update visit counts and expected rewards. Fully denoised states meeting the success criteria are added to a success set S , similar to beam search. Here, the probability ϵ plays a role analogous to L : instead of deterministically expanding ev ery possible child, we stochastically control the growth of the tree. Specifically , if a node is visited V times, it will hav e, in expectation, V · ϵ children ov er the course of the search. Thus, ϵ acts as a soft, probabilistic branching factor , balancing between aggressive e xploration (large ϵ ) and focused refine- ment of promising states (small ϵ ). The constant C , by contrast, governs the e xploration-exploitation trade-off among the children that ha ve already been expanded. A larger C encourages exploration of less-visited children, while a smaller C prioritizes selecting the currently best-performing states based on their rew ards. T ogether , ϵ and C jointly determine the behavior of the search. Another adv antage of the modified stochastic MCTS is its compatibility with batched infer ence . In practice, our neural network implementation only supports denoising a batch of samples at the same time step. In standard MCTS, dif ferent trajectories in the search tree may be at dif ferent time steps, making it difficult to perform batched denoising efficiently . By using the same ϵ across all states in a batch, our algorithm ensures that the denoising steps for multiple nodes can be executed in parallel, e ven when e xpanding different children stochastically . This design not only preserves the exploration–e xploitation behavior of MCTS b ut also maintains the computational efficiency required for diffusion models that rely on synchronized batched e valuation. In practice, we set K = 100 , ϵ = 0 . 5 , C = 1 . 0 , and run N sim = 20 simulations per decision step. After each round of simulations, the current node is moved to the child with the highest expected rew ard, and the global time indices t x , t z are updated to match the selected child. This procedure repeats until the state reaches ( t x , t z ) = 1 , producing a set of successful fully denoised states. Pseudo-code of the algorithm is presented in Algorithm 4 . H . 5 G E N E R A T E & H A L L U C I NAT E ( G & H ) In addition to search-based inference methods, we explore a simpler approach that combines genera- tiv e initialization with sequence refinement, which we call Generate & Hallucinate (G&H) . W e first generate candidate binder sequences using our generativ e model, and then refine these sequences using the BindCraft hallucination framew ork ( Pacesa et al. , 2025 ). BindCraft optimizes binder sequences using the ColabDesign implementation of AlphaFold2 (AF2), minimizing a multi-term loss that accounts for binder confidence, interface confidence, predicted alignment errors (pAE) within the binder and between binder and target, residue contact constraints, radius of gyration, backbone helicity , and optional termini constraints. Gradients are backpropagated through AF2 to iterati vely update the sequence, with loss weights set according to the original BindCraft. The number of recycles in AF2 prediction is set to 3 to ensure accurate approximation. After generating sequences with our model, BindCraft is initialized in “wild type” mode using the generated sequence and refines it through staged optimization. W e focus on the last three stages, which are used in our implementation: • Stage 2 (Softmax normalization): Sequence logits are con verted to probabilities via softmax with temperature annealing, softmax ( logits /T ) , where the temperature T decreases ov er iterations as T = 1 × 10 − 2 + (1 − 1 × 10 − 2 ) · (1 − ( step + 1) / iterations ) . Learning rates are scaled according to this schedule. This stage runs for 45 iterations. • Stage 3 (Straight-through estimator): One-hot discrete representations are used for forward passes while gradients are backpropagated through softmax. This allows refinement of discrete sequences while maintaining differentiability . This stage runs for 5 iterations. • Stage 4 (Discrete sequence optimization): Sequences are fully discretized. Random muta- tions are sampled from the softmax distribution of the pre vious stage, and only mutations that 33 Published as a conference paper at ICLR 2026 T able 6: T ranslation noise and T eddymer ablation studies. average unique successes and the number of times each method ranks best across 19 targets. Results are shown for Complexa, and v ariants without T eddymer data and without translation noise. Model # Unique Successes ↑ # T imes Best Method ↑ Self MPNN-FI MPNN Self MPNN-FI MPNN Complexa 9.10 13.5 14.4 19 13 14 Complexa w/o T eddymer 0.15 1.68 3.84 0 0 0 Complexa w/o T ranslation Noise 1.47 3.89 3.73 0 6 5 T able 7: T ranslation noise and T eddymer ablation studies (detailed results). Unique successes across 19 targets for Comple xa and its variants without T eddymer data or translation noise, ev aluated with different sequence redesign methods. Results are visualized in Fig. 13 . T arget Complexa Complexa w/o T eddymer Complexa w/o T ranslation Noise MPNN MPNN-FI Self MPNN MPNN-FI Self MPNN MPNN-FI Self PD-L1 16 14 9 8 4 0 1 4 1 BHRF1 26 29 21 18 7 2 28 28 14 IFN AR2 51 52 39 5 2 0 4 2 0 BBF14 24 25 20 12 10 0 4 4 0 DerF7 24 16 10 7 0 0 1 2 2 DerF21 35 31 22 5 4 1 3 4 1 PD1 27 21 15 2 0 0 6 4 4 Insulin 16 20 8 1 0 0 3 1 0 IL7RA 6 12 6 2 2 0 1 0 0 Claudin1 4 4 2 0 0 0 2 0 0 CrSAS6 12 8 3 4 1 0 0 1 0 T rkA 25 20 16 4 1 0 3 4 0 SC2RBD 0 0 0 0 0 0 1 1 0 CbAgo 0 0 0 0 0 0 1 2 0 CD45 0 0 0 2 0 0 3 3 0 BetV1 0 0 0 1 0 0 2 1 2 HER2-AA V 0 0 0 0 0 0 0 0 0 SpCas9 3 4 1 0 0 0 2 7 1 VEGF A 5 2 1 0 1 0 2 3 1 improv e the design loss are retained. The number of mutations per iteration is proportional to the binder length ( 0 . 01 × binder length). This stage is performed for 15 iterations. In our implementation, generative samples are refined using either (i) stages 2+3+4 or (ii) stage 4 alone. In the main paper, we use (ii) as our optimization method and we show a comparison between the two choices in Sec. I.3 . This approach provides a simple and computationally ef ficient combination of generati ve initialization with principled hallucination-based sequence refinement, and can be flexibly inte grated with any of the search algorithms described above. I A B L A T I O N S T U D I E S A N D A D D I T I O N A L E X P E R I M E N T S In this section, we present our ablation studies and additional experimental results. W e perform two experiments to assess the importance of two core choices in our Complexa model training: the impact of the T eddymer training data in Sec. I.1 , and translation noise in Sec. I.2 . W e also explore different choices of using BindCraft optimization stages in Generate and Hallucinate in Sec. I.3 . Finally , we provide detailed results for designing binders for very challenging targets in Sec. I.4 , generative model benchmark results in Sec. I.5 , inference-time scaling results across all tar gets in Sec. I.6 and Sec. I.7 , and interface hydrogen-bond optimization results for all tar gets in Sec. I.8 . I . 1 A B L A T I O N E X P E R I M E N T : T R A I N I N G W I T H O U T T E D D Y M E R T o assess the impact of T eddymer on model performance, we conduct an ablation study in which the model is trained only on the PDB data described in Sec. D.1 . All other model configurations and training hyperparameters are kept identical to the base Complexa; only the dataset is changed. The model is trained for 235k steps with 64 GPUs and batch size 6 per GPU and ev aluated using the same protocol as the base model. Overall results are reported in T ab. 6 , per-tar get results are shown in 34 Published as a conference paper at ICLR 2026 0 10 20 30 40 50 Unique successes MPNN 0 10 20 30 40 50 Unique successes MPNN-FI IFNAR2 BHRF1 BBF14 DerF21 T rkA PD1 Insulin DerF7 PDL1 IL7R A CrS AS6 Claudin1 VEGF A SpCas9 SC2RBD CbAgo CD45 BetV1 HER2_A A V 0 10 20 30 40 Unique successes Self Comple xa Comple xa w/o T eddymer Comple xa w/o T ranslation Noise Figure 13: T ranslation noise and T eddymer ablation studies. Unique successes of different Complexa variants (base, without T eddymer data, and without translation noise) on easy tar gets (left of the dashed line) and hard targets (right of the dashed line). T ab . 7 and visualized in Fig. 13 . As clearly shown, the number of unique successes drops significantly across most targets, demonstrating the critical importance of including T eddymer in training. I . 1 . 1 T E D DY M E R A B L A T I O N W I T H S E P A R A T E F O L D I N G M O D E L S F O R S C O R I N G T eddymer is deri ved from structures from the AFDB ( V aradi et al. , 2021 ), which were predicted using AlphaFold2 ( Jumper et al. , 2021 ), and our main in-silico success criteria use ipAE scores calculated with the ColabDesign implementation ( Ovchinniko v et al. , 2025 ) of AlphaFold2-Multimer ( Ev ans et al. , 2022 ) (follo wing Zambaldi et al. ( 2024 )). T o alle viate concerns that we are overly relying on AlphaFold2-type models for both data creation (T eddymer) and model e valuation, we additionally assess the benefit of including T eddymer in the training data by ev aluating two variants of our model (training with and without T eddymer) with distinct third-generation structure prediction models RosettaFold-3 ( Corle y et al. , 2025 ) (RF3) and Boltz-2 ( Passaro et al. , 2025 ), instead of AlphaFold2- Multimer . In other words, we are repeating the T eddymer ablation study using ipAE scores from RF3 and Boltz-2 for e valuation, applied to the same generated binders as before. See detailed per target unique success results in Fig. 14 for Boltz-2 and Fig. 15 for RF3. The aggregated results across all targets are in T ab . 8 for Boltz-2 and T ab . 9 for RF3. Multiple sequence alignments (MSAs) ( Steinegger & S ¨ oding , 2017 ) are used for the tar gets when folding with Boltz-2. T emplate conditioning is used for targets when folding with RF3. W e use the same threshold values for all metrics described in Sec. F to filter for success. W e find through this additional ablation study that training on T eddymer shows clear benefits, e ven when e valuating with either RF3 or Boltz-2, thereby alleviating concerns about relying too much on AlphaFold2-type models specifically . Note that overall absolute success rates v ary between the e valuations with the different folding models. This is in part because we used the same ipAE success thresholds as before, without recalibration specifically for RF3 and Boltz-2. Also the use of MSAs and target structure templates af fects ipAE scores. W e did not bother to adjust and re-calibrate ipAE thresholds here, because we are only 35 Published as a conference paper at ICLR 2026 0 20 40 60 80 # Unique successes MPNN 0 20 40 60 # Unique successes MPNN-FI IFNAR2 BHRF1 BBF14 DerF21 T rkA PD1 Insulin DerF7 PDL1 IL7R A CrS AS6 Claudin1 VEGF A SpCas9 SC2RBD CbAgo CD45 BetV1 HER2_A A V 0 10 20 30 40 # Unique successes Self Comple xa (Boltz2) Comple xa w/o T eddymer (Boltz2) Figure 14: T eddymer ablation study with Boltz-2. Boltz-2-based ev aluation of the unique successes of different Complexa variants (trained with vs. without T eddymer data) on easy tar gets (left of the dashed line) and hard targets (right of the dashed line). T able 8: T eddymer ablation study with Boltz-2. Boltz-2-based ev aluation of the av erage unique successes across 19 targets. Results are shown for Complexa and the v ariant trained without T eddymer data. Model # Unique Successes ↑ MPNN MPNN-FI Self Complexa 34.4 31.4 14.8 Complexa w/o T eddymer 16.2 13.1 2.2 interested in the relative performance of models trained with and without T eddymer for the same RF3-based or Boltz-2-based ev aluations. I . 2 A B L A T I O N E X P E R I M E N T : T R A I N I N G W I T H O U T T R A N S L AT I O N N O I S E As introduced in Sec. 3.2 , the base Comple xa objective incorporates translation noise to encourage reasoning ov er the global positioning of the protein. T o ev aluate its effect, we train the model using the same dataset and hyperparameters as the base model, b ut without the translation noise. Evaluation results are reported in T ab . 6 , with per-target results in T ab . 7 and visualizations in Fig. 13 . The results sho w that, in general, removing translation noise substantially reduces the number of unique successes across most targets. Some hard targets show minor improvements, likely because the additional noise in the objecti ve can make learning already dif ficult structures ev en harder . Overall, these findings confirm that the translation noise component is beneficial for model generalization. I . 3 A B L A T I O N E X P E R I M E N T : B I N D C R A F T S TAG E S I N G E N E R A T E & H A L L U C I NATE For our Generate & Hallucinate algorithm proposed in Sec. 3.2 (detailed in Sec. H.5 ), we choose to use our generated sequence as a prior instead of from some random initialization and run discrete sequence optimization through mutation (stage 4 only) with BindCraft. Here we explore the options 36 Published as a conference paper at ICLR 2026 0 10 20 30 # Unique successes MPNN 0 5 10 15 # Unique successes MPNN-FI IFNAR2 BHRF1 BBF14 DerF21 T rkA PD1 Insulin DerF7 PDL1 IL7R A CrS AS6 Claudin1 VEGF A SpCas9 SC2RBD CbAgo CD45 BetV1 HER2_A A V 0 5 10 15 # Unique successes Self Comple xa (RF3) Comple xa w/o T eddymer (RF3) Figure 15: T eddymer ablation study with RF3. RF3-based ev aluation of the unique successes of dif ferent Complexa v ariants (trained with vs. without T eddymer data) on easy tar gets (left of the dashed line) and hard targets (right of the dashed line). T able 9: T eddymer ablation study with RF3. RF3-based ev aluation of the av erage unique successes across 19 targets. Results are shown for Complexa and the v ariant trained without T eddymer data. Model # Unique Successes ↑ MPNN MPNN-FI Self Complexa 4.9 4.3 2.4 Complexa w/o T eddymer 1.2 0.5 0.1 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 20 40 60 80 # Unique Successes Easy T argets 5 10 15 20 25 30 GP U Hours 2 4 6 8 10 12 # Unique Successes Hard T argets Generate and Hallucinate Generate and Hallucinate w/ logit opt. BindCraft Figure 16: Generate & Hallucinate ablation study . Numbers of unique successes against inference-time GPU hours, for Generate & Hallucinate, its variant with logit optimization, and BindCraft, av eraged ov er 12 easy and 7 hard targets. whether to do the sequence logits optimization (stages 2+3+4). W e run the experiments with the same setting as Generate & Hallucinate, but also add the logit optimization stages. W e compare the two methods under the same inference-time compute b udget following our e xperiments in Sec. 4.2 . The av eraged results are shown in Fig. 16 and per -target results are sho wn in Fig. 17 and Fig. 18 . On easy targets, we see that our Generate & Hallucinate outperforms its v ariant with logit optimization 37 Published as a conference paper at ICLR 2026 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 5 10 15 20 25 30 35 40 # Unique Successes PDL1 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 50 100 150 200 250 300 350 # Unique Successes BHRF1 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 20 40 60 80 100 # Unique Successes IFNAR2 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 25 50 75 100 125 150 # Unique Successes DerF21 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 10 20 30 40 50 # Unique Successes T rkA 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 10 20 30 40 # Unique Successes DerF7 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 10 20 30 40 50 60 70 # Unique Successes BBF14 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 10 20 30 40 50 # Unique Successes PD1 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 10 20 30 40 50 60 # Unique Successes Insulin 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 5 10 15 20 # Unique Successes IL7RA 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 10 20 30 40 # Unique Successes CrSAS6 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 5 10 15 20 # Unique Successes Claudin1 Generate and Hallucinate Generate and Hallucinate w/ logit opt. BindCraft Figure 17: Generate & Hallucinate ablation study (easy targets). Number of unique successes over 12 easy targets against inference-time GPU hours, for Generate & Hallucinate, its variant with logit optimization, and BindCraft. Successes are measured with self-generated sequences and inference-time compute includes both generation and ev aluation time. 5 10 15 20 25 30 GP U Hours 0 5 10 15 20 # Unique Successes SC2RBD 5 10 15 20 25 30 GP U Hours 0.0 2.5 5.0 7.5 10.0 12.5 15.0 # Unique Successes CbAgo 5 10 15 20 25 30 GP U Hours 2 4 6 8 10 12 # Unique Successes BetV1 5 10 15 20 25 30 GP U Hours 0 5 10 15 20 # Unique Successes HER2_AA V 5 10 15 20 25 30 GP U Hours 0 2 4 6 8 10 # Unique Successes CD45 5 10 15 20 25 30 GP U Hours 0 5 10 15 20 25 30 # Unique Successes SpCas9 5 10 15 20 25 30 GP U Hours 0 2 4 6 8 10 12 # Unique Successes VEGF A Generate and Hallucinate Generate and Hallucinate w/ logit opt. BindCraft Figure 18: Generate & Hallucinate ablation study (hard targets). Number of unique successes over 7 hard targets against inference-time GPU hours, for Generate & Hallucinate, its variant with logit optimization, and BindCraft. Successes are measured with self-generated sequences and inference-time compute includes both generation and ev aluation time. and BindCraft on most targets and by a lar ge margin on the av erage performance, which prov es that our generativ e model provides a strong prior for sequence optimization without the need to run logit optimization. On hard targets, these three methods compare on par in terms of average performance and shine on different tar gets. W e argue that this is highly affected by whether our generati ve model can provide a good prior on these challenging targets. Overall, we find adding logit optimization stages does not show clear adv antages, so we decide not to include them. 38 Published as a conference paper at ICLR 2026 Having presented our numerical ablation results on the hallucination stages of Generate & Hallucinate, we would no w like to discuss them in more detail. Why is logit optimization not uni versally helpful, e ven on easy tar gets when starting from a good initial binder candidate? The logit optimization stages of hallucination are intrinsically slo w as they rely on gradients that need to be backpropagated through the structure prediction model that provides the re ward. Moreover , the logit optimization corresponds to an approximate discrete sequence relaxation, which is necessary for the gradient-based updates. Therefore, the logit optimization is an approximate and coarse process, but it can help steering a random input towards reasonable sequence logits relatively quickly , and these logits can then be further refined with the later stages. But in our case, especially for easy targets, we already start the optimization from a good sequence generated by the base model. The approximate early logit optimization stages of hallucination therefore do not offer additional benefits, and the corresponding compute is better spent on carefully refining the sequence via stage 4 mutations instead. For hard targets, the situation can change: The initial binder candidate by Complexa’ s generativ e model is often not strong and significant refinement is necessary , for which the early logit optimization stages that more radically update the sequence can be helpful as well, before switching to stage 4 mutation- based refinement. Note, howe ver , that these mutations are random and unguided. This stands in contrast to the efficient inference-time search algorithms de veloped for Complexa. The optimization in algorithms like Beam Search is effecti vely guided by Complexa’ s pre-trained generative prior , and the generation trajectories are steered early on. Hallucination can only ev er modify a potentially flawed initial sample. This is exactly a key point of Complexa’ s framew ork: Directly integrating generativ e pre-training with inference-time optimization for binder design enables enhanced and more scalable binder design compared to previous hallucination methods. I . 4 I N F E R E N C E - T I M E S C A L I N G F O R V E RY H A R D T A R G E T S In Sec. 4.2 , we explore inference-time search for three highly challenging multi-chain targets: TNF- α , H1, and IL17A. For these targets, we observe that AlphaFold2-Multimer often produces low-confidence predictions, resulting in many binders with ipAE < 7 but plddt < 90 . T o address this issue, we incorporate plddt into our rew ard function. Specifically , we normalize the ipAE score to the range [0 , 1] by dividing it by 31, then add it to plddt to form the final rew ard. For TNF- α and H1, we run MCTS with parameters K = 100 , ϵ = 0 . 5 , C = 1 , and N sim = 20 . After the MCTS search, we apply Generate and Hallucinate for local sequence optimization, increasing the number of mutations per iteration to 0 . 05 × the binder length to fully explore the local sequence neighborhood. For IL17A, we use K = 50 , ϵ = 0 . 5 , C = 1 , and N sim = 20 , allowing for a deeper search tree that more ef fectiv ely lev erages the av ailable inference-time compute. The subsequent Generate and Hallucinate steps use the same 0 . 05 × binder length mutation rate. Using this approach, we identify 15 unique successes for TNF- α within 475 GPU hours, 7 unique successes for H1 within 604 GPU hours, and 1 unique success for IL17A within 387 GPU hours. The inference-time search scaling curv es for the three very hard tar gets are shown in Fig. 19 . Note that for IL17A, we found one in-silico success early on during the process, but not another one. W e show visualizations of these binders in Fig. 30 . Note that as all samples go through BindCraft refinement for sequence optimization, which changes the underlying sequences, we visualize the refolded structures predicted by AlphaFold2-Multimer instead of our generated structure. These results highlight the strength and flexibility of our framework for tackling extremely challenging binder design tasks through inference-time search and optimization. I . 5 E X T E N D E D R E S U LT S : G E N E R A T I V E M O D E L B E N C H M A R K W I T H S E Q U E N C E R E - D E S I G N In the generati ve model benchmark we generate 200 samples per model for each of the 19 tar gets and ev aluate them in three different w ays: for backbone design models RFDiffusion and Protapardelle-1c we just run ProteinMPNN eight times for each backbone and choose the best refolded sequence; for APM and Complexa we also test MPNN-redesign but k eeping the interface fixed or just e valuation of the full generated sequence. For MPNN-redesign (Fig. 20 top plot), one can see that Complexa clearly outperforms the baselines on most tasks. For MPNN-redesign with fixed interface, performance for Complexa is similar , while APM performs significantly worse, indicating that our generated interfaces are of higher quality . Finally , for model generated sequences the relev ant APM baseline collapses for most targets to zero 39 Published as a conference paper at ICLR 2026 0 100 200 300 400 500 600 GP U Hours 0 2 4 6 8 # Unique Successes H1 0 100 200 300 400 GP U Hours 0 3 6 9 12 15 # Unique Successes T N F - 0 100 200 300 400 GP U Hours 0 1 2 3 # Unique Successes IL17A Figure 19: H1, TNF- α , and IL17A targets. Inference-time optimization compute scaling plots for three very hard targets. See Sec. I.4 for details. T able 10: De novo binder design performance f or all protein targets . Unique successes for different models and three sequence redesign methods per target. T arget RFDiffusion Protpardelle-1c APM Complexa (Ours) MPNN MPNN MPNN MPNN-FI Self MPNN MPNN-FI Self PD-L1 8 2 1 0 0 16 14 9 BHRF1 5 4 20 10 5 26 29 21 IFN AR2 11 2 3 3 1 51 52 39 BBF14 2 0 0 0 0 24 25 20 DerF7 5 0 0 0 0 24 16 10 DerF21 4 4 11 8 0 35 31 22 PD1 7 0 1 0 0 27 21 15 Insulin 8 1 5 3 0 16 20 15 IL7RA 4 0 1 1 0 6 12 6 Claudin1 2 0 11 1 0 4 4 2 CrSAS6 8 0 1 1 0 12 8 3 T rkA 7 0 0 0 0 25 20 16 SC2RBD 2 0 0 0 0 0 0 0 CbAgo 0 0 3 1 0 0 0 0 CD45 1 0 0 1 0 0 0 0 BetV1 3 0 3 0 0 0 0 0 HER2-AA V 1 0 0 0 1 0 0 0 SpCas9 3 1 0 0 0 3 4 1 VEGF A 0 0 0 0 0 5 2 1 T able 11: De novo binder design perf ormance of models for all small molecule targets . RFdiffusionAA only produces backbones and as a result can only be evaluat ed under full LigandMPNN re-design. W e report the number of FoldSeek clusters out of the successful subset of 200 samples of length 100. T arget Model # Unique Successes ↑ Self MPNN-FI MPNN SAM RFdiffusionAA - - 2 Complexa 10 8 15 OQO RFdiffusionAA - - 3 Complexa 6 12 9 F AD RFdiffusionAA - - 5 Complexa 17 14 17 IAI RFdiffusionAA - - 8 Complexa 19 14 22 successes, while Complexa still produces many div erse successes for a v ariety of targets, highlighting that it is actually a practically useful co-design model. T ab . 11 provides the per-tar get unique successes for Complexa compared to RFDif fusion-AllAtom. Here we report the unique successes for a single LigandMPNN re-design (both full and fixed interface). For some tasks, using Lig andMPNN to re-design the interface or the entire sequence is beneficial, howe ver the sequences generated by Complexa always perform well and outperform the baseline. RFDiffusion-AllAtom can only be ev aluated with full sequence re-design since the model only generates backbones. Overall we find that the sequences generated from Complexa score well, with performance scaling as a function of inference-time compute (see Fig. 23 ). 40 Published as a conference paper at ICLR 2026 0 10 20 30 40 50 Unique successes MPNN 0 10 20 30 40 50 Unique successes MPNN-FI IFNAR2 BHRF1 BBF14 DerF21 T rkA PD1 Insulin DerF7 PDL1 IL7R A CrS AS6 Claudin1 VEGF A SpCas9 SC2RBD CbAgo CD45 BetV1 HER2_A A V 0 10 20 30 40 Unique successes Self Comple xa APM RFDiffusion P r otpar delle-1c Figure 20: Unique successes for differ ent models with thr ee sequence redesign methods on easy tar gets (left of the dashed line) and hard targets (right of the dashed line). Detailed results are in T ab . 10 I . 6 E X T E N D E D R E S U LT S : I N F E R E N C E - T I M E S E A R C H F O R A L L P R OT E I N T A R G E T S Accompanied by the averaged inference-scaling results sho wn in Sec. 4.2 , we present per-target results for 12 easy targets (Fig. 21 ) and 7 hard targets (Fig. 22 ). W e use the ipAE value from AlphaFold2-Multimer as the re ward and run different algorithms to a fix ed inference-time compute budget. W e use 16 GPU hours for easy targets and 32 GPU hours for hard targets at maximum. For the easy targets, our inference-time search algorithms consistently outperform the three hallu- cination baselines. This is expected: while hallucination-based methods such as BindCraft often achiev e high success rates, they require significantly more compute due to the large number of backpropagations through AlphaFold2. Among the 12 easy targets, the Best-of-N strategy achie ves the highest number of unique successes on 8 targets. This suggests that for easy cases where our base model already performs well, advanced algorithms like beam search may be unnecessary , as they risk wasting compute on generating redundant successes. In these scenarios, simply sampling repeatedly with Best-of-N is both efficient and ef fectiv e. For moderately challenging targets, such as BHRF1, IL7RA, and Claudin1, where the base model produces fewer successes under a fixed b udget, more sophisticated algorithms become beneficial. By fully exploiting promising regions of the search space, methods like Beam Search or Monte Carlo T ree Search can discov er additional high-quality solutions. This effect is e ven more pronounced on the 7 hard targets (Fig. 22 ), where the base model alone struggles to generate successes. Here, advanced search algorithms, including Beam Search, Fe ynman-Kac Steering, and MCTS, demonstrate clear advantages by systematically e xploring and refining candidate solutions. Howe ver , on two extremely difficult targets, SARS-CoV -2 RBD and HER2-AA V , our approach yields fe wer unique successes than BindCraft, and on BetV1, BoltzDesign is more ef ficient. These results highlight the complementary strengths of different methods. While our inference-time search framew ork is the most effecti ve across most targets, hallucination-based approaches remain v aluable. Because they do not require training and directly optimize for the final success criteria, they a void potential ov erfitting and can occasionally outperform trained models on highly challenging targets. 41 Published as a conference paper at ICLR 2026 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 20 40 60 80 100 # Unique Successes PDL1 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 50 100 150 200 250 300 350 # Unique Successes BHRF1 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 50 100 150 200 250 300 # Unique Successes IFNAR2 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 50 100 150 # Unique Successes DerF21 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 20 40 60 80 100 120 # Unique Successes T rkA 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 20 40 60 80 100 120 140 # Unique Successes DerF7 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 25 50 75 100 125 150 # Unique Successes BBF14 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 20 40 60 80 100 120 140 # Unique Successes PD1 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 25 50 75 100 125 150 175 # Unique Successes Insulin 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 10 20 30 # Unique Successes IL7RA 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 20 40 60 80 # Unique Successes CrSAS6 2.5 5.0 7.5 10.0 12.5 15.0 GP U Hours 0 10 20 30 40 # Unique Successes Claudin1 Best-of -N (ours) Beam Search (ours) F eynman Kac Steering (ours) Monte Carlo Tree Search (ours) Generate and Hallucinate (ours) BoltzDesign BindCraft AlphaDesign Figure 21: Unique successes against inference-time GPU hours on 12 easy targets. Successes are measured with self-generated sequences and inference-time compute includes both generation and ev aluation time. 5 10 15 20 25 30 GP U Hours 0 5 10 15 20 # Unique Successes SC2RBD 5 10 15 20 25 30 GP U Hours 0.0 2.5 5.0 7.5 10.0 12.5 15.0 # Unique Successes CbAgo 5 10 15 20 25 30 GP U Hours 0 2 4 6 8 10 12 14 # Unique Successes BetV1 5 10 15 20 25 30 GP U Hours 0 5 10 15 20 # Unique Successes HER2_AA V 5 10 15 20 25 30 GP U Hours 0 5 10 15 20 25 # Unique Successes CD45 5 10 15 20 25 30 GP U Hours 0 10 20 30 40 50 60 # Unique Successes SpCas9 5 10 15 20 25 30 GP U Hours 0 20 40 60 80 # Unique Successes VEGF A Best-of -N (ours) Beam Search (ours) F eynman Kac Steering (ours) Monte Carlo Tree Search (ours) Generate and Hallucinate (ours) BoltzDesign BindCraft AlphaDesign Figure 22: Unique successes against inference-time GPU hours on 7 hard targets. Successes are measured with self-generated sequences and inference-time compute includes both generation and ev aluation time. I . 7 E X T E N D E D R E S U LT S : I N F E R E N C E - T I M E S E A R C H F O R A L L L I G A N D T A R G E T S In addition to the target-a veraged inference-scaling results shown in Fig. 9 , we plot the per-tar get results for four ligand tar gets in Fig. 23 . While the SAM target pro ves challenging in T able 11 , we observe significant success for inference-scaling methods best-of-N, beam search, and FK-steering. For the other three targets, best-of-N yields the highest unique success rate, with beam search as a close second. Notably , Monte Carlo tree search performs the worst across all targets. Overall across all targets both Comple xa with best-of-N and beam-search significantly outperform BoltzDesign. W e hypothesize that best-of-N performs the strongest due to the re ward function used in all Complexa ligand-conditional inference-scaling e xperiments, which is the 0-1 normalized RF3 min ipAE, and the 42 Published as a conference paper at ICLR 2026 5 10 15 20 GP U Hours 0 20 40 60 80 100 120 # Unique Successes SAM 5 10 15 20 GP U Hours 0 20 40 60 80 100 120 # Unique Successes F AD 5 10 15 20 GP U Hours 0 20 40 60 80 100 120 # Unique Successes IAI 5 10 15 20 GP U Hours 0 10 20 30 40 50 60 70 # Unique Successes OQO Best-of -N (ours) Beam Search (ours) F eynman Kac Steering (ours) Monte Carlo Tree Search (ours) BoltzDesign Figure 23: Unique successes against inference-time GPU hours on 4 small molecule targets. Successes are measured with self-generated sequences and inference-time compute includes both generation and ev aluation time. 0 50 100 150 200 250 300 Unique Successes Comple xa (no r ewar d) Comple xa (ipAE) Comple xa (H- Bond) Comple xa (ipAE+H- Bond) IFNAR2 BHRF1 BBF14 DerF21 T rkA PD1 Insulin DerF7 PDL1 IL7R A CrS AS6 Claudin1 VEGF A SpCas9 SC2RBD CbAgo CD45 BetV1 HER2_A A V 0 2 4 6 8 10 12 Interface H-bonds Figure 24: Unique successes and number of interface hydrogen bonds on 19 targets using Beam Search with differ ent combinations of folding and h ydrogen bond rewards. Successes are measured with self-generated sequences and hydrogen bond counts are computed on refolded structures ov er successful designs. fact that, despite passing min ipAE and binder backbone scRMSD, the majority of samples fail due to having a ligand scRMSD ≥ 5 . As a result, our reward function, while a component of the success criteria, does not fully capture the primary challenge of protein-lig and co-design: maintaining binding site specificity . Generating protein binders with good pLDDT and P AE is relati vely straightforward, as seen in Cho et al. ( 2025 ). Ho wever , imposing a stronger interface criterion via min ipAE and enforcing ligand binding site conserv ation during refolding significantly filters out samples. W e leave extending re wards to incorporate both confidence and structural metrics as a direction for future work. I . 8 E X T E N D E D R E S U LT S : I N T E R FAC E H Y D R O G E N B O N D O P T I M I Z A T I O N F O R A L L T A R G E T S In addition to the standard folding reward-guided inference-time optimization, we further in vestigate interface hydrogen bond (H-Bond) optimization, as introduced in Sec. 4.2 . Alongside the av eraged results reported in T ab . 3 , we pro vide per-tar get visualizations of both the number of unique successes and the number of interface hydrogen bonds in Fig. 24 . For this e xperiment, we run beam search with a fixed initial b udget of 200 samples per target, using the standard setup of N = 4 , L = 4 , and K = 100 as described in Sec. H.2 . This ensures that each target produces the same total number of generated samples, prev enting any single target from dominating the hydrogen bond metric. Howev er, because of differences in sequence lengths, this setup naturally results in varying inference-time compute across tar gets. 43 Published as a conference paper at ICLR 2026 T o establish a fair comparison, we also include a zero-reward baseline, which is conceptually equiv alent to Best-of-N but follows the same sampling protocol to maintain consistency across methods. W e then ev aluate beam search with four reward configurations: (1) ipAE-only , (2) H- Bond-only , (3) a combined reward of ipAE and H-Bond with equal weights, and (4) the zero-rew ard baseline. Here, the H-Bond rew ard is defined as the number of interface hydrogen bonds. As shown in Fig. 24 , optimizing solely for the H-Bond reward yields the highest number of inter- face hydrogen bonds. Moreover , combining the H-Bond re ward with the ipAE re ward consistently increases the number of hydrogen bonds compared to ipAE-only optimization. Interestingly , this com- bined objectiv e sometimes also improves the number of unique successes, suggesting that optimizing for hydrogen bonds can indirectly encourage successful binder generation and increase diversity . Across most targets, all three rew ard-guided strategies outperform the zero-rew ard baseline, showing the effecti veness of incorporating hydrogen bond information into inference-time optimization. Overall, these results highlight the potential of interface h ydrogen bond optimization as a comple- mentary objectiv e, improving both interface quality and design success rates. I . 9 E X T E N D E D R E S U LT S : E N Z Y M E D E S I G N Atomic Motif Enzyme Design. Building on the strong performance of Complexa in small molecule- conditioned tasks, we extend its capabilities to the more challenging domain of atomic motif enzyme (AME) design. Follo wing Ahern et al. ( 2025 ), we assess the ef ficacy of our model on a div erse set of 41 tasks, which target the complexity and variability of the theozyme (acti ve site) scaffolding problem. Specifically , we generate a protein binder conditioned on the ligand target(s), protein functional groups, and relev ant reaction cofactors. The goal is to generate unique successful binders that preserve the enzymatic geometry of the theozyme. Modeling Details. Follo wing Sec. G.2.3 , we extend the conditioning in the sequence dimension to accommodate the catalytic residue fragments, similar to our approach for the target. This results in a total sequence length of L binder + L target + L catalytic . In contrast to the target featurization, we only require the Atom37 coordinates of the catalytic residue fragments, along with their corresponding amino acid types. The pair representation remains unchanged. By extending the sequence condition- ing, Complexa is able to jointly infer the optimal positions in both sequence and 3D space to place and complete these fragments, while preserving co-designability and ov erall binder success. T raining Details. Similar to the base Complexa, we follow a 3-stage training protocol. In the first stage, we reuse the original atomistic autoencoder used to train the base protein-conditioned model, which has a maximum protein length of 256. W e then pretrain the flow model on the AFDB cluster representativ es, randomly selecting 1-8 pseudo-catalytic residues to condition on for 400k steps on 96 NVIDIA A100-80GB GPUs. Next, we fine-tune on an e ven ratio of AFDB and PLINDER with 50% target dropout for 100k steps on 48 NVIDIA A100-80GB GPUs. During this final fine-tuning stage, when a ligand is present, we restrict the choice of catalytic residues to those with C α atoms within 10 ˚ A of any ligand atom. T o accurately scaf fold a div erse set of residue fragments, we randomly sample one of the possible functional groups for each residue in the set defined by the AME benchmark. While we condition on raw Atom37 representations, we found that restricting to a specific diverse subset of atom configurations works better in practice than randomly sampling a subset of the residue’ s atoms. The specific configurations used are provided belo w: 44 Published as a conference paper at ICLR 2026 PHE: { C , O } { CB , CD1 , CD2 , CE1 , CE2 , CG , CZ } ALA: { C , CA , CB , N } { CA , N } { CA , CB } { C , CA , O } { C , O } HIS: { CD2 , CE1 , CG , ND1 , NE2 } { CB , CD2 , CE1 , CG , ND1 , NE2 } { CD2 , CE1 , NE2 } { CE1 , CG , ND1 } { CE1 , ND1 , NE2 } GL Y : { CA , N } { C , CA , N } TRP: { CD1 , CD2 , CE2 , CG , CZ2 , NE1 } { CB , CD1 , CD2 , CE2 , CE3 , CG , CH2 , CZ2 , CZ3 , NE1 } ASP: { CB , CG , OD1 , OD2 } { CG , OD2 } { CG , OD1 } { C , CA , CB , N } { C , CA , CB , CG , N , O } ARG: { CZ , NE , NH1 , NH2 } { C , CA , CB , N } { CZ , NH1 } { CD , CZ , NE } { CD , CG , CZ , NE , NH1 , NH2 } { CB , CD , CG , CZ , NE } { CZ , NH2 } { C , O } { CD , CZ , NE , NH1 , NH2 } L YS: { CD , CE , NZ } { CE , NZ } THR: { CA , CB , CG2 , OG1 } { C , CA , CB , N } { CB , OG1 } GLU: { C , CA , O } { CA , N } { CD , OE2 } { CD , OE1 } { CD , CG , OE1 , OE2 } ILE: { C , CA , O } { C , CA , CB , N } { CB , CD1 , CG1 , CG2 } SER: { CB , OG } { C , CA , O } { CA , CB , OG } { C , O } { CA , N } { C , CA , CB , N } ASN: { CB , CG , ND2 , OD1 } { CA , N } { CG , ND2 } CYS: { CA , CB , SG } { CB , SG } GLN: { CA , N } { CD , OE1 } { CD , CG , NE2 , OE1 } TYR: { CE1 , CE2 , CZ , OH } { CZ , OH } { CB , CD1 , CD2 , CE1 , CE2 , CG , CZ , OH } PRO: { CD , CG , N } { CA , CB , CD , CG , N } LEU: { CB , CD1 , CD2 , CG } MET : { CE , CG , SD } V AL: { CB , CG1 , CG2 } 45 Published as a conference paper at ICLR 2026 Figure 25: Enzyme Design Benchmark—Best-of-8 Re-designs. Complexa significantly outperforms RFDif- fusion2 in 38/41 AME enzyme design benchmark tasks. Both methods use best-of-8 re-designed sequences. AME Benchmark Results. Follo wing the success criteria introduced in Ahern et al. ( 2025 ), we consider an enzymatic design a success if it meets the following criteria. Note that we use the recent RosettaFold-3 ( Corle y et al. , 2025 ) (RF3) for co-folding due to its ability to template the input ligand conformation, which is critical for maintaining the geometry of the theozyme that the models are conditioned on. Criteria: 1. The catalytic residue types are recovered. 2. The generated binder backbone has a scRMSD of ≤ 2 ˚ A after refolding with RF3. 3. The all-atom catalytic functional groups are recovered with a scRMSD of ≤ 1 . 5 ˚ A after refolding with RF3. 4. There are no clashes with the ligand(s), defined as no binder atom within 1 . 5 ˚ A. In this uninde xed task, we infer catalytic residue indices from the generated output using a greedy matching procedure ( Chen et al. , 2025 ). Specifically , for each residue in the ground-truth theozyme, we find its closest structural counterpart in the generated protein and compute the RMSD using this matched set of residues. This matching process is performed independently for each generated protein, accounting for potential shifts in motif placement across samples. As in our prior e valuations, we cluster the generated proteins with FoldSeek to report the number of unique successful designs. W e ev aluate our model against RFDiffusion2, which generates protein backbones without sequences. T o obtain full-atom complexes, RFDif fusion2 relies on LigandMPNN ( Dauparas et al. , 2025 ) for se- quence generation, followed by refolding with RF3. W e compare our model, using both self-generated sequences and single-shot LigandMPNN, to RFDiffusion2 with LigandMPNN. Additionally , we report the best-of-8 LigandMPNN re-designs, as per the original e v aluation protocol ( Ahern et al. , 2025 ). Follo wing Ahern et al. ( 2025 ), we separate the design tasks by the number of residue islands, i.e. the number of contiguous segments of catalytic residues in the original PDB structure. As shown in T able 12 , our model achiev es a success rate of 41/41 with self-generated sequences and 40/41 with a single LigandMPNN re-design, outperforming RFDif fusion2’ s 30/41. Moreov er, when considering the best-of-8 LigandMPNN sequence re-designs, our model surpasses RFDif fusion2 on 38/41 tasks, including all tasks with ≥ 4 residue islands. Notably , in the three tasks where we underperform, our model achie ves higher raw success rates, with the dif ference attrib uted to only 1-2 unique sample- lev el losses. W e visualize these results in Fig. 10 (single sequence), Fig. 25 (best-of-8), and Fig. 26 (av erage results by residue island count). Note that we did not sample RFDiffusion2 ourselv es, but were provided the generated RFDif f usion2 samples directly by the RFDiffusion2 authors ( Ahern et al. , 2025 ). 46 Published as a conference paper at ICLR 2026 T able 12: Enzyme Design Benchmark—Detailed Quantitative Results. “ All” indicates total number of successes produced by the model (we produce 100 samples per task), while “Unique” indicates number of unique successes, obtained by clustering all successes. The method with the most unique successes for 8 sequence re-designs (LigandMPNN(8)) is highlighted in blue and for single sequence methods is highlighted in red. AME T ask # residue islands RFDiffusion2 LigandMPNN(8) RFDiffusion2 LigandMPNN(1) Complexa (ours) LigandMPNN(8) Complexa (ours) LigandMPNN(1) Complexa (ours) Self-Generated All Unique All Unique All Unique All Unique All Unique M0630 1 78 37 55 22 88 38 76 32 70 27 M0663 1 1 1 0 0 62 38 36 26 31 24 M0710 2 72 42 39 22 91 40 67 34 64 31 M0717 2 55 28 27 12 65 26 39 16 28 11 M0664 2 72 38 36 19 88 49 70 47 60 42 M0555 2 59 23 11 6 80 39 38 20 42 19 M0636 3 39 25 17 16 52 34 24 18 19 17 M0674 3 11 10 2 2 42 25 20 13 19 12 M0739 3 13 8 4 3 52 22 17 7 13 7 M0096 3 39 22 11 6 49 29 15 10 20 14 M0738 3 31 21 8 8 59 39 29 19 23 16 M0209 3 40 23 9 9 92 36 37 18 43 25 M0054 3 31 17 7 4 49 31 24 16 15 13 M0188 3 15 7 2 2 49 39 15 14 24 17 M0711 3 10 5 3 3 57 42 35 29 31 25 M0179 3 15 13 6 4 45 36 18 15 17 15 M0110 3 19 16 6 6 35 23 15 12 14 11 M0731 3 5 5 1 1 6 4 1 1 2 2 M0097 4 41 24 17 12 34 30 19 18 16 15 M0349 4 17 13 5 4 30 28 8 8 7 7 M0129 4 20 15 6 6 26 22 13 12 5 5 M0255 4 17 12 5 4 58 42 32 24 26 20 M0500 4 6 5 1 1 34 25 14 14 16 13 M0151 4 9 9 1 1 25 15 16 11 3 3 M0058 4 2 2 0 0 12 11 1 1 2 2 M0584 4 6 4 1 1 32 17 7 3 9 5 M0040 4 7 2 3 1 32 15 6 4 5 2 M0093 4 8 8 2 2 20 18 6 6 5 4 M0315 4 3 3 0 0 18 18 4 4 6 6 M0907 5 2 2 0 0 11 11 3 3 1 1 M0870 5 7 7 2 2 34 14 11 5 9 4 M0732 5 4 3 1 1 32 17 11 8 4 4 M0552 5 0 0 0 0 9 6 1 1 1 1 M0078 5 2 2 1 1 6 6 1 1 5 4 M0904 6 2 2 0 0 12 5 2 2 3 1 M0157 6 1 1 0 0 16 10 9 7 5 5 M0050 6 5 5 1 1 23 16 7 5 6 5 M0365 6 1 1 0 0 21 11 6 2 7 3 M0024 6 0 0 0 0 22 20 5 5 7 7 M0092 6 0 0 0 0 10 9 1 1 2 2 M0375 7 0 0 0 0 3 1 0 0 1 1 Figure 26: Enzyme Design Benchmark—Aggregated Results. Complexa significantly outperforms RFDiffu- sion2 across all sequence modes. I . 1 0 C A S E S T U DY : L O N G I N F E R E N C E - T I M E S E A R C H A N D P L A T E AU B E H A V I O R In addition to the relativ ely short-term inference-time optimization analysis on easy and hard targets shown in Fig. 7 , we further in vestigate long-term behavior and explore whether it is possible to exhausti vely generate and search for valid binders for a hard target. For this computationally expensi ve case study , we selected a single target, SpCas9 . In Fig. 27 , we analyze the solution space and long-term scaling by examining unique success counts using the standard template modeling score (TM-score) clustering threshold of 0.5, alongside coarser thresholds. W e conducted an extensi ve beam search on the SpCas9 target to generate binders of v arying lengths. W e use the standard setup 47 Published as a conference paper at ICLR 2026 0 200 400 600 800 1000 1200 GP U Hours 0 50 100 150 200 250 # Unique Successes SpCas9 Beam Search (0.4) Beam Search (0.45) Beam Search (0.5) Figure 27: Extensive Sear ch Case Study . Number of unique successes on the target SpCas9 against inference- time GPU hours with dif ferent TM-score clustering thresholds used in the unique success calculation (green: 0 . 4 , red: 0 . 45 and blue: 0 . 5 ). Successes are measured with self-generated sequences and inference-time compute includes both generation and ev aluation time. Figure 28: Interface hydrogen bonding and interface predicted aligned error corr elations. The number of interface hydrogen bonds increases with decreasing ipAE values of ColabDesign during inference-time search with negati ve ipAE as a rew ard (Sec. I.11 ). of N = 4 , L = 4 , and K = 100 , as detailed in Sec. H.2 . W ithin a computational budget of 1,100 GPU hours for optimization and ev aluation, the model produced 13,596 successful binders, resulting in 246 unique successes under the abov e default unique success clustering criteria. As illustrated in Fig. 27 , the number of unique successes continues to increase up to 1,100 GPU hours when using tighter clustering thresholds, indicating a vast potential solution space. Interestingly , when the threshold is lo wered to 0.4 for coarser clustering, a plateau slo wly emerges after 600 GPU hours (at which point ov er 8,000 successful binders had been generated). While the total number of successful binders continues to rise, the discovery of structurally distinct clusters slo ws. Overall, we find that while Complexa can generate di verse successes, it e ventually approaches saturation within the solution space. I . 1 1 I P A E S C O R E S A N D H Y D R O G E N B O N D C O R R E L AT I O N S T o in vestigate whether inference-time optimization of one property like ipAE score can lead to adversarial ef fects such as degradation in biophysical metrics like hydrogen bonds, we in vestigated both ipAE scores and the number of h ydrogen bonds in Complexa samples that were generated by optimisation for low ipAE only (Fig. 28 ). The results show that no adversarial optimization seems to 48 Published as a conference paper at ICLR 2026 T able 13: Binding affinity comparison of unique small molecule binders. W e report the a verage and best pK d in kcal/mol predicted by FLOWR-R OO T . ‡ denotes redesign of the binder sequence using LigandMPNN. † denotes co-folding with RosettaF old3, otherwise the model-generated structure is used. RFDiffusionAA requires both LigandMPNN redesign and co-folding as it generates a backbone without sequence. T arget Model pK d ↑ # of Samples Mean ± Std Best SAM RFDiffusionAA ‡† 5.65 ± 0.15 5.76 2 Complexa (ours) 4.81 ± 0.49 5.49 10 Complexa (ours) † 4.94 ± 0.54 5.74 10 OQO RFDiffusionAA ‡† 7.01 ± 0.31 7.35 3 Complexa (ours) 7.19 ± 0.14 7.37 6 Complexa (ours) † 7.51 ± 0.28 8.05 6 F AD RFDiffusionAA ‡† 6.49 ± 0.76 7.39 5 Complexa (ours) 6.52 ± 0.59 7.39 17 Complexa (ours) † 6.66 ± 0.59 7.65 17 IAI RFDiffusionAA ‡† 5.67 ± 0.40 6.47 8 Complexa (ours) 5.58 ± 0.49 6.71 19 Complexa (ours) † 5.77 ± 1.05 8.25 19 take place, b ut rather the opposite: reducing the ipAE scores leads to an increase in hydrogen bond interactions between binder and target, with a Spearman correlation of -0.69. I . 1 2 B I N D I N G A FFI N I T I E S F O R S M A L L M O L E C U L E T A R G E T S T o further assess the physical v alidity of the ligand-conditioned protein binders generated by Com- plexa, we e valuate the predicted binding af finity of the unique protein-ligand complexes reported in T able 1 . W e utilize FLO WR.ROO T ( Cremer et al. , 2025 ), a recent machine learning-based af finity predictor trained on one of the largest combinations of datasets to date, offering state-of-the-art accuracy while being orders of magnitude faster than Boltz-2 ( Passaro et al. , 2025 ). Additionally , FLO WR.ROO T can predict affinity for gi ven structures without requiring re-co-folding. In T ab . 13 , we compare our results with RFDif fusion-AllAtom, using the same generated samples that also underly the results presented in the table in the main text. W e outperform RFDiffusion- AllAtom on 3 out of 4 targets, generating the highest affinity binders on a verage. Notably , for SAM, RFDiffusion-AllAtom only generates 2 successful binders, and our best binder matches their best, demonstrating that we o verall generate ph ysically plausible binders. It worth noting that, as sho wn in T ab . 1 , Complexa outperforms RFDif fusion-AllAtom by a large mar gin in terms of ipAE-based success metrics and does so while maintaining a much higher sampling efficienc y . I . 1 3 B I O P H Y S I C A L I N T E R FAC E A NA LY S E S F O R P RO T E I N T A R G E T S T o enrich the analysis of the generated Complexa binders be yond folding score metrics, we conducted an analysis of the biophysical properties of the generated binder-target interf aces and compared them to the interfaces in the PDB multimers we used for training, specifically the same subset as depicted in Fig. 12 for the T eddymer analysis (this is, we used the same 2,000 PDB multimer samples as reference and analyze the same metrics). Please see Fig. 29 for the results. As one can see in these metrics, for most properties the generated binders follo w the same trend as the PDB reference set, with the general trend being that the generated interfaces and binders are slightly less hydrophobic and smaller , which is also reflected in the reduced interface shape complementarity . Overall, this indicates that our model generates realistic tar get-binder interfaces. J B A S E L I N E E V A L UA T I O N S BindCraft W e used the code and checkpoints provided in the public BindCraft repository . For binder generation, we directly used the default 4stage multimer hardtarget configuration for the adv anced setting and the default filter provided in the repository . Hotspot conditioning was set according to T able 4 . The original full pipeline of BindCraft included re-designing the hallucinated 49 Published as a conference paper at ICLR 2026 0 10 20 30 40 50 60 # Interface H- Bonds 0.00 0.05 0.10 0.15 0.20 0.25 F raction 0 20 40 60 80 100 Binder interface hydr ophobicity % 0.00 0.02 0.04 0.06 0.08 0.10 0.12 0.14 F raction 0.0 0.2 0.4 0.6 0.8 1.0 Binder surface hydr ophobicity % 0.00 0.05 0.10 0.15 0.20 0.25 F raction 0.0 0.2 0.4 0.6 0.8 1.0 Binder interface shape complementarity 0.00 0.05 0.10 0.15 0.20 0.25 F raction 0 2k 4k 6k 8k 10k Binder interface dS AS A (Ų) 0.00 0.05 0.10 0.15 0.20 0.25 F raction 0 25 50 75 100 125 150 # Binder interface r esidues 0.00 0.05 0.10 0.15 0.20 0.25 F raction Comple xa (ours) PDB Multimer Figure 29: Generated binder interfaces vs. PDB multimer inferfaces. Comparison of bioinformatic metrics between the PDB Multimer reference distribution and successful binders generated by Comple xa. sequence with ProteinMPNN 20 times and filtering for the best. In our ev aluation pipeline, we had a similar workflo w of re-designing the sequence 8 times and filtered for the best. Therefore, we skipped the BindCraft built-in sequence re-designing stage after structure relaxation, so that the ev aluation of BindCraft samples was consistent with our model and other baselines. The reported sampling time of BindCraft did not include the sequence re-designing stage. RFdiffusion W e used the code and checkpoint provided in the public RFdif fusion repository . W e used the default setting in the repository for tar get-conditioned binder generation. Hotspot conditioning was turned on. W e sampled with noise scale ca and noise scale frame set to both 0 and 1 and report the results for noise 0 since these consistently outperformed the noise 1 setting. ProtP ardelle-1c W e used the code and checkpoints provided on the public Protpardelle-1c repository . Specifically , we used the cc83 epoch2616 checkpoint for target-conditioned binder generation. The sampling parameters were default: step scale=1.2 , schurns=200 , crop cond starts=0.0 , and translations=[0.0, 0.0, 0.0] . Hotspot conditioning was set according to T able 4 . APM W e used the code and checkpoints provided on the public APM repository . W e set both the direction condition and direction surface to null , following the instruction from authors for binder generation. BoltzDesign1 W e used the code and checkpoints pro vided in the public BoltzDesign1 repository . For protein targets, we set the flag use msa=true while generating binders. W e turned off the b uilt-in LigandMPNN ( Dauparas et al. , 2025 ) sequence re-design and AlphaFold ( Abramson et al. , 2024 ) and rather used the identical fix ed interface LigandMPNN re-design in our ev aluation pipeline. W e turned off the Rosetta ( Alford et al. , 2017 ) energy scoring because it was redundant in our ev aluation. AlphaDesign W e used the AlphaDesign code released publicly on Zenodo. W e used the default setting provided for the target-conditioned binder generation. W e set the parameter design max iter=200 as instructed. W e skipped the AlphaDesign built-in sequence re-designing model and used the best-of-8 ProteinMPNN strategy in our ev aluation pipeline to be consistent with other baselines. For the sampling of all baselines and our Complexa model, we set a computation budget of 4 NVIDIA A100 hours for each sample. AlphaDesign could exceed the b udget for large targets. For example, only 1 of the 200 samples of target VEGF A finished within 4 hours. Those unfinished samples were unfortunately considered as fail cases during the benchmark of # unique successes with certain GPU hours budget (e.g. T able 7 ). 50 Published as a conference paper at ICLR 2026 Figure 30: Binders generated by Complexa for the challenging lar ge multi-chain targets TNF- α (three-chain target), IL17A (two-chain tar get), and H1 (two-chain target; two dif ferent binders visualized). All shown binders meet the in-silico success criteria. Binders are visualized in cartoon representation, including side chains, multi-chain targets in surf ace representation. See Sec. I.4 . K A D D I T I O N A L V I S U A L I Z A T I O N S In Fig. 30 , we show successful de novo binders generated by Complexa for the challenging multi-chain targets TNF- α (three-chain target), IL17A (two-chain tar get), and H1 (two-chain tar get). In Fig. 31 , we show successful small molecule binders generated by Complexa for the four small molecules SAM, IAI, F AD and OQO. In Fig. 32 , we show fold class-conditioned binder generation with Complexa for five dif ferent targets, controlling secondary structure content. L D E C L A R AT I O N O N U S A G E O F L A R G E L A N G UA G E M O D E L S Large Language Models were used during the preparation of the manuscript exclusi vely to catch typographical and grammatical errors and to improve writing style. Beyond that, no LLMs were in volved in the research or the project in an y way . 51 Published as a conference paper at ICLR 2026 Figure 31: Binders generated by Complexa for small molecule targets SAM, IAI, F AD and OQO (see Sec. E ). All shown binders meet the in-silico success criteria. 52 Published as a conference paper at ICLR 2026 Figure 32: Fold class-conditioned binders generated by Complexa for five dif ferent targets. W e control the secondary structure content through conditioning on C-lev el CA T labels “Mainly Alpha”, “Mainly Beta” or “Mixed Alpha Beta” ( Dawson et al. , 2016 ; Geffner et al. , 2025 ). W e can observe that the generated binders follow the conditioning, exhibiting primarily alpha helices, beta sheets, or both. All shown binders meet the in-silico success criteria. Binders are visualized in cartoon representation, including side chains, targets in surface representation. 53 Published as a conference paper at ICLR 2026 T able 14: Hyperparameters for Complexa model training. W e denote two versions of Comple xa that specialized in generating binders for protein targets and small molecule ligand target as Protein- Protein and Ligand-Protein , respectiv ely . Protein-Pr otein CA T refers to the model that can generate binders to protein target conditioned on CA T labels. Hyperparameters Complexa Protein-Pr otein Protein-Protein CA T Ligand-Pr otein Model Architectur e Partially Latent Flow Matching sequence repr dim 768 768 768 sequence cond dim 256 256 256 t sinusoidal enc dim 256 256 256 idx. sinusoidal enc dim 256 256 256 fold class cond dim 0 256 0 pair repr dim 256 256 256 seq separation dim 128 128 128 pair distances dim ( x t ) 30 30 30 pair distances dim ( ˆ x ( x t ) ) 30 30 30 pair distances min ( ˚ A) 1 1 1 pair distances max ( ˚ A) 30 30 30 # attention heads 12 12 12 # transformer layers 14 14 14 # trainable parameters 159 M 159 M 159 M V AE enc/dec sequence repr dim 768 768 768 enc/dec # attention heads 12 12 12 enc/dec # transformer layers 12 12 12 enc/dec sequence cond dim 128 128 128 enc/dec idx. sinusoidal enc dim 128 128 128 enc/dec pair repr dim 256 256 256 latent dimension 8 8 8 # trainable parameters 256 M 256 M 256 M T raining Details V AE AFDB training Dataset AFDB monomer AFDB monomer AFDB monomer max sequence length 256 256 512 # train steps 500k 500k 140K train batch size per GPU 14 14 5 learning rate 1e-4 1e-4 1e-4 optimizer Adam Adam Adam # GPUs 16 16 32 V AE PDB fine-tuning Dataset PDB PDB N/A # train steps 40k 40k N/A train batch size per GPU 12 12 N/A learning rate 1e-4 1e-4 N/A optimizer Adam Adam N/A # GPUs 16 16 N/A Partially Latent pre-training Dataset AFDB monomer AFDB monomer AFDB monomer max sequence length 256 256 512 # train steps 540K 540K 270K train batch size per GPU 12 12 5 learning rate 1e-4 1e-4 1e-4 optimizer Adam Adam Adam # GPUs 32 32 48 Partially Latent fine-tuning Dataset T eddymer+PDB T eddymer PLINDER+AFDB monomer # train steps 290K 200K 60K target dropout 0% 0% 50% use LoRA False False T rue LoRA rank N/A N/A 32 LoRA α N/A N/A 64 train batch size per GPU 6 6 5 learning rate 1e-4 1e-4 1e-4 optimizer Adam Adam Adam # GPUs 96 96 96 54
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment