Making Multi-Axis Models Robust to Multiplicative Noise: How, and Why?
In this paper we develop a graph-learning algorithm, MED-MAGMA, to fit multi-axis (Kronecker-sum-structured) models corrupted by multiplicative noise. This type of noise is natural in many application domains, such as that of single-cell RNA sequenci…
Authors: Bailey Andrew, David R. Westhead, Luisa Cutillo
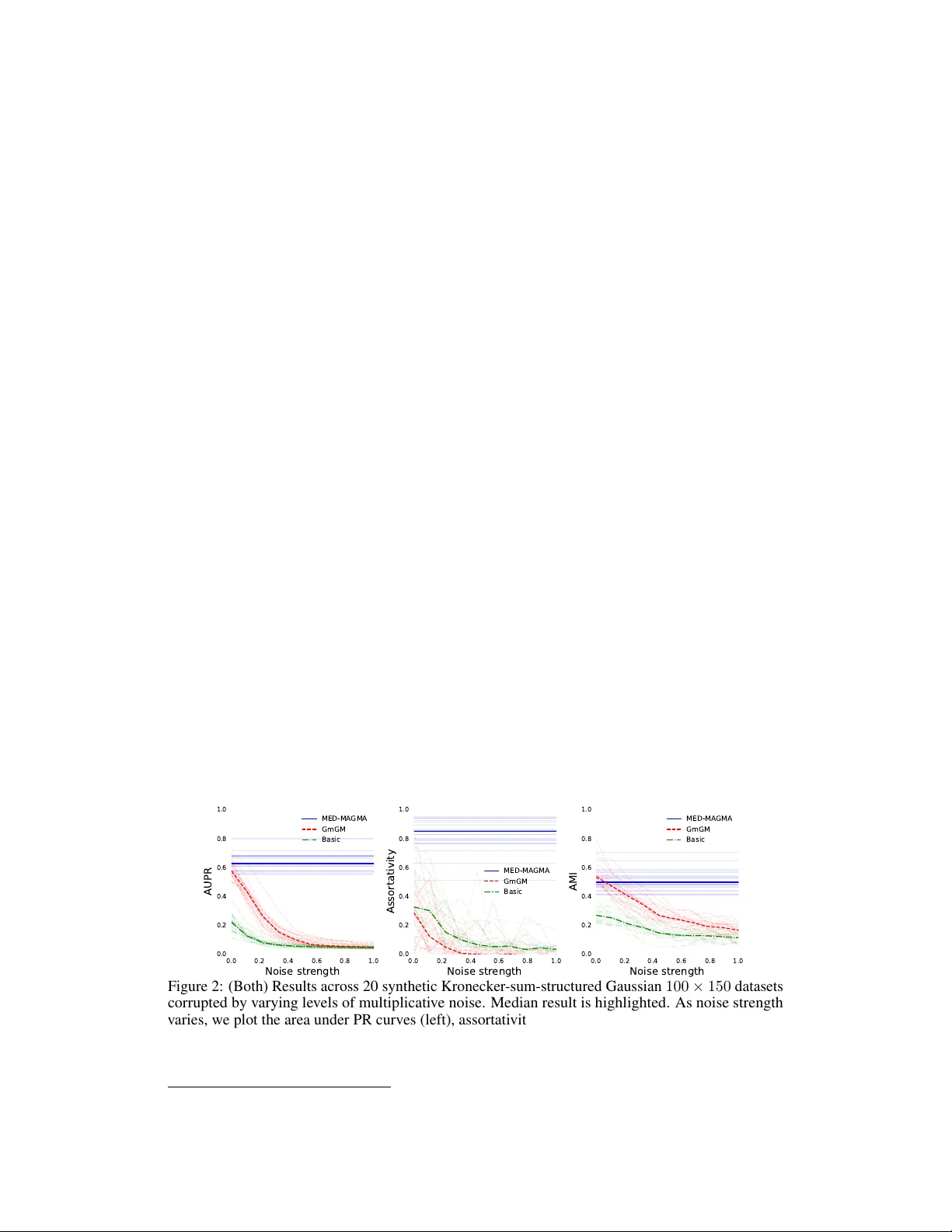
Making Multi-Axis Models Rob ust to Multiplicativ e Noise: How , and Wh y? Bailey Andrew School of Computer Science Univ ersity of Leeds W oodhouse, Leeds, UK; LS2 9JT sceba@leeds.ac.uk David R. W esthead School of Molecular and Cellular Biology Univ ersity of Leeds Luisa Cutillo School of Mathematics Univ ersity of Leeds Abstract In this paper we dev elop a graph-learning algorithm, MED-MA GMA, to fit multi- axis (Kronecker -sum-structured) models corrupted by multiplicativ e noise. This type of noise is natural in many application domains, such as that of single-cell RN A sequencing, in which it naturally captures technical biases of RNA sequencing platforms. Our work is ev aluated against prior work on each and ev ery public dataset in the Single Cell Expression Atlas under a certain size, demonstrating that our methodology learns networks with better local and global structure. MED- MA GMA is made a vailable as a Python package ( MED-MAGMA ). 1 Introduction This paper deals with the problem of ho w to mak e Kronecker -structured (or ‘multi-axis’) methods robust to a form of noise common in applied domains such as those using single-cell RN A sequencing data (scRN A-seq; matrix-v ariate data in which ro ws represent cells, columns represent genes, and entries represent the amount a giv en gene is expressed in a giv en cell). Multi-axis models aim to simultaneously learn matrices Ψ rows , Ψ cols that encode dependency networks (‘graphs’) between the rows and the columns, respectiv ely . F or scRN A-seq, this would yield both cell and gene netw orks, useful for tasks such as cell subtyping and gene regulatory network inference. In particular, we aim to fit models of the follo wing form for matrix-variate data, where ζ is some function whose role is to mix the O ( d rows ) row and O ( d cols ) column dependencies into a network of O ( d rows d cols ) matrix entry dependencies. laten t data ∼ N 0 , ζ ( Ψ rows , Ψ cols ) − 1 observ ed data ij = r rows i r cols j (laten t data ij ) (multiplicativ e noise) r rows i , r cols j ∼ an y strictly positiv e distribution There are many methods to fit the abo ve model without noise under a v ariety of choices of ζ , which we will discuss in Section 2. Ho we ver , no such method e xists to fit the distributions under this noise regime. This is unfortunate, as not only is this type of noise common, but taking it into account also dramatically increases the flexibility of the model. Preprint. In scRN A-seq, this type of noise arises from two simple observ ations. Some genes are more likely to be measured than others due to biases such as GC-bias [10, 3], and cells ha ve less measured gene expression than reality due to the incompleteness of the measuring process. If the true measurement of a gene g in cell c is x , but only 0 ≤ r cells c ≤ 1 of the expression in that cell is collected, and gene g is r genes g times as likely to be collected than the other genes, then we should expect to observe a measurement of r genes g r cells c x rather than x itself. Even in cases where this noise type does not exist, the flexibility of models accounting for this noise is beneficial. This is because our model is a natural generalization of the class of elliptical distrib utions to the matrix-variate case. If a dataset follo ws the model below , then it has an elliptical distribution: laten t data ∼ N 0 , Ω − 1 observ ed data = r · (latent data) r ∼ any strictly positi ve distrib ution Elliptical distributions encompass many other distributions, such as the normal distrib ution (when r = 1 ), the multi v ariate t distrib ution (when r ∼ q ν χ 2 ν ), and the multiv ariate Laplace distribution (when r 2 ∼ Exp onential(1) ). In fact, any distribution whose density has elliptical contours can, under mild conditions, be expressed in this form. Most multi-axis models either assume the dataset is Gaussian, or make the weaker ‘Gaussian copula’ assumption (i.e. arbitrary marginals b ut the variables ‘interact Gaussian-ly’); the latter is often fit via the use of the nonparanormal skeptic [26]. Gaussian copulas are restricti ve in terms of the types of dependencies they allo w . They do not allo w ‘tail dependence’ - extreme e vents are alw ays asymptotically uncorrelated. Howe ver , Figure 1 implies that tail dependence does e xist in scRN A-seq data. Elliptical distributions allo w for such extremal dependencies. 0.00 0.25 0.50 0.75 1.00 T ail dependence R eal genes Gaussian copula 0.00 0.25 0.50 0.75 1.00 T ail dependence R eal cells Gaussian copula Figure 1: Estimated tail dependence across 1000 randomly selected pairs of genes (left) and cells (right) from the E-MT AB-7249 scRNA-seq dataset discussed in Section 4.3, compared to random data generated from a Gaussian copula. Thus, it should be clear why our proposed model is useful: it is robust to a natural type of noise, and strongly increases the e xpressiv e po wer of multi-axis models. In this paper , we will produce an algorithm (MED-MAGMA: the m atrix e lliptical d istrib ution’ s m ulti- a xis g raphical m odelling a lgorithm) to learn Ψ rows , Ψ cols under the presented model (Chapter 3) and in Chapter 4 comprehen- si vely v alidate its performance across every sub-1000-cell dataset in the public Single Cell Expression Atlas database [30]. In the next section we giv e the moti vation and history behind such models. 2 Background Multi-axis models were introduced to solve the problem of inference when samples are not indepen- dent and identically distributed (i.i.d.). If one is willing to assume that the dataset { d i } is i.i.d, then a simple formulation would be: d i ∼ N 0 , Ψ − 1 cols In this case, Ψ cols encodes the graph of conditional dependencies: 2 Ψ cols ij = 0 ⇐ ⇒ feature (column) i is conditionally independent from feature j T wo variables being conditionally independent means that, after conditioning out the rest of the dataset, they are statistically independent from one another . In other words, conditional dependencies intuitiv ely capture the concept of ‘direct correlations’: if A is correlated with B , which in turn is correlated with C , then A and C will almost always also be correlated to some degree, via their indirect connection through B . Howe ver , A and C will not be conditionally dependent , as their indirect connection will hav e been conditioned out. This connection moti vates the use of conditional dependencies: knowledge of direct ef fects is much more valuable than indirect ef fects. For more complex datasets, the i.i.d. assumption breaks down. In particular , the ‘samples’ of a scRN A-seq dataset are cells of v arious different cell types, which had been interacting with other cells in the dataset for their entire life-cycle, until being destroyed and collected for data analysis. In this scenario, not only are the cells not independent, b ut the dependencies between the cells are also of interest in their own right. Compiling the dataset into a d rows × d cols matrix D , we could address this by using the follo wing distribution, where v ec is the vectorization operation that stacks the columns of a matrix into a single vector: v ec [ D ] ∼ N 0 , Ω − 1 Howe v er , Ω would be a d rows d cols × d rows d cols matrix capturing the dependencies between (cell, gene) pairs - it is computationally intractible to store and statistically intractible to learn. What we want is to decompose Ω into tractable chunks directly representing the ro ws and columns: Ω = ζ ( Ψ rows , Ψ cols ) . Rather than O ( d 2 rows d 2 cols ) parameters, this framework only requires O ( d 2 rows + d 2 cols ) . There are many choices for ζ , but the y are all based on the Kronecker product ⊗ : h a 11 a 12 ··· a 21 ··· ··· i ⊗ B = h a 11 B a 12 B ··· a 21 B ··· ··· i The following are the choices already considered in the literature: ζ ( Ψ rows , Ψ cols ) = Ψ cols ⊗ I (Rows are i.i.d, same as before) ζ ( Ψ rows , Ψ cols ) = I ⊗ Ψ rows (Columns are i.i.d) ζ ( Ψ rows , Ψ cols ) = Ψ cols ⊗ Ψ rows (‘Strong product’ assumption) ζ ( Ψ rows , Ψ cols ) = Ψ cols ⊗ I + I ⊗ Ψ rows (‘Cartesian product’ assumption) ζ ( Ψ rows , Ψ cols ) = ( Ψ cols ⊗ I + I ⊗ Ψ rows ) 2 (Sylvester generati ve model) There are also models which break Ω up into three axes, distinguishing between dif ferent types of ro w dependencies [1]. The strong product assumption is the oldest [9], although it is often misreported as the ‘tensor product’ assumption. The Cartesian product assumption was introduced by Kalaitzis et al. [21] and the Sylv ester generati ve model by W ang, Jang, and Hero [39]. For scRN A-seq data, the assumptions abov e correspond to the following statements: Assumption (Strong product assumption) . The str ength of the dependency between the expr ession of a gene g 1 in a cell c 1 and the expr ession of a gene g 2 in a cell c 2 is equal to the pr oduct of the dependencies between c 1 and c 2 , and g 1 and g 2 . In other wor ds, genes depend on the e xpr ession of other genes within the cell and g enes in other cells. Assumption (Cartesian product assumption) . A gene g 1 may only be conditionally dependent ( ∼ ) on other genes g 2 within the same cell c 1 , or the same gene in other cells c 2 : ( c 1 , g 1 ) ∼ ( c 1 , g 2 ) , ( c 1 , g 1 ) ∼ ( c 2 , g 1 ) , and ( c 1 , g 1 ) ≁ ( c 2 , g 2 ) . Just because the y may be dependent does not mean the y must be. In other wor ds, genes can only dir ectly affect the expr ession of other genes within the same cell (or the same gene in dif fer ent cells). 3 Assumption (Sylvester generati ve model) . Ψ rows D + DΨ cols ∼ N ( 0 , I ) This model naturally arises when the dataset is gener ated fr om certain partial differ ential equations. For scRN A-seq data, the Cartesian product is the most natural. While genes can interact with genes in other cells in certain scenarios, such interactions will be much weak er than within-cell interactions; the strong product assumption would gi ve equal weight to within- and between-cell interactions. The operation A ⊗ I + I ⊗ B is denoted the ‘Kronecker sum’: A ⊕ B . Many algorithms exist to fit Kronecker -sum-structured normal distributions, the most scalable being GmGM [2]. Ho we ver , none fit such distributions under the scenario of multiplicati v e noise. 3 Methodology T o fit the model, we will perform maximum likelihood estimation (MLE) using e xpectation maxi- mization (EM). Let us rephrase our model for a dataset x = v ec [ X d rows × d cols ] as follows: x = ( r cols ⊗ r rows ) ◦ z z ∼ N 0 , ( Ψ cols ⊕ Ψ rows ) − 1 r rows , r cols ∼ an y strictly positiv e distribution The r v ariables are allo wed to ha ve dependencies among themselv es, b ut they must be independent of the latent variable z . As a shorthand, we will let Ω = Ψ cols ⊕ Ψ rows . As further shorthand, we will implicitly let capital variables V and lowercase v ariables of the same name v follow the relationship v = v ec [ V ] . Our final algorithm is summarized belo w . In the follo wing sections, we will deriv e the algorithm step-by-step. Algorithm 1 MED-MA GMA Require: A d rows × d cols dataset X with vectorization x y ← f ( x ) ▷ Remov e noisy directions (Section 3.2) Ψ rows ← I ▷ Initialize estimates Ψ cols ← I repeat Find z ∗ by flip-flopping axes while solving Equation 3 ▷ Section 3.5 Approximate S rows and S cols using Ψ rows , Ψ cols , and z ∗ with Equation 2 ▷ Section 3.4 ( Ψ rows , Ψ cols ) ← GmGM ( S rows , S cols ) ▷ Section 3.3 until Ψ rows and Ψ cols con ver ge Output: Ψ rows and Ψ cols , which encode the conditional dependencies of the data. 3.1 Handling non-identifiability Our model generalizes the class of elliptical distrib utions, and thus cannot discriminate between Ω and r Ω for r > 0 . This is not a critical issue - we are only interested in the sparsity structure of Ω , not its scale, as it is the sparsity structure that encodes the conditional dependencies. Howe ver , it must be addressed to ensure proper behavior of our algorithm. Observe the follo wing, for some measure of magnitude ∥·∥ : x = ( r cols ⊗ r rows ) ◦ z = r ( r ′ cols ⊗ r ′ rows ) ◦ z where ∥ r ′ cols ⊗ r ′ rows ∥ = 1 This is why the non-identifiablilty arises: due to being free to choose r , x and r x are indistinguishable by the model, and hence so are Ω and r Ω . As we don’ t care about this difference, we can assume w .l.o.g. that r = 1 ; in other words, w .l.o.g. ∥ r cols ⊗ r rows ∥ = 1 . It will be con venient later if we choose ∥·∥ to be a geometric mean constraint: ( Q i r rows i ) 1 d rows Q j r cols j 1 d cols = 1 . 4 W e also ha ve to address the non-identifiability of Kronecker products: a ⊗ b = c a ⊗ 1 c b . This non-identifiability is an artifact of the parameterization, and does not af fect the extent to which Ω can be learned. W e can simply modify our constraint to get rid of this artifact: Q i r rows i = 1 = Q j r cols j . Thus, we aim to fit the following: x = ( r cols ⊗ r rows ) ◦ z z ∼ N 0 , ( Ψ cols ⊕ Ψ rows ) − 1 r rows , r cols ∼ an y strictly positiv e distribution such that Y i r rows i = 1 = Y j r cols j 3.2 Removing the effect of the noise This model is a mixture of distrib utions (the normal distribution and an y arbitrary strictly positi ve distribution), and thus does not directly yield a likelihood to estimate. In fact, no likelihood will be a v ailable as long as we have such a general assumption on the r variables. What we need is a function f such that f ( x ) = f ( y ) ⇐ ⇒ x = ( r cols ⊗ r rows ) ◦ y . Such a function thro ws out all the information x provides that has become corrupted by the noise, lea ving only the information upon which it is safe to do inference; we can then deduce a likelihood ov er f ’ s image. It is worth thinking about the class of d -dimensional elliptical distributions again, in which we ha ve x = r z . The arbitrariness of r ensures we can ne ver kno w the magnitude of z , but we can kno w its orientation. In particular, x ∥ x ∥ = r z ∥ r z ∥ = z ∥ z ∥ ; f ( y ) = y ∥ y ∥ maps the data to a circle. The probability distribution at an y point p along this circle is obtained by integrating over e v ery point y such that f ( y ) = f ( p ) . This space of points, denoted generically F (and denoted f − 1 ( p ) when considering a specific circle point p ), is called the ‘fiber space’, and the remaining space Q (the circle) is the ‘quotient space’; R d ∼ = F × Q . W e obtain the likelihood ov er Q by integrating out F : p df ( x | Ω ) = Z y ∈ f − 1 ( x ) p df N ( y | Ω ) d y For the standard class of elliptical distributions, this integral is actually tractable, yielding the angular Gaussian distribution, whose MLE turns out to be T yler’ s M estimator [38]. For our distribution, the scenario is significantly more complicated, as normalizing to the unit sphere does not remove the noise. Y et the procedure stays the same: we will produce a function f that thro ws away unreliable information while preserving everything else, and then inte grate out the fibers induced by f to produce a formula for the likelihood ov er Q . f ( X ) ij = x ij Q i ′ j ′ x i ′ j ′ 1 d rows d cols Q j ′ x ij ′ 1 d cols Q i ′ x i ′ j 1 d rows (1) Proposition (Goodness of f ) . The function f described in Equation 1 has the following pr operty: f ( X ) = f ( Y ) if and only if one can find strictly positive r rows , r cols such that we have v ec [ X ] = ( r cols ⊗ r rows ) ◦ v ec [ Y ] . Pr oof. See Appendix B. Handling zeros Man y datasets, such as most scRN A-seq datasets, contain zeros. Equation 1 as written is undefined for such zeros. W e present a zero-handling modification of f in Appendix C. From here onwards, we will assume that f works with zeros. 5 3.3 Perf orming expectation maximization Now that we ha v e f , we can express the likelihood of our distrib ution as: p df ( x | Ω ) = Z z ∈ f − 1 ( x ) p df N ( z | Ω ) d z ∝ √ det Ω Z z ∈ f − 1 ( x ) e − 1 2 z T Ωz d z Sadly , this integral is likely intractable due to the complicated structure of f − 1 . This motiv ates the use of the EM algorithm, frequently used in settings with latent variables such as this. Letting K ⊕ ++ be the space of positi ve definite Kronecker -sum-separable matrices (i.e. the space of valid Ω ), the EM algorithm is as follows: let y = f ( x ) = f ( z ) be our denoised observ ed variable let Q Ω Ω ( t ) = E z ∼N 0 , Ω − 1 ( t ) and z ∈ f − 1 ( y ) [log p df N ( z | Ω )] Ω ( t +1) = argmax Ω ∈K ⊕ ++ Q Ω Ω ( t ) (iterate until con ver gence) Thus we hav e reduced our problem to the (admittedly intimidating) problem of maximizing Q . Proposition (Maximizing Q ) . Optimizing Q corr esponds to the same optimization problem as GmGM . GmGM uses the sufficient statistics XX T and XX T . T o maximize Q , we r eplace these with: S rows = E z ∼N ( 0 , Ω − 1 ( t ) ) z ∈ f − 1 ( y ) ZZ T and S cols = E z ∼N ( 0 , Ω − 1 ( t ) ) z ∈ f − 1 ( y ) Z T Z Mor e gener ally , any method to fit unr e gularized Kr onec ker -sum-structur ed models without noise that operates on the suf ficient statistics can be used in place of GmGM. Pr oof. The proof is giv en in Appendix D. This is excellent: GmGM is a fast algorithm. All that remains is to ev aluate the expectations. 3.4 Evaluating S rows and S cols with Laplace’ s approximation Let g ( z ) be either ZZ T or Z T Z ; the logic behind ev aluating the e xpectation is the same. W e can express the e xpectation as follows: E z ∼N ( 0 , Ω − 1 ( t ) ) z ∈ f − 1 ( y ) [ g ( z )] = R z ∈ f − 1 ( y ) g ( z ) e − 1 2 z T Ω ( t ) z d z R z ∈ f − 1 ( y ) e − 1 2 z T Ω ( t ) z d z (2) It may appear that we have sw apped one hard problem for another - we were trying to av oid a nasty integral-over -fiber in Section 3.3, yet now we ha ve a ratio of them! Ho we ver , this ratio can be well-approximated by Laplace’ s method [36]. The intuition behind Laplace’ s method is that the inte gral is dominated by the exponential term, whose maximum occurs at z ∗ (in other words, the minimum of z T Ω ( t ) z occurs at z ∗ ). By focusing on local approximations about z ∗ , we can well-approximate the integral in the re gion where it has the most mass. Letting P be the matrix that projects onto the tangent space about z ∗ in the fiber , Laplace’ s method tells us that: R z ∈ f − 1 ( y ) g ( z ) e − 1 2 z T Ω ( t ) z d z R z ∈ f − 1 ( y ) e − 1 2 z T Ω ( t ) z d z ≈ g ( z ∗ ) + 1 2 tr h PΩ ( t ) P T − 1 P ∇ 2 g ( z ∗ ) P T i 6 The trace term above looks intimidating, b ut in practice it can be ev aluated efficiently; we sho w how in Appendix E. Thus, all that remains is to find the point z ∗ ∈ f − 1 ( y ) that minimizes z T Ω ( t ) z . 3.5 Finding z ∗ with quadratic programming In order to approximate S rows and S cols statistics, we need to solve the following optimization problem: z ∗ = argmin z ∈ f − 1 ( y ) z T Ω ( t ) z = argmin r rows , r cols > 0 Q i r rows i = 1 = Q j r cols j X ik r rows i X j ℓ r cols j Y ij Ω ( t ) ( ij ) , ( k ℓ ) r cols ℓ Y kℓ r rows k This is equiv alent to minimizing an order -4 polynomial with constraints - not a trivial task at all. Howe v er , our problem naturally lends itself to a flip-flop algorithm over two quadratic sub-problems; the terms in the parentheses may be aggregated into a d rows × d rows matrix ˜ Ω not depending on r rows . This is a quadratic form in r rows , and so for fixed r cols we can reduce our problem: argmin r T rows ˜ Ωr rows such that r rows > 0 and Y i r rows i = 1 (3) This is still non-con ve x 1 due to the nonlinear multiply-to-one constraint, but in practice is efficiently optimizable. An analogous formulation holds for r cols ; we can flip-flop between them to find the optimal r rows , r cols , and hence z ∗ . 4 Results In this section, we ev aluate our method, MED-MA GMA, in three ways: by comparing it to GmGM on synthetic data (Section 4.1), comparing it to GmGM systematically across all public datasets of a certain type (Section 4.2), and finally by exploring a specific example (Section 4.3). The code to run these experiments is a v ailable on GitHub (https://github .com/MED-MA GMA-NeurIPS2026/MED- MA GMA-NeurIPS2026) and our model is a vailable on PyPI ( pip install MED-MAGMA ). 4.1 Synthetic validation 0.0 0.2 0.4 0.6 0.8 1.0 Noise str ength 0.0 0.2 0.4 0.6 0.8 1.0 AUPR MED-MA GMA GmGM Basic 0.0 0.2 0.4 0.6 0.8 1.0 Noise str ength 0.0 0.2 0.4 0.6 0.8 1.0 Assortativity MED-MA GMA GmGM Basic 0.0 0.2 0.4 0.6 0.8 1.0 Noise str ength 0.0 0.2 0.4 0.6 0.8 1.0 AMI MED-MA GMA GmGM Basic Figure 2: (Both) Results across 20 synthetic Kronecker -sum-structured Gaussian 100 × 150 datasets corrupted by v arying lev els of multiplicati ve noise. Median result is highlighted. As noise strength varies, we plot the area under PR curv es (left), assortati vity (center), and AMI (right). T o v alidate our method on synthetic data, we first generated ground truth graphs from the Barabasi- Albert distrib ution, and con verted these to positiv e definite matrices Ψ rows and Ψ cols . W e then 1 W e did experiment with a sum-to-one constraint, which would be con ve x; it performed well on synthetic data but not real data, due to the constraint not respecting the geometry of the problem nor the structure of f . 7 generated 20 samples from N 0 , ( Ψ cols ⊕ Ψ rows ) − 1 . T o corrupt the samples with multiplicativ e noise, we generated each r ( ℓ ) variable as chi-squared (df=1) and raised them to the po wer α ∈ [0 , 1] ; α represents the noise strength ( α = 0 yields no noise). For two of our metrics (assortativity and AMI), comparison requires a ground truth clustering - we used Leiden clustering [37] with resolution 1 on the ground truth graphs to create this. For AMI, we also need a clustering on the learned graphs - we measured AMI using Leiden clustering at v arious resolutions (from 0.02 to 2) and recorded the result with the best AMI. The generated datasets presented here are 100 × 150 matrices; in Appendix A we show results on 200 × 200 matrices. W e compare MED-MA GMA and GmGM on this synthetic dataset in Figure 2, using the metrics of precision and recall, assortati vity , and AMI. Assortati vity measures the tendency of vertices of the same type to be connected together , whereas AMI measures the agreement between two clusterings. Intuitiv ely , these three metrics measure the ability of methods to capture structure at different scales, with precision and recall measuring the recovery of specific edges, AMI measuring the recovery of clusterings, and assortativity sitting in between. W e also compare the results against a baseline single-axis precision-matrix estimator (the MLE, which for a dataset X is just XX T − 1 ). W e can see that MED-MA GMA is unaffected by noise strength, whereas GmGM and our baseline are quite sev erely af fected. 4.2 Systematic validation T o v alidate MED-MA GMA on real data, we systematically do wnloaded ev ery dataset in the Single Cell Expression Atlas less than a thousand cells which contained a categorical variable and referenced a source publication. There were 30 such datasets in total; we list these in Appendix G. By e v aluating our methodology in this manner , we ensure that we ev aluate over a representati ve sample of all scRN A-seq datasets. The datasets receiv ed minimal preprocessing, described in Appendix F. Each dataset contained a categorical v ariable, typically ‘cell type’. For comparison, we used both assortati vity and adjusted mutual information [29]. W e thresholded the learned graphs to keep only the top k edges per verte x, with k varying from 1 to 40. W e used Leiden clustering, varying the resolution parameter from 0 . 02 to 2 . 0 to obtain clusterings of v arious sizes - we only report the best AMI across all clusterings and thresholds. W e compared against both standard GmGM, GmGM equipped with the nonparanormal skeptic, and our single-axis baseline; Figure 3 demonstrates that assortativity and AMI are higher in MED-MA GMA than the other methods, with or without the nonparanormal skeptic. 30 28 26 24 22 20 18 16 14 12 10 8 6 4 2 R ank 0.4 0.2 0.0 0.2 0.4 0.6 Assortativity 30 28 26 24 22 20 18 16 14 12 10 8 6 4 2 R ank 0.0 0.2 0.4 0.6 0.8 1.0 AMI MED-MA GMA GmGM GmGM/nonpara Single Axis median Figure 3: (Both) The datasets are ordered by increasing performance along the x-axis. GmGM/nonpara refers to GmGM equipped with the nonparanormal skeptic. The median per- formance is highlighted. (Left) Assortativities belo w 0 represent a statistical tendenc y for cells of differ ent categories to connect, which indicates the netw ork does capture information about the dataset, but it is harder to interpret. 4.3 Specific evaluation Both prior sections serve to sho w that MED-MA GMA yields better results than GmGM. Thus, models that tak e into account multiplicativ e noise perform better on scRN A-seq data. In this section, we 8 briefly pick one of the datasets from earlier and show that the cell and gene networks are robust, and that the gene network learned is also reasonable. In particular , we pick the ‘median dataset’ when ranking datasets by assortativity . By picking the median dataset, it giv es us a generic view into what we can expect the a verage performance of our methodology to be. The chosen dataseet, E-MT AB-7249, contains 185 cells from a patient with high-grade serous ov arian carcinoma (HGSOC) [31]. The source paper in vestigated the biology of HGSOC through se veral modalities of data from multiple patients. Here, we are using the portion of this dataset av ailable on the Single Cell Expression Atlas, comprised only of the cells of patient ‘OCM.38a’. The data from the atlas had 7 clusters representing cell subtypes: SA-a, SA-b, SB, T A-a, T A-b, TB, and a final small ‘N/A ’ cluster of unlabeled cells. From the source paper , it is clear that these are subtypes of the following cell types: stromal (SA-a, SA-b, SB) and neoplastic (T A-a, T A-b, TB). neoplastic stromal m0 per centage >30% m3 per centage >30% Figure 4: (All) A UMAP plot of E-MT AB-7249 cells based on gene expression. (Left) Cell type. (Middle, Right) The percentage of gene expression from genes in modules m0 and m3, respecti vely . W e clustered the learned gene network (Leiden, resolution=1), yielding 8 ‘modules’. Figure 4 shows that two of our gene modules strongly correspond to stromal and neoplastic cells. T o quantify the relation between our gene modules and the cell subtypes, we performed a one-w ay ANO V A testing whether the percentage e xpression of each gene module dif fered across cell subtypes. All modules showed significant v ariation between clusters ( p = 2 × 10 − 29 to 1 × 10 − 2 ) except one ( p = 0 . 11 ), indicating that MED-MA GMA learned gene modules capturing processes associated with specific cell subtypes. For one final test, we ev aluated the robustness of our cell and gene networks using Robin [34]. Robin measures the tendency for clusterings on a network to change after an increasing proportion of the edges are randomly perturbed: networks whose clusterings change significantly after a small clustering are not robust. Robin outputs Bayes factors; our cell netw ork has factor of 108 and our gene network has a factor of 59, indicating v ery strong evidence that clusterings on MED-MAGMA ’ s networks are rob ust to perturbations. 5 Discussion and limitations W e have de veloped a model that allo ws multi-axis estimation under multiplicativ e noise, and an algorithm to fit the model. Such a model is important; man y datasets, such as scRN A-seq datasets, are af fected by this type of noise. It also significantly increases the flexibility of multi-axis models; we did not make a Gaussian copula assumption, allo wing our model to be suitable for datasets that contain more complicated modes of dependenc y (such as tail dependence). W e demonstrated that our methodology outperforms prior work on synthetic data as well as through a systematic ev aluation of all public scRN A-seq datasets belo w a certain size on a public database. Despite the promising results, our model does hav e limitations. Currently , our method can handle matrices with thousands of rows/columns, but would struggle with tens of thousands. Both the approximation of ( S rows , S cols ) and the GmGM sub-problem dominate the runtime, so improv ements in either of those would be significant. Additionally , elliptical-style distributions such as ours (despite their generality) still make some tangible assumptions about the dependencies in the dataset - namely that dependencies are linear and radially symmetric. It would be a w orthwhile future direction to further weaken the assumptions made here. 9 Acknowledgments and Disclosur e of Funding Bailey Andrew is supported by the UKRI Engineering and Physical Sciences Research Council (EPSRC) [EP/S024336/1]. David W esthead is supported in part by the National Institute for Health and Care Research (NIHR) Leeds Biomedical Research Centre (BRC) (NIHR203331). The vie ws expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. References [1] Bailey Andre w, Da vid Robert W esthead, and Luisa Cutillo. “The Strong Product Model for Network Inference without Independence Assumptions”. en. In: Pr oceedings of The 28th International Confer ence on Artificial Intelligence and Statistics . PMLR, Apr . 2025, pp. 5230– 5238. U R L : https : / / proceedings . mlr . press / v258 / andrew25a . html (visited on 03/12/2026). [2] Ethan B. Andre w, David W esthead, and Luisa Cutillo. “GmGM: a fast multi-axis Gaussian graphical model”. en. In: Pr oceedings of The 27th International Confer ence on Artificial Intelligence and Statistics . PMLR, Apr . 2024, pp. 2053–2061. U R L : https://proceedings. mlr.press/v238/andrew24a.html (visited on 03/12/2026). [3] Y uval Benjamini and T erence P . Speed. “Summarizing and correcting the GC content bias in high-throughput sequencing”. eng. In: Nucleic Acids Resear c h 40.10 (May 2012), e72. I S S N : 1362-4962. D O I : 10.1093/nar/gks001 . [4] Åsa K Björklund et al. “The heterogeneity of human CD127(+) innate lymphoid cells re vealed by single-cell RN A sequencing”. eng. In: Natur e immunology 17.4 (Apr . 2016), pp. 451–460. I S S N : 1529-2916. D O I : 10. 1038 / ni .3368 . U R L : https: / / doi .org / 10 . 1038/ ni . 3368 (visited on 03/13/2026). [5] Gaëlle Breton et al. “Human dendritic cells (DCs) are deriv ed from distinct circulating precursors that are precommitted to become CD1c+ or CD141+ DCs”. eng. In: The J our - nal of experimental medicine 213.13 (Dec. 2016), pp. 2861–2870. I S S N : 1540-9538. D O I : 10 . 1084 / jem . 20161135 . U R L : https : / / europepmc . org / articles / PMC5154947 (visited on 03/13/2026). [6] J Gray Camp et al. “Human cerebral organoids recapitulate gene expression programs of fetal neocortex de v elopment”. eng. In: Pr oceedings of the National Academy of Sciences of the United States of America 112.51 (Dec. 2015), pp. 15672–15677. I S S N : 1091-6490. D O I : 10 . 1073 /pnas . 1520760112 . U R L : https : / /europepmc . org / articles / PMC4697386 (visited on 03/13/2026). [7] Y i-Ling Chen et al. “Re-evaluation of human BDCA-2+ DC during acute sterile skin inflam- mation”. eng. In: The Journal of e xperimental medicine 217.3 (Mar . 2020), e20190811. I S S N : 1540-9538. D O I : 10 . 1084 / jem . 20190811 . U R L : https : / / europepmc . org/ articles / PMC7062525 (visited on 03/13/2026). [8] W oosung Chung et al. “Single-cell RN A-seq enables comprehensi ve tumour and immune cell profiling in primary breast cancer”. eng. In: Nature communications 8 (May 2017), p. 15081. I S S N : 2041-1723. D O I : 10 . 1038 / ncomms15081 . U R L : https : / / europepmc . org/articles/PMC5424158 (visited on 03/13/2026). [9] A. P . DA WID. “Some matrix-variate distribution theory: Notational considerations and a Bayesian application”. In: Biometrika 68.1 (Apr . 1981), pp. 265–274. I S S N : 0006-3444. D O I : 10 . 1093 / biomet / 68 . 1 . 265 . U R L : https : / / doi . org / 10 . 1093 / biomet / 68 . 1 . 265 (visited on 02/26/2026). [10] Juliane C. Dohm et al. “Substantial biases in ultra-short read data sets from high-throughput DN A sequencing”. In: Nucleic Acids Researc h 36.16 (Sept. 2008), e105. I S S N : 0305-1048. D O I : 10.1093 /nar/ gkn425 . U R L : https:/ /doi. org/ 10 .1093 /nar/ gkn425 (visited on 03/04/2026). [11] Auda A Eltahla et al. “Linking the T cell receptor to the single cell transcriptome in antigen- specific human T cells”. eng. In: Immunology and cell biology 94.6 (July 2016), pp. 604–611. I S S N : 1440-1711. D O I : 10. 1038/ icb. 2016. 16 . U R L : https: // doi. org/ 10 .1038 /icb . 2016.16 (visited on 03/13/2026). 10 [12] Jean F an et al. “Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RN A-seq data”. eng. In: Genome resear ch 28.8 (Aug. 2018), pp. 1217–1227. I S S N : 1549-5469. D O I : 10 . 1101 / gr . 228080 . 117 . U R L : https : / / europepmc . org / articles/PMC6071640 (visited on 03/13/2026). [13] Joannah R Fergusson et al. “Maturing Human CD127+ CCR7+ PDL1+ Dendritic Cells Express AIRE in the Absence of Tissue Restricted Antigens”. eng. In: F r ontiers in immunology 9 (Jan. 2018), p. 2902. I S S N : 1664-3224. D O I : 10.3389/fimmu.2018.02902 . U R L : https: //europepmc.org/articles/PMC6340304 (visited on 03/13/2026). [14] Cornelius Fischer et al. “Signals trigger state-specific transcriptional programs to support div ersity and homeostasis in immune cells”. eng. In: Science signaling 12.581 (May 2019), eaao5820. I S S N : 1937-9145. D O I : 10 . 1126 / scisignal . aao5820 . U R L : https : / / doi . org/10.1126/scisignal.aao5820 (visited on 03/13/2026). [15] T obias Gerber et al. “Mapping heterogeneity in patient-deriv ed melanoma cultures by single- cell RN A-seq”. eng. In: Oncotar get 8.1 (Jan. 2017), pp. 846–862. I S S N : 1949-2553. D O I : 10. 18632 / oncotarget . 13666 . U R L : https : / / europepmc . org / articles / PMC5352202 (visited on 03/13/2026). [16] Monica Golumbeanu et al. “Single-Cell RN A-Seq Reveals T ranscriptional Heterogeneity in Latent and Reacti vated HIV-Infected Cells”. eng. In: Cell r eports 23.4 (Apr . 2018), pp. 942– 950. I S S N : 2211-1247. D O I : 10.1016/j.celrep.2018.03.102 . U R L : https://doi.org/ 10.1016/j.celrep.2018.03.102 (visited on 03/13/2026). [17] Lara Hemeryck et al. “Or ganoids from human tooth sho wing epithelial stemness phenotype and differentiation potential”. eng. In: Cellular and molecular life sciences 79.3 (Feb. 2022), p. 153. I S S N : 1420-9071. D O I : 10.1007/s00018- 022- 04183- 8 . U R L : https://europepmc.org/ articles/PMC8881251 (visited on 03/13/2026). [18] Aaron M Horning et al. “Single-Cell RN A-seq Reveals a Subpopulation of Prostate Cancer Cells with Enhanced Cell-Cycle-Related T ranscription and Attenuated Androgen Response”. eng. In: Cancer r esear c h 78.4 (Feb. 2018), pp. 853–864. I S S N : 1538-7445. D O I : 10. 1158/ 0008 - 5472 . can - 17 - 1924 . U R L : https : / / europepmc . org / articles / PMC5983359 (visited on 03/13/2026). [19] Laura Jardine et al. “Blood and immune dev elopment in human fetal bone marrow and Do wn syndrome”. eng. In: Natur e 598.7880 (Oct. 2021), pp. 327–331. I S S N : 1476-4687. D O I : 10. 1038 / s41586 - 021 - 03929 - x . U R L : https : / / europepmc . org / articles / PMC7612688 (visited on 03/13/2026). [20] Nicole V incent Jordan et al. “HER2 e xpression identifies dynamic functional states within circulating breast cancer cells”. eng. In: Natur e 537.7618 (Sept. 2016), pp. 102–106. I S S N : 1476-4687. D O I : 10 . 1038 / nature19328 . U R L : https : / / europepmc . org / articles / PMC5161614 (visited on 03/13/2026). [21] Alfredo Kalaitzis et al. “The Bigraphical Lasso”. en. In: Pr oceedings of the 30th International Confer ence on Machine Learning . PMLR, May 2013, pp. 1229–1237. U R L : https : / / proceedings.mlr.press/v28/kalaitzis13.html (visited on 03/12/2026). [22] Dimitris Karamitros et al. “Single-cell analysis reveals the continuum of human lympho- myeloid progenitor cells”. eng. In: Natur e immunology 19.1 (Jan. 2018), pp. 85–97. I S S N : 1529-2916. D O I : 10 . 1038 / s41590 - 017 - 0001 - 2 . U R L : https : / / europepmc . org / articles/PMC5884424 (visited on 03/13/2026). [23] Joakim Karlsson et al. “Transcriptomic Characterization of the Human Cell Cycle in Individual Unsynchronized Cells”. eng. In: Journal of molecular biolo gy 429.24 (Dec. 2017), pp. 3909– 3924. I S S N : 1089-8638. D O I : 10.1016/j.jmb.2017.10.011 . U R L : https://doi.org/10. 1016/j.jmb.2017.10.011 (visited on 03/13/2026). [24] Kyu-T ae Kim et al. “Application of single-cell RN A sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma”. eng. In: Genome biology 17 (Apr . 2016), p. 80. I S S N : 1474-760X. D O I : 10 . 1186 / s13059 - 016 - 0945 - 9 . U R L : https : //europepmc.org/articles/PMC4852434 (visited on 03/13/2026). [25] Marios Koutsakos et al. “Human CD8 + T cell cross-reactivity across influenza A, B and C viruses”. eng. In: Natur e immunology 20.5 (May 2019), pp. 613–625. I S S N : 1529-2916. D O I : 10.1038/s41590- 019- 0320- 6 . U R L : https://doi. org/10.1038/ s41590- 019- 0320- 6 (visited on 03/13/2026). 11 [26] Han Liu et al. “The nonparanormal skeptic”. In: Pr oceedings of the 29th International Cofer - ence on International Confer ence on Machine Learning . ICML’12. Madison, WI, USA: Omnipress, June 2012, pp. 1731–1738. I S B N : 978-1-4503-1285-1. (V isited on 03/12/2026). [27] Shutong Liu et al. “Single cell sequencing rev eals gene expression signatures associated with bone marro w stromal cell subpopulations and time in culture”. eng. In: Journal of translational medicine 17.1 (Jan. 2019), p. 23. I S S N : 1479-5876. D O I : 10 . 1186 / s12967 - 018 - 1766- 2 . U R L : https://europepmc.org/articles/PMC6330466 (visited on 03/13/2026). [28] Junjie Lu et al. “Single-cell RN A sequencing rev eals metallothionein heterogeneity during hESC dif ferentiation to definiti ve endoderm”. eng. In: Stem cell r esear ch 28 (Apr . 2018), pp. 48–55. I S S N : 1876-7753. D O I : 10 . 1016 / j . scr .2018 . 01 .015 . U R L : https : / / doi . org/10.1016/j.scr.2018.01.015 (visited on 03/13/2026). [29] Hassaan Maan et al. “Characterizing the impacts of dataset imbalance on single-cell data integration”. en. In: Nature Biotechnology 42.12 (Dec. 2024), pp. 1899–1908. I S S N : 1546-1696. D O I : 10 . 1038 / s41587 - 023 - 02097 - 9 . U R L : https : / / www . nature . com / articles / s41587- 023- 02097- 9 (visited on 03/24/2026). [30] Pedro Madrigal et al. “Expression Atlas in 2026: enabling F AIR and open e xpression data through community collaboration and inte gration”. In: Nucleic Acids Resear ch 54.D1 (Jan. 2026), pp. D147–D157. I S S N : 1362-4962. D O I : 10 . 1093 / nar / gkaf1238 . U R L : https : //doi.org/10.1093/nar/gkaf1238 (visited on 03/12/2026). [31] Louisa Nelson et al. “A living biobank of ov arian cancer e x vi vo models reveals profound mitotic heterogeneity”. eng. In: Natur e Communications 11.1 (Feb . 2020), p. 822. I S S N : 2041-1723. D O I : 10.1038/s41467- 020- 14551- 2 . [32] Quy H Nguyen et al. “Profiling human breast epithelial cells using single cell RN A sequencing identifies cell diversity”. eng. In: Natur e communications 9.1 (May 2018), p. 2028. I S S N : 2041-1723. D O I : 10 . 1038 / s41467 - 018 - 04334 - 1 . U R L : https : / / europepmc . org / articles/PMC5966421 (visited on 03/13/2026). [33] M Joseph Phillips et al. “A Nov el Approach to Single Cell RN A-Sequence Analysis F acilitates In Silico Gene Reporting of Human Pluripotent Stem Cell-Derived Retinal Cell T ypes”. eng. In: Stem cells (Dayton, Ohio) 36.3 (Mar . 2018), pp. 313–324. I S S N : 1549-4918. D O I : 10 . 1002 / stem . 2755 . U R L : https : / / europepmc . org / articles / PMC5823737 (visited on 03/13/2026). [34] V aleria Policastro et al. “R OBustness In Netw ork (robin): an R Package for Comparison and V alidation of Communities”. In: The R J ournal 13.1 (June 2021), pp. 276–293. I S S N : 2073- 4859. D O I : 10.32614/RJ- 2021- 040 . U R L : https://doi.org/10.32614/RJ- 2021- 040/ (visited on 03/12/2026). [35] Florian Rambo w et al. “T ow ard Minimal Residual Disease-Directed Therapy in Melanoma”. eng. In: Cell 174.4 (Aug. 2018), 843–855.e19. I S S N : 1097-4172. D O I : 10 . 1016 / j . cell . 2018 . 06 . 025 . U R L : https : / / doi . org / 10 . 1016 / j . cell . 2018 . 06 . 025 (visited on 03/13/2026). [36] Luke T ierne y and Joseph B. Kadane. “ Accurate Approximations for Posterior Moments and Marginal Densities”. In: J ournal of the American Statistical Association 81.393 (Mar . 1986). _eprint: https://www .tandfonline.com/doi/pdf/10.1080/01621459.1986.10478240, pp. 82– 86. I S S N : 0162-1459. D O I : 10 . 1080 / 01621459 . 1986 . 10478240 . U R L : https : / / www . tandfonline . com / doi / abs / 10 . 1080 / 01621459 . 1986 . 10478240 (visited on 03/12/2026). [37] V . A. T raag, L. W altman, and N. J. van Eck. “From Louv ain to Leiden: guaranteeing well- connected communities”. en. In: Scientific Reports 9.1 (Mar . 2019), p. 5233. I S S N : 2045-2322. D O I : 10 . 1038 / s41598 - 019 - 41695 - z . U R L : https : / / www . nature . com / articles / s41598- 019- 41695- z (visited on 03/12/2026). [38] David E. T yler. “A Distrib ution-Free $M$-Estimator of Multi variate Scatter”. en. In: The Annals of Statistics 15.1 (Mar . 1987), pp. 234–251. I S S N : 0090-5364, 2168-8966. D O I : 10 . 1214 / aos / 1176350263 . U R L : https : / / projecteuclid . org / journals / annals - of - statistics / volume - 15 / issue - 1 / A - Distribution - Free - M - Estimator - of - Multivariate- Scatter/10.1214/aos/1176350263.full (visited on 03/12/2026). 12 [39] Y u W ang, Byoungwook Jang, and Alfred Hero. “The Sylv ester Graphical Lasso (SyGlasso)”. en. In: Pr oceedings of the T wenty Thir d International Confer ence on Artificial Intellig ence and Statistics . PMLR, June 2020, pp. 1943–1953. U R L : https : / / proceedings . mlr . press / v108/wang20d.html (visited on 03/12/2026). [40] Y ue J W ang et al. “Single-Cell T ranscriptomics of the Human Endocrine P ancreas”. eng. In: Diabetes 65.10 (Oct. 2016), pp. 3028–3038. I S S N : 1939-327X. D O I : 10.2337/db16- 0405 . U R L : https://europepmc.org/articles/PMC5033269 (visited on 03/13/2026). [41] Y an Xu et al. “Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis”. eng. In: JCI insight 1.20 (Dec. 2016), e90558. I S S N : 2379- 3708. D O I : 10. 1172/ jci. insight. 90558 . U R L : https: // europepmc. org/ articles/ PMC5135277 (visited on 03/13/2026). [42] Liying Y an et al. “Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells”. eng. In: Natur e structural & molecular biology 20.9 (Sept. 2013), pp. 1131–1139. I S S N : 1545-9985. D O I : 10.1038/nsmb.2660 . U R L : https://doi.org/10. 1038/nsmb.2660 (visited on 03/13/2026). [43] Xin Zhao et al. “Single-cell RN A-seq re veals a distinct transcriptome signature of aneuploid hematopoietic cells”. eng. In: Blood 130.25 (Dec. 2017), pp. 2762–2773. I S S N : 1528-0020. D O I : 10 . 1182 / blood - 2017 - 08 - 803353 . U R L : https : / / europepmc . org / articles / PMC5746166 (visited on 03/13/2026). A Synthetic results f or 200 × 200 datasets 0.0 0.2 0.4 0.6 0.8 1.0 Noise str ength 0.0 0.2 0.4 0.6 0.8 1.0 AUPR MED-MA GMA GmGM Basic 0.0 0.2 0.4 0.6 0.8 1.0 Noise str ength 0.0 0.2 0.4 0.6 0.8 1.0 Assortativity MED-MA GMA GmGM Basic 0.0 0.2 0.4 0.6 0.8 1.0 Noise str ength 0.0 0.2 0.4 0.6 0.8 1.0 AMI MED-MA GMA GmGM Basic Figure 5: (Both) Results across 20 synthetic Kronecker -sum-structured Gaussian 200 × 200 datasets corrupted by v arying lev els of multiplicati ve noise. Median result is highlighted. As noise strength varies, we plot the area under PR curv es (left), assortati vity (center), and AMI (right). W e giv e the results for 200 × 200 synthetic datasets, generated the same way as in Section 4.1, in Figure 5. This was omitted from the main paper for space reasons; we include it here to demonstrate that our method works re gardless of whether the data is lopsided or more square. The basic single- axis baseline performs poorly on this test; this is because such methods work best when one has sufficiently more samples relati v e to features, whereas here we hav e equal samples/features. B Proof that f remov es only the corrupted directions In this section, we will demonstrate that our definition of f (Equation 1) removes all directions corrupted by noise from our data, and nothing else. Proposition (Goodness of f ) . The function f described in Equation 1 has the following pr operty: f ( X ) = f ( Y ) if and only if one can find strictly positive r rows , r cols such that we have v ec [ X ] = ( r cols ⊗ r rows ) ◦ v ec [ Y ] . Pr oof. Recall the following direction of f : 13 f ( X ) ij = x ij Q i ′ j ′ x i ′ j ′ 1 d rows d cols Q j ′ x ij ′ 1 d cols Q i ′ x i ′ j 1 d rows (Equation 1) Note that this has the following equi v alent, more complicated formulation: f ( X ) ij = sgn ( x ij ) exp( log | x ij | + 1 d rows d cols X i ′ j ′ log | x i ′ j ′ | − 1 d rows X i ′ log | x i ′ j | − 1 d cols X j ′ log | x ij ′ | ) Letting L = log ◦ | X | be the elementwise logarithm of the absolute v alues of X , we can see that the core of our function is ultimately just a linear function on L , L : L ( L ) ij = ℓ ij + mean( L ) − row means ( L ) i − col means ( L ) j This is a well-known function (double-centering of a matrix) and it is easy to e xpress it in matrix form: L ( L ) = L + 1 T L1 d cols d rows 11 T − 1 d cols L1 1 T − 1 1 d rows 1 T L = L − L 1 d cols 11 T − 1 d rows 11 T L + 1 d rows 11 T L 1 d cols 11 T = L I − 1 d cols 11 T + 1 d rows 11 T L I − 1 d cols 11 T = I − 1 d rows 11 T L I − 1 d cols 11 T In other words: f ( X ) = sgn ( X ) ◦ exp ◦ I − 1 d rows 11 T log ◦ | X | I − 1 d cols 11 T f ( x ) = sgn ( x ) ◦ exp ◦ I − 1 d cols 11 T ⊗ I − 1 d rows 11 T log ◦ | x | (where x = v ec [ X ] ) The benefit of expressing our function in this form is that we can fully describe what information is lost and kept by the eigenv ectors of h I − 1 d cols 11 T i ⊗ h I − 1 d rows 11 T i . This is the Kronecker product of two centering matrices - the n × n centering matrix has a single zero eigenv alue, with the remaining n − 1 eigen v alues being 1. The eigen v alues of a Kronecker product are all the products of the eigen v alues, giving d rows + d cols − 1 zero eigen v alues (the rest being one). If we can show that our noise gets mapped to zero and spans a ( d rows + d cols − 1) -dimensional subspace, we will ha ve prov en that our function works. log ◦ | r ( r cols ⊗ r rows ) ◦ x | = log ◦ r cols ⊕ log ◦ r rows + log ◦ | x | + log( r ) 1 (Noise is linear in logspace) 14 I − 1 d cols 11 T ⊗ I − 1 d rows 11 T (log ◦ r cols ⊕ log ◦ r rows + log( r ) 1 ) = I − 1 d cols 11 T log ◦ r cols ⊗ I − 1 d rows 11 T + I − 1 d cols 11 T ⊗ I − 1 d rows 11 T log ◦ r rows + log( r ) I − 1 d cols 11 T 1 ⊗ I − 1 d rows 11 T 1 Our constraint that Q i r ( ℓ ) i = 1 is equi valent to P i log r ( ℓ ) i = 0 . In other w ords, in logspace our vectors ha ve mean zero, and are thus annihilated by the centering matrix. The whole equation then becomes zero: our noise is indeed removed by our function. Finally , note that we parameterize r , r rows and r cols with 1 + d rows + d cols parameters. Howe ver , our two geometric mean constraints further decrease the dimensionality; our noise is parameterized by d rows + d cols − 1 parameters. This is exactly the size of the nullspace of our double-centering matrix, guaranteeing that only noise-corrupted directions are remov ed from x . This completes the proof C A version of f that handles zero v alues 0.0 0.2 0.4 0.6 0.8 1.0 Density 0.0 0.2 0.4 0.6 0.8 1.0 AUPR MED-MA GMA GmGM 0.0 0.2 0.4 0.6 0.8 1.0 Noise str ength 0.0 0.2 0.4 0.6 0.8 1.0 AUPR MED-MA GMA GmGM Figure 6: (Both) Area under PR curves across 20 synthetic 200 × 201 Kronecker -sum-structured Gaussian datasets corrupted by v arying le vels of multiplicati v e noise and sparsity . Median result is highlighted. (Left) As percentage of nonzero entries v ary (noise strength 0.2) (Right) Area under PR curves as noise strength v aries (density 0.5). When the input X contains zeros, the function f breaks - either one must di vide by zero or take the logarithm of it. Howe ver , zeros are unaffected by multiplicati ve noise; intuiti vely , they should not cause such a catastrophic failure. W e use the obvious modification (with Figure 6 demonstrating the performance): we will skip o ver the zero entries when doing products. Let nnz be a function that counts the number of nonzero entries in a v ector , and Q =0 be a product over only nonzero terms; we hav e the follo wing: f ( X ) ij = x ij Q =0 i ′ j ′ x i ′ j ′ 1 nnz( x ) Q =0 j ′ x ij ′ 1 nnz ( x row i ) Q =0 i ′ x i ′ j 1 nnz ( x col j ) D Proof that optimizing Q is equi valent to GmGM Recall the problem of optimizing Q : 15 let Q Ω Ω ( t ) = E z ∼N 0 , Ω − 1 ( t ) and z ∈ f − 1 ( y ) [log p df N ( z | Ω )] Ω ( t +1) = argmax Ω ∈K ⊕ ++ Q Ω Ω ( t ) (iterate until con ver gence) As claimed in the main paper, maximization of Q requires solving the same optimization problem as GmGM. Below , let J ij be the matrix of zeros except for the ( i, j ) entry being one. Definition (GmGM [2]) . GmGM solves the con vex minimization pr oblem of minimizing the ne gative log likelihood, with gr adients: −∇ Ψ rows log p df N ( z | Ω ) = XX T − tr Ω − 1 J ij ⊗ I ij −∇ Ψ cols log p df N ( z | Ω ) = X T X − tr Ω − 1 I ⊗ J ij ij It only r elies on the data thr ough the terms XX T and X T X ; we will denote minimum point as GmGM XX T , X T X . Proposition (Maximizing Q ) . Optimizing Q corr esponds to the same optimization problem as GmGM . GmGM uses the sufficient statistics XX T and XX T . T o maximize Q , we r eplace these with: S rows = E z ∼N ( 0 , Ω − 1 ( t ) ) z ∈ f − 1 ( y ) ZZ T and S cols = E z ∼N ( 0 , Ω − 1 ( t ) ) z ∈ f − 1 ( y ) Z T Z Mor e gener ally , any method to fit unr e gularized Kr onec ker -sum-structur ed models without noise that operates on the suf ficient statistics can be used in place of GmGM. Pr oof. W e will first show that the gradient w .r .t. Ψ rows yields a very similar gradient to GmGM; the argument is analogous for Ψ cols . Note also that Q is being maximized, whereas GmGM frames the problem as a minimization problem, so we expect the gradients to ha ve opposite signs. ∇ Ψ rows Q Ω Ω ( t ) = ∇ Ψ rows E z ∼N 0 , Ω − 1 ( t ) and z ∈ f − 1 ( y ) [log p df N ( z | Ω )] = E z ∼N 0 , Ω − 1 ( t ) and z ∈ f − 1 ( y ) [ ∇ Ψ rows log p df N ( z | Ω )] (By dominated con ver gence theorem) = E z ∼N 0 , Ω − 1 ( t ) and z ∈ f − 1 ( y ) h tr Ω − 1 J ij ⊗ I ij − ZZ T i (GmGM definition) = tr Ω − 1 J ij ⊗ I ij − E z ∼N 0 , Ω − 1 ( t ) and z ∈ f − 1 ( y ) ZZ T = tr Ω − 1 J ij ⊗ I ij − S rows ( S rows definition) It should no w be clear by inspection that the maximum of Q corresponds with GmGM ( S rows , S cols ) . This completes the proof. E Efficient ev aluation of 1 2 tr h PΩ ( t ) P T − 1 P ( ∇ 2 g ( z ∗ )) P T i W e will assume g ( z ) = ZZ T , as g ( z ) = Z T Z follows analogously . As in the previous section, let J ( ij ) be the matrix of zeros except for the ( i, j ) entry being one. Let us start by deri ving the Hessian of g , by in vestigating the directional deri v ativ e: 16 ∇ J ( kℓ ) ∇ J ( ij ) ZZ T = ∇ J ( kℓ ) ZJ T ( ij ) + J ( ij ) Z T = J ( kℓ ) J T ( ij ) + J ( ij ) J T ( kℓ ) = J ( kℓ ) J ( j i ) + J ( ij ) J ( ℓk ) = δ j ℓ J ( ki ) + J ( ik ) ∇ J ( kℓ ) ∇ J ( ij ) ZZ T ab = δ j ℓ ( δ ak δ bi + δ ai δ bk ) ∇ 2 v ec ZZ T = I d cols ⊗ J ( ab ) d rows + J ( ba ) d rows W e will no w work out the matrix P which projects onto the tangent space of the fiber . As we hav e seen in Section B, the fiber is linear -in-logspace with d rows + d cols − 1 basis vectors of the form v cols ⊕ v rows in which 1 T v rows = 0 = 1 T v cols . Note then that: 1 d rows I ⊗ 1 T 1 d cols 1 T ⊗ I ( v cols ⊕ v rows ) = " v cols + 1 T v rows d rows 1 d rows v rows + 1 T v cols d cols 1 # = v cols v rows Thus P = 1 d rows I ⊗ 1 T 1 d cols 1 T ⊗ I projects onto a d rows + d cols space, from which the tangent space can be reached by just dropping the last row from the matrix. It should be clear that P ∇ 2 g ( z ) P T is easy , as all matrices in volv ed are conformable block matrices of Kronecker products. Note that the 1 d cols and 1 d rows factors cancel out in the trace expression, so we will drop them: P = I ⊗ 1 T 1 T ⊗ I . All that remains is to efficiently find the in verse of PΩ ( t ) P T . Note the following: PΩ ( t ) P T = Ψ cols ⊗ 1 T + I ⊗ 1 T Ψ rows 1 T Ψ cols ⊗ I + 1 T ⊗ Ψ rows P T = d rows Ψ cols + 1 T Ψ rows 1 I Ψ cols 1 ⊗ 1 T + 1 ⊗ 1 T Ψ rows 1 T Ψ cols ⊗ 1 + 1 T ⊗ Ψ rows 1 d cols Ψ rows + 1 T Ψ cols 1 I Removing the last row of P amounts to remo ving the last row and column of this matrix. It can easily be in verted by the block matrix in version formula, requiring inv ersions of only d ℓ × d ℓ matrices rather than a d rows d cols × d rows d cols matrix. Thus, our trace term can be evaluated ef ficiently . From this point on, all steps require standard (if tedious) linear algebra, which we leav e to the reader . Our code contains a working implementation for those who wish to reference it. F Description of the systematic approach to pr epr ocessing and ev aluating datasets As mentioned in the main paper, we systematically do wnloaded every dataset in the Single Cell Expression Atlas, subject to three criteria (belo w). One of the main difficulties in e v aluating methods is the ease in which authors can (intentionally or unintentionally) end up cherry-picking e xamples that make their model look good. By being systematic, we can guarantee that our results represent the typical outputs of our model. Criterion 1. The dataset must be at most 1000 cells large. W e chose this threshold as it was lar ge enough to include man y datasets (40 before applying the remaining criteria) while still including only datasets small enough for our algorithm to run quickly on. It is also a nice round number . The next ‘nice round number’, in our opinion, is 5000 - which w ould be 63 datasets. As our algorithm scales cubically , the largest datasets would take 125 times as long as before. 17 Criterion 2. The dataset must contain at least one categorical variable. This is necessary to have a ground truth to compare against for AMI and assortativity . When there were multiple, we picked the first one we noticed; these are giv en in Appendix G. Criterion 3. The dataset must clearly reference a source paper in the atlas. This rule was for our con venience; we did not w ant to waste time hunting do wn a paper to cite for the dataset. 30 datasets satisfied all criteria. W e then had to design a standard preprocessing regime that worked for all datasets, choosing to do the minimal preprocessing necessary to get the algorithm to run. As such, our only requirement was to subset the total number of genes in each dataset; by default, the y all had tens of thousands. W e chose to pick the genes so that each dataset became roughly square, to av oid potentially introducing ‘extreme lopsidedness’ as a confounding factor . Our process was as follows: Step 1. Select the top 2000 most v ariable genes. This is a v ery common default preprocessing step and threshold. Step 2. Iterate from s = 1% to s = 100% in increments of 1% . For each v alue of s , select only genes with at least s nonzero v alues. Whichever value of s leads to the most square matrix, pick those genes. W e chose to pick genes based on sparsity to mirror another standard preprocessing step (throw out ‘low-quality’ genes that do not appear much), and we quantized our v alues of s to ensure that not all of our matrices were perfectly square (which might also confound the results). G Description of datasets used A brief description and citation has been giv en in T able 1 for e v ery used dataset. Note that E-CURD-7 and E-EN AD-21 appear to be the same dataset; it is not a mistake. Both appear separately in the scRN A-seq database; a dif ferent categorical v ariable has been used for each. 18 Dataset Categorical V ariable Categories Cells Genes E-MT AB-4850 [11] Sample Characteristic[cell line] 2 60 63 E-GEOD-75367 [20] Sample Characteristic[facs marker] 4 72 70 E-MT AB-6142 [23] Sample Characteristic[cell cycle phase] 3 96 91 E-GEOD-36552 [42] Sample Characteristic[cell type] 6 115 127 E-CURD-10 [24] Sample Characteristic[growth condition] 3 118 104 E-GEOD-99795 [18] Factor V alue[compound] 2 144 128 E-GEOD-110499 [12] Sample Characteristic[organism part] 2 168 169 E-MT AB-7249 [31] Sample Characteristic [inferred cell type - authors labels] 7 185 185 E-MT AB-7606 [25] Sample Characteristic[phenotype] 2 209 203 E-GEOD-81383 [15] Sample Characteristic[cell line] 3 226 226 E-MT AB-6075 [14] F actor V alue[compound] 3 236 231 E-GEOD-124858 [27] Factor V alue[passage] 5 241 239 E-MT AB-7381 [13] Sample Characteristic[individual] 3 287 291 E-GEOD-111727 [16] Sample Characteristic[disease] 2 320 324 E-GEOD-109979 [28] Factor V alue[time] 4 329 324 E-GEOD-100618 [22] Sample Characteristic[cell type] 3 414 418 E-MT AB-8498 [7] Sample Characteristic[cell type] 3 448 452 E-MT AB-9801 [19] Sample Characteristic [inferred cell type - authors labels] 12 484 501 E-GEOD-75688 [8] Sample Characteristic[histology] 4 520 517 E-GEOD-86618 [41] Sample Characteristic[disease] 2 537 538 E-GEOD-98556 [33] Sample Characteristic[cell type] 2 545 550 E-GEOD-83139 [40] Sample Characteristic [inferred cell type - ontology labels] 8 617 612 E-CURD-7 [32] Sample Characteristic[sampling site] 2 631 627 E-EN AD-21 [32] Sample Characteristic[cell type] 2 631 627 E-GEOD-70580 [4] Sample Characteristic[cell type] 4 648 643 E-EN AD-20 [35] Factor V alue[time] 4 667 673 E-GEOD-75140 [6] Sample Characteristic[cell type] 3 734 728 E-GEOD-89232 [5] Sample Characteristic[cell type] 2 957 926 E-CURD-6 [43] Sample Characteristic[disease] 4 986 1031 E-MT AB-10596 [17] Sample Characteristic[growth condition] 3 993 984 T able 1: All datasets analyzed during Section 4.2. The ‘genes’ column counts genes after preprocess- ing. 19
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment