A Priori Sampling of Transition States with Guided Diffusion
Transition states, the first-order saddle points on the potential energy surfaces, govern the kinetics and mechanisms of chemical reactions and conformational changes. Locating them is challenging because transition pathways are topologically complex…
Authors: Hyukjun Lim, Soojung Yang, Lucas Pinède
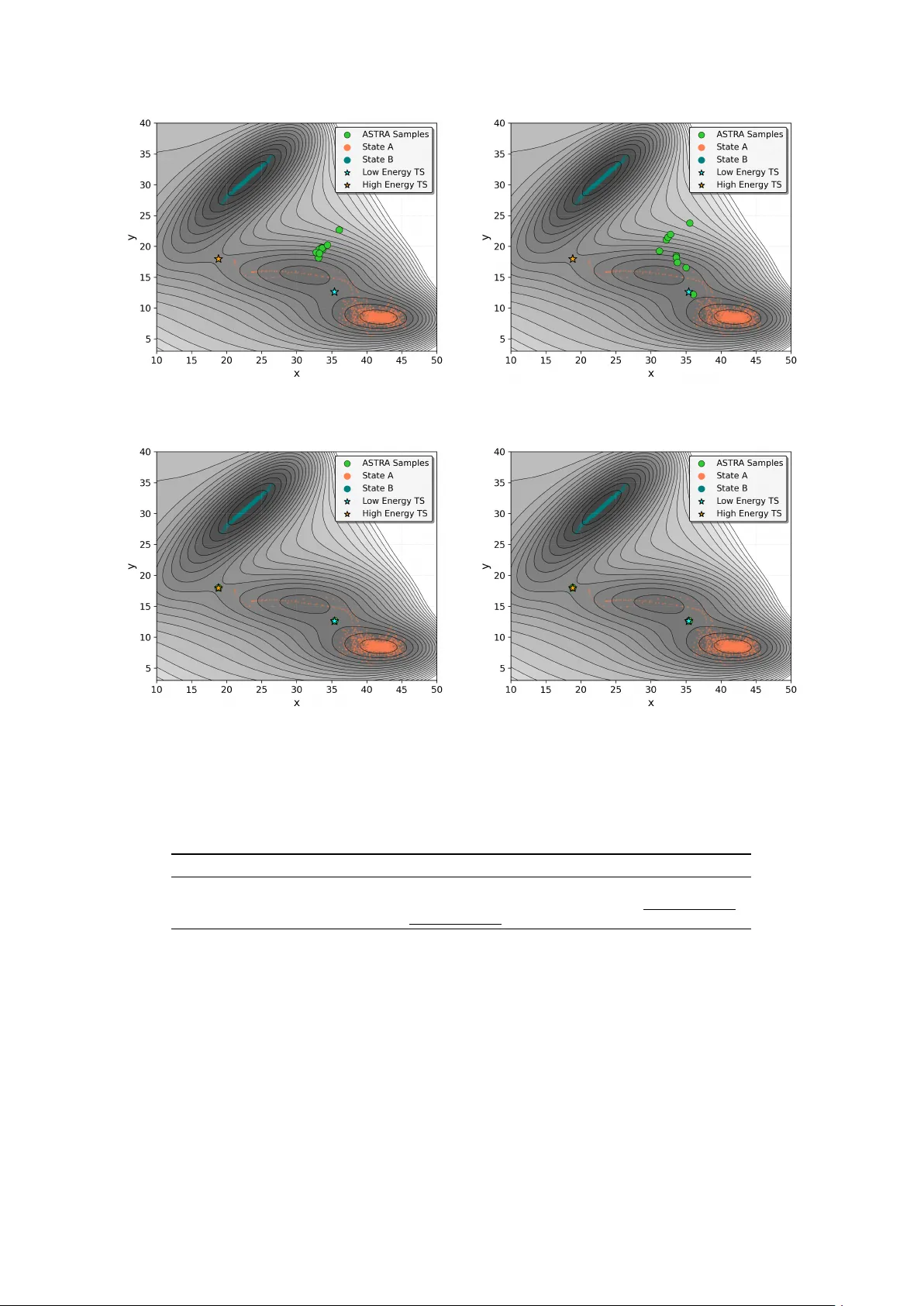
A Priori Sampling of T ransition States with Guided Diffusion Hyukjun Lim 1 † , So o jung Y ang 2* † , Lucas Pinède 3 † , Miguel Steiner 4 , Y uanqi Du 5 , Rafael Gómez-Bom barelli 4* 1 Departmen t of Materials Science and Engineering, Seoul National Univ ersity , 1 Gw anak-ro, Gwanak-gu, Seoul, 08826, Republic of Korea. 2 Computational and Systems Biology , MIT, 77 Massach usetts A ven ue, Cambridge, 02139, Massac husetts, USA. 3 Chimie P arisT ech, PSL Univ ersity , 11 rue Pierre et Marie Curie, P aris, 75005, F rance. 4 Departmen t of Materials Science and Engineering, MIT, 77 Massac husetts A ven ue, Cam bridge, 02139, Massac husetts, USA. 5 Departmen t of Computer Science, Cornell Universit y , 127 Hoy Road, Ithaca, 14850, NY, USA. *Corresp onding author(s). E-mail(s): so o jungy@mit.edu ; rafagb@mit.edu ; Con tributing authors: h yukjunlim@snu.ac.kr ; lucas.pinede@etu.c himieparistech.psl.eu ; steinmig@mit.edu ; yd392@cornell.edu ; † These authors contributed equally to this w ork. Abstract T ransition states, the first-order saddle p oin ts on the p oten tial energy surfaces, gov ern the kinetics and mec hanisms of c hemical reactions and conformational changes. Lo cating them is challenging b ecause transition path wa ys are topologically complex and can pro ceed via an ensem ble of div erse routes. Existing metho ds address these challenges by in tro ducing heuristic assumptions about the pathw ay or reaction co ordinates, which limits their applicabilit y when a go od initial guess is una v ailable or when the guess precludes alternative, potentially relev ant pathw ays. W e propose to bypass such heuristic limitations b y introducing ASTRA, A Priori S ampling of TRA nsition States with Guided Diffusion, which reframes the transition state search as an inference-time scaling problem for generative mo dels. ASTRA trains a score-based diffusion mo del on configura- tions from kno wn metastable states. Then, ASTRA guides inference tow ard the isodensity surface separating the basins of metastable states via a principled composition of conditional scores. A Score-Aligned Ascent (SAA) process then appro ximates a reaction co ordinate from the differ- ence b et ween conditioned scores and combines it with ph ysical forces to drive conv ergence onto first-order transition states. V alidated on b enc hmarks from 2D p oten tials to biomolecular confor- mational c hanges and chemical reaction, ASTRA lo cates transition states with high precision and disco vers multiple reaction pathw ays, enabling mechanistic studies of complex molecular systems. Keyw ords: T ransition State Search, Score-Based Diffusion Mo dels, Guided Diffusion, Computational Chemistry 1 1 In tro duction Understanding the dynamics of molecular systems, from c hemical reactions to protein folding, requires c haracterizing their transition states (TSs) [ 1 , 2 ]. Defined as first-order saddle p oin ts on the p oten- tial energy surface (PES), TSs represen t the highest energy configurations along a minimum energy path and act as kinetic b ottlenec ks that determine reaction rates and mechanisms. Despite their cen tral imp ortance, lo cating TSs remains notoriously difficult: they are to o short-lived for atomistic exp erimen tal c haracterization, and to o rarely visited for unbiased molecular dynamics simulation [ 3 ]. V arious enhanced sampling techniques hav e therefore b een developed to circumv ent these timescale limitations [ 4 – 7 ], but their efficacy relies on defining collective v ariables (CV s) that accurately describ e the relev an t slo w degrees of freedom . Identifying appropriate CV s is challenging when the transi- tion mec hanism is unknown; requiring iterative refinemen t from an initial guess that can become computationally exp ensiv e. Time-indep enden t TS optimization (TSO) metho ds offer an alternativ e by finding individual TS structures and describing a series of transformations within the framework of TS theory [ 1 , 2 ], though their fo cus on individual structures initially limited their application to systems with limited con- formational flexibilit y . Over the past decade, these metho ds hav e b een increasingly automated and scaled up to mo del complex reaction netw orks across diverse c hemical systems [ 8 – 13 ]. How ever, TSO metho ds remain computationally in tensive, and require chemically informed initial guesses that are difficult to generalize across c hemical systems [ 14 ]. The reliance of b oth enhanced sampling MD and TSO metho ds on heuristics makes the TS search tedious and difficult to scale to complex systems. In recent y ears, data-driv en strategies hav e aimed to accelerate MD and TSO approac hes b y learn- ing CV s automatically [ 15 – 22 ], but the transferabilit y of learned manifolds remains limited for systems outside the training distribution – a limitation shared across data-driv en approac hes. A prominent example is the committor function — the probability of reaching the product state b efore the reactant from a giv en configuration — which can be learned from transition path data via maximum-lik eliho od metho ds [ 23 – 25 ], or from equilibrium and short-tra jectory MD data via v ariational [ 26 , 27 ] and self- consistency formulations [ 28 – 30 ]. In either case, success requires either sufficient transition path data or substan tial computational inv estment in iterative sampling [ 31 – 34 ] and activ e learning [ 35 , 36 ], and the need for iterative enhanced sampling to co ver the transition region p ersists regardless. Generativ e ML mo dels offer another alternative: conditional mo dels sample TSs given the reactant and product states [ 37 – 40 ], while unconditional mo dels target the equilibrium distribution [ 41 – 44 ] and require p ost-hoc iden tification of TSs. Both remain limited by their training data distribution. Recent w orks hav e partially addressed this by using geometric optimization and energy ev aluations during training to find transition paths and extract TSs without a pre-curated TS dataset [ 45 , 46 ] or b y lever- aging MD-derived metrics [ 47 ] and the Onsager–Mac hlup functional with a force field approximated from a generative mo del trained on equilibrium distribution to find transition paths [ 48 ]. Neverthe- less, these approaches still rely on either accurately learning the PES near the TS or constructing mappings to pre-computed TS distributions [ 48 , 49 ], limiting their utility to underexplored systems. T o go b ey ond sampling the learned distribution, inference-time control in diffusion mo dels offers a promising av enue for steering generative pro cesses tow ard desired regions of chemical and conforma- tional space. Steering the sampling of generative mo dels based on ob jectiv es, constraints, or rew ards has b een an active area of researc h, spanning earlier approaches suc h as classifier-free guidance [ 50 ] to rew ard-titled, annealed, and equal-densit y distribution sampling [ 51 – 53 ]. Ho wev er, steered sampling outside the training PES distribution ha ve not b een exte nsiv ely explored. Recent attempts to in te- grate enhanced sampling with generativ e mo dels remained limited b y p oor transferability to unseen systems or reliance on predefined CV s [ 54 ]. Given that diffusion mo dels can learn to sample div erse states and that atomistic simulations pro vide ground truth energetics, combining them offer a natural framew ork for TS ensem ble generation with low data requirements. W e in tro duce ASTRA ( A Priori S ampling of TRA nsition States with Guided Diffusion), a system- indep enden t workflo w to lo cate and optimize transition states of arbitrary molecular systems without iterativ e enhanced sampling, heuristics, or TS training data (see Figure 1 ). Our approac h com bines recen t adv ances in generativ e ML with principles from computational chemistry . The central idea is that a useful TS ensemble guess can b e inferred from the learned probability distributions of the sur- rounding metastable states. ASTRA therefore employs a conditionally trained score-based diffusion mo del [ 50 , 55 ] that learns the data manifold of reactan t and product basins from short MD tra jecto- ries. At inference, the rev erse diffusion pro cess is guided to sample configurations on the iso densit y surface where the probabilit y of belonging to either state is equal, via a principled comp osition of 2 conditional scores [ 52 ] – which w e term Score-Based Interpolation (SBI) . These samples are then refined b y force-based up dates on the PES, ascending along the reaction co ordinate and descending along orthogonal directions, leveraging our second key insight that the reaction coordinate can b e effectiv ely appro ximated from the conditional diffusion scores. This process, whic h w e call Score- Aligned Ascen t (SAA) , alleviates the need for the diffusion model to learn the PES p erfectly . Crucially , ASTRA op erates en tirely a priori , requiring only samples from the reactant and pro duct states with no knowledge of the transition region, yielding high-quality TS guesses ideally suited for rapid conv ergence with single-ended optimization metho ds. ASTRA is thus uniquely p ositioned to replace con ven tional double-ended TS guess methods in standard hierarchical search proto cols [ 56 – 59 ], pro viding a univ ersal and a priori approac h to saddle point lo cation. Our contributions are threefold: (1) W e prop ose a workflo w for direct TS sampling that leverages guided diffusion, eliminating the need for path-finding algorithms or prior TS data. (2) W e introduce the com bination of Score-Based Interpolation and Score-Aligned Ascen t as a principled mechanism for guiding a diffusion pro cess to sample first-order saddle p oin ts. (3) W e sho w that ASTRA successfully iden tifies kno wn transition states and competing path wa ys in systems ranging from t wo-dimensional p oten tials to high-dimensional chemical systems. Fig. 1 : Overview of ASTRA . The metho d consists of three stages: (1) training a conditional gener- ativ e model, (2) sampling from an iso densit y surface, and (3) inference-time guidance that com bines a reaction co ordinate appro ximated from score differences of the tw o conditional mo dels θ A , θ B ( R = S A θ − S B θ ) with physical forces to rapidly sample transition states defined as first-order saddle p oin ts. 2 Results T o assess the p erformance of the ASTRA algorithm rigorously , we established a unified ev aluation framew ork consistent across all test systems. This is designed to prob e the qualit y of the TS ensembles generated b y our metho d. F or each system, a score-based diffusion mo del is first trained on configura- tions, from kno wn metastable states, obtained from short MD tra jectories. The ASTRA algorithm is then e mplo y ed to generate a candidate TS ensemble. Detailed exp erimen tal parameters are provided in App endix C , and the results of all ablation studies are presented in Appendix H.1 . F or the 2D analytical potential energy surfaces, the ASTRA-generated samples are directly com- pared to the kno wn transition states using the L2 distance metric. In the higher-dimensional chemical systems, we quantify the quality of the samples with committor v alues. The committor function q ( x ) defines the probability that a tra jectory initiated from a given state x will reach the designated pro d- uct state b efore returning to the reactant state. By definition, the true TS ensemble corresp onds to the q ( x ) = 0 . 5 isosurface. W e compute this metric using a pretrained ML committor mo del or MD- based ev aluations when a force field is a v ailable (i.e. for all-atoms systems). In the latter case, w e appro ximate committor v alues by initiating multiple replicas of Langevin dynamics simulations from eac h structure with random initial velocities sampled from the Maxw ell–Boltzmann distribution. 3 A dditionally , for chemical s ystems with a force field av ailable, w e assess the quality of the TS guesses provided by the different approac hes with the conv ergence of the Dimer algorithm, a single- ended TS optimization method. This setup mirrors standard practice for obtaining an optimized saddle p oin t structure, where double-ended metho ds, such as Nudged Elastic Band (NEB), typically provide a robust initial guess for the conv ergence of a subsequent single-ended TS optimization metho d. In that context, we ev aluate ASTRA sp ecifically as a TS guess generation metho d. 2.1 Analytical Poten tials (a) Double well p oten tial. (b) Müller-Brown p oten tial. (c) Double path p oten tial. Fig. 2 : Our metho d disco vers transition state regions with high prec ision. It finds a transition state for the (a) double well p otential, and multiple transition states for the (b) Müller-Brown p oten tial and (c) double path p oten tial. On a one-dimensional double well p oten tial, w e first demonstrate the fundamental capability of our metho d to sample TSs ov er a simple energy barrier. The mo del is trained on data from tw o distinct energy w ells, designated as State A and State B. As illustrated in Figure 2a , our algorithm identifies the TS by generating samples densely clustered at the p eak of the p oten tial barrier separating the t wo minima. T o further assess our method’s p erformance, w e emplo y the Müller-Brown p oten tial [ 60 ], whic h is a common t w o-dimensional benchmark for ev aluating TS searc h algorithms. Its energy surface features three lo cal minima connected b y tw o first-order saddle p oin ts, offering a controlled en viron- men t to assess a metho d’s ability to identify non-linear path wa ys and distinct saddle points. F or our analysis, we assigned the t wo low est-energy minima as the initial and final states (State A and State B in Figure 2b ). The ASTRA algorithm successfully lo cates configurations clustering around b oth accessible transition states connecting A and B. Ablation studies shown in Figure H6 indicate that this ability to discov er multiple, top ologically distinct saddle p oin ts is primarily driven by the Score-Aligned Ascent (SAA) comp onen t of our metho d. A critical test for adv anced sampling metho ds is the ability to iden tify multiple, top ologically distinct path wa ys for a giv en transition. W e ev aluate this specific capability on a tw o-dimensional p oten tial engineered to include tw o comp eting reaction channels connecting the same reactant and pro duct states. As shown in Figure 2c , the ASTRA algorithm successfully p opulates selectively b oth TS regions corresponding to the t wo comp eting pathw a ys. This simultaneous discov ery of distinct reac- tion channels, ac hieved without any explicit path-based guidance, demonstrates a k ey adv an tage of our metho d. Whereas traditional algorithms are designed to conv erge to a single pathw a y , our approac h can explore the entire TS manifold, thereby providing mechanistic insights in to complex energy land- scap es that can b e easily ov erlo ok ed with conv en tional techniques without extensive sampling, and a voids b eing trapp ed in lo cal maxima that are irrelev ant for the studied c hemical pro cess. 2.2 Conformational Isomerization in Common Protein Mo del Systems Ha ving demonstrated proficiency on low-dimensional analytical p oten tials, we tested our algorithm’s p erformance on molecular systems. The conformational dynamics of alanine dip eptide, a 22-atom molecular system, provide a widely adopted mo del and benchmark for studying conformational ensem- bles of biomolecules. The dynamics are conv entionally analyzed through a Ramachandran plot, defined 4 (a) Alanine dip eptide. (b) Chignolin. Fig. 3 : Application of our metho d to chemical systems. (a) F or the alanine dip eptide, the generated samples (circles) lo calize the transition region betw een tw o states. (b) F or c hignolin, the free energy landscap e is pro jected onto the tw o slow est time-indep enden t components learned from a conv erged sim ulation [ 3 ]. The background densities in b oth plots corresp ond to the training data distribution where distinct colors indicate the defined State A and State B. The color scale for the alanine dipeptide plot represents the MD-based committor v alue while the scale for chignolin indicates the machine- learned committor v alue [ 36 ]. b y the dihedral angles ϕ and ψ . W e fo cus on the isomerization b et ween the tw o most p opulated con- formational states, the C5 and C7ax conformations, which represent distinct lo cal minima separated b y free energy barriers [ 61 ]. W e highligh t the absence of mo del knowledge in the transition region in Figure 3a a), showing the distribution of the ASTRA-generated structures o verlaid on the distribution of the training set. W e observe that they are lo calized precisely within three narrow regions of the Ramachandran plot, whic h corresp ond to the kno wn transition states for the C5-C7ax intercon version. This is v alidated b y committor analysis, where short Langevin Dynamics simulations initiated from these configurations yield a distribution of committor v alues sharply p eak ed at 0.5 (Figure 4 ) for all transition states. This highlights the key ability of ASTRA to identify distinct and structurally diverse path wa ys for a transition in a chemical system, without any input or prior knowledge of the mec hanisms at play . A dditionally , we assess the quality of our samples with a single-ended TS optimization pro cedure, whic h t y pically requires a high-qualit y initial guess, geometrically close to the saddle point. The ob jective is to ev aluate ho w similar the ASTRA-generated samples are to the reference structures obtained b y an optimization metho d. Specifically , we rep ort the con v ergence rate, the ro ot mean square deviation (RMSD) of atomic p ositions, and the energy differences b et w een the initial and optimized structures. As illustrated in Figure 4 , and in greater detail in T able H2 and T able H3 , we find that our metho d is able to pro vide a ma jorit y of samples that conv erge to a saddle p oin t within a few optimization steps. Additionally , final optimized structures remain close to the original ASTRA samples, as evidenced by the low RMSD v alues and small c hanges in energy barriers. The v alidity of the transition state guesses generated by ASTRA is further confirmed by comparing them to structures obtained by NEB, a reference metho d for finding minimum energy paths. W e show in Figure 4 that the configurations optimized b y NEB are almost indistinguishable from the ones generated b y our algorithm. Additional details and metrics are provided in T able H4 . W e also refer to App endix G for complete analysis and details of the metho d and the key comp onen ts of its success in this system. W e extend our analysis to chignolin, a 10-residue fast-folding protein with multiple comp eting folding pathw ays, and transition timescales spanning microseconds. F or this task, we initialized our score-based diffusion mode l from the publicly a v ailable c heckpoint of Ref. 62 , which was pretrained on coarse grained c hignolin tra jectories. W e then fine-tuned the mo del using our training protocol to enable Classifier-F ree Guidance on the MD tra jectory dataset b y D. E. Sha w Rese a rch [ 3 ]. T o prev ent direct sup ervision on the transition region during fine-tuning, all frames corresp onding to the 5 Fig. 4 : Detailed p erformance of ASTRA on alanine dipeptide. a) The ASTRA samples cov ering all three TSs are compared to the Nudged Elastic Band reference metho d. One ASTRA sample is selected randomly for each TS for readabilit y . The bac kground sho ws the training data distribution and col- ors represent the tw o defined states for the classifier free guidance training. The three-dimensional structures are ov erlay ed for each identified TS. b) shows the distribution of committor v alue for the stable ASTRA samples p eak ed around 0.5. c-e) characterize ho w close ASTRA samples are from their Dimer-optimized coun terpart. c) shows the distribution of num b er of iterations necessary to conv erge Dimer from the stable ASTRA samples. d) displays the Ro ot Mean Square Deviation (RMSD) of ASTRA samples after Dimer optimization compared to b efore, while e) sho ws that difference in terms of the energy of the structures. W e indicate the num b er of stable ASTRA samples, compared to the total num b er of structures drawn from our algorithm for that analysis, alongside the Dimer conv er- gence rate from ASTRA stable samples. Stable ASTRA samples corresp ond to structures sampled by ASTRA for which running 2ps MD from is stable. transition region w ere remov ed from the fine-tuning set. This region was iden tified using a pre-trained, mac hine learning-based committor mo del [ 36 ], where frames with ML committor v alues q in the range of 0 . 0001 ≤ q ≤ 0 . 9999 w ere excluded. The res ulting configurations generated by our method were then pro jected onto a tw o-dimensional space defined by time-lagged indep enden t comp onen t analysis (TICA) for visualization [ 63 , 64 ]. The generated configurations are concentrated along the q ≈ 0 . 5 isosurface, providing strong quantitativ e evidence that our method concen trates probability mass on the TS ensemble (Figures 3b and H12 ). F urthermore, these samples are not concentrated on one sp ot but are spread across the TICA space within the transition region. Structural analysis rev eals that the generated samples can b e classified into the tw o known comp eting folding pathw a ys for chignolin, commonly referred to as TS down and TS up (Figure 5 ). This confirms our metho d’s ability to accurately describ e transition state diversit y in folding mechanisms. 2.3 Chemical Reaction Bey ond conformational sampling of biomolecules, TS are of ma jor imp ortance in the study of chemical reaction mechanisms. How ev er, learning high energy structures gov erned b y cov alent b ond formation and clea v age without prior data of suc h structures has b een a ma jor challenge. W e test our algorithm on a first-generation donor-acceptor Stenhouse adduct, which has already b een studied in the context of learning collective v ariables [ 36 ]. The molecule consists of 42 atoms. It transitions from a linear, conjugated (op en) state to a cyclic (closed) state through a multi-step reaction path wa y that includes Z-E isomerization, conformational rotation, and a thermally driv en 4 π -electrocyclization [ 65 ]. W e fo cus on the TS sampling of the 4 π -electrocyclization that in volv es a concerted formation of tw o and breaking of one co v alent b ond in tw o distinct regions of the molecule, demonstrating if our algorithm can learn to interpolate the reactant and product state for multiple b onds at once. Analogous to alanine dip eptide, w e constructed a training dataset b y sampling configurations from the metastable transient isomer and stable enol closed isomer with conv entional MD sim ulations. Both sim ulations were not transitory and we further ensured that our dataset excludes any state close the 6 (a) ASTRA-generated samples. (b) References. Fig. 5 : Visualization of c hignolin transition state mechanisms. Represen tative structures from ASTRA-sampled TS down and TS up ( left ) ensembles are o verlaid with transparent tub es on the refer- ence conformations from Ref. 36 ( right ). The structural agreement v alidates our metho d’s abilit y to resolv e distinct folding path wa ys. transition region with a pre-trained ML committor mo del [ 36 ] that predicted all v alues to b e either b elo w 0.0001 or ab o ve 0.9999. Fig. 6 : Lewis structure representation of the chemical reaction and three dimensional reactant struc- ture with the t wo co ordinates highligh ted that span the plot. The t wo c hosen degrees of freedom separate the reactant and pro duct state w ell, but were only used for visualization and not included in any part of our algorithm. The MD simulation data p oin ts are plotted as densities. The structures generated by the ASTRA algorithm and the structures optimized with the Dimer algorithm and con- v erged to a true transition state are shown as differently shap ed p oin ts that are colored based on the committor v alue predicted by the pretrained mo del. The ASTRA algorithm at inference then consistently generated structures within the TS region, whic h are shown in Figure 6 , indicated by committor v alues predicted by the pretrained ML mo del differen t from 0 or 1. How ev er, the ML committor v alues do not p eak at 0.5 for b oth the structures generated by ASTRA or structures optimized with the Dimer algorithm that are confirmed to b e TS structures with a single imaginary normal mo de. A p ossible reason is that the ML committor mo del 7 w as not trained on optimized TS structures, but on frames of MD sim ulations. Hence, w e do not rely on the committor v alues in this analysis when comparing conv erged TS structures obtained with differen t metho ds and instead fo cus our analysis on the cov erage of the v arious p ossible TS conformers and compare this co verage to other single- and double-ended TS search algorithms. V arious TS conformers are possible for this reaction due to i) the different out-of-plane p ositions of the geminal methyl groups as shown in a previous study [ 36 ], ii) the dihedral angle of the 6-membered ring with the formed 5-mem b ered ring, and iii) the orientation of the eth yl groups of the amine group. Generating v arious TS structures from baseline workflo ws and ASTRA, rev eals a clear clustering of data p oin ts on the tw o-dimensional subspace, whic h is depicted in Figure D3 . The differen t clusters rev eal a large structural div ersity of p oten tial TS conformers that is successfully co v ered by the ASTRA samples and required a multi-lev el workflo w of conformer generation and TS guess generation with existing approaches. 3 Discussion In this work, we in tro duce ASTRA, a workflo w that reconceptualizes TS sampling as an inference- time guidance task of generative mo dels by leveraging a pretrained score-based mo del conditioned on known metastable states and ph ysical forces. Our method composes learned distributions with Score-Based Interpolation (SBI) to target directly the dividing surface of equal densit y , while the Score-Aligned Ascent (SAA) mec hanism steers the sampling pro cess tow ards transition states. This approac h successfully lo cates the TS ensemble regions with high precision and discov ers comp eting reaction path wa ys in c hemical systems, as demonstrated on alanine dip eptide, chignolin, and an electro cyclical reaction, without any prior kno wledge of the underlying transition mechanisms. The a priori sampling of transition states is primarily enabled by the synergetic use of SBI and SAA. The implicit probabilistic definition of the transition state region b y SBI provides a clear adv an tage o ver classical in terp olation metho ds, such as linear or geo desic in terp olation, that require carefully selected starting structures to ensure div ersity . Y et, in the alanine dip eptide benchmark only a guidance scale of 1.0 with Iso densit y Interpolation enables the sampling of all three transition states, b ecause a larger guidance scale tends to collapse the samples in high-density mo des. Enhancing the div ersity of samples on the PES during SBI could further improv e the robustness, particularly in systems exhibiting complex, multi-path wa y reaction dynamics. T o guarantee conv ergence, traditional minimum energy path algorithms suc h as the NEB require b oth a go o d initial mechanistic path and high-quality structures. F or example, NEB fails to con verge a linearly in terp olated path betw een the t wo states. In con trast, SAA lev erages the difference of scores, em ulating a reaction co ordinate that connects the tw o basins at a low latent level. As such, the optimization is less b ound by the physicalit y of the guess structures as we map them back to the data manifold with subsequen t denoising. This robustness enables SAA to con verge the tw o main TS in the alanine dip eptide system, even when initialized from a linear interpolation in Cartesian space. W e refer to Appendix G for an extended analysis and key metho dological details of the metho d. While ASTRA is not y et able to substitute all existing TS exploration workflo ws with a drop-in replacemen t, we show that (i) diffusion mo dels can infer reliable TS structures from adjacent states and (ii) our inference-time guidance with SBI and SAA enables massive shortcuts in the generation of out-of-distribution samples. The concurrent work by T ang et al. [ 66 ] learns the committor function of a given system b y launching simulations from structures generated with a diffusion mo del that was also trained only on metastable states at first. They do not mo dify the reverse-diffusion pro cess and hence require a computationally intensiv e iterative approach to conv erge on a reliable mo del of the committor function. In con trast, ASTRA acts as a “single-shot” generator. While it remains to b e seen if one-shot generation can fully replace iterative enhanced sampling in large biological systems, it is straigh tforward to integrate our ASTRA approach into existing iterativ e approac hes as a p ow erful ini- tialization strategy . This could p oten tially eliminate the reliance on heuristic collective v ariables and reduce the n umber of iterations needed to unco ver complex reaction mechanisms. Contrary , in the application of ASTRA as a TS guess generation mo del in time-independent TS optimization tasks, our generative workflo w is computationally more exp ensiv e than existing traditional computational c hemistry w orkflows b ecause of the system-focused training data generation and training ov erhead. Ho wev er, existing metho ds can often become trapp ed in single path wa ys due to heuristic initialization strategies without massiv e scale-up and automation of the TS search, while ASTRA enables direct 8 broad and diverse TS guess generation free from heuristic assumptions. This diversit y is k ey in confor- mational flexible systems, where ASTRA’s computational ov erhead could b e offset b y the increased sampling efficiency at inference. A key adv antage of our approach is its compatibility with increasingly p o werful pretrained genera- tiv e mo dels for molecular conformational sampling. F or instance, models suc h as BioEm u [ 41 ] generate protein ensembles consistent with the Boltzmann distribution and are therefore biased to ward low- energy states, with potentially limited accuracy near transition regions. When combined with our inference-time, force-guided scaling strategy , ho wev er, such mo dels can still serv e as strong priors. Starting from these pretrained priors rather than from scratch further improv es the data efficiency of the search. F uture work will extend ASTRA to ward a general-purp ose inference-time guidance framework for rare ev ent discov ery across div erse chemical systems. Although this study emplo ys classical force fields and semi-empirical quan tum chemistry metho ds, the framework easily generalizes to emplo ying higher- fidelit y force predictions such as mac hine learning interatomic p oten tials (MLIPs). Beyond transition state disco very based on saddle p oin t definitions, ASTRA can b e adapted to explore rare ev ent regions more broadly , for example through uncertaint y-aw are guidance [ 22 , 67 ] that targets configurations with high predictiv e uncertaint y under the generative mo del or MLIPs. Such a strategy can b e used for autonomous data acquisition and self-refinement steps in the active learning framework to impro ve the mo dels. More broadly , the principle of SBI and SAA in generative mo deling ma y extend b ey ond c hemistry and offer a foundation for rare-even t discov ery in complex dynamical systems. 4 Metho ds 4.1 Score-Based Interpolation of Diffusion Mo dels for Initial Guess Our ob jective is to infer the TS, giv en short MD data of eac h metastable state. In particular, our aim was to sample the TS from a generativ e mo del that learned only the resp ectiv e distribution of eac h metastable state, without any kno wledge of the TS region and without further training on TS structures. W e model the metastable distributions with a single system-specific diffusion mo del. In particular, follo wing the denoising score matching framew ork [ 55 ], w e formulate a mapping betw een the target data distribution p 0 ( x 0 ) and a simple Gaussian prior p T ( x T ) ∼ N (0 , I ) , through a Sto c hastic Differen tial equation: d x = f ( x , t ) dt + g ( t ) d w , (1) where d w is a Wiener pro cess, f ( x , t ) is the drift co efficien t, and g ( t ) is the diffusion co efficien t. The rev erse process can b e written d x = [ f ( x , τ ) − g ( τ ) 2 ∇ x log p τ ( x )] dτ + g ( τ ) d ¯ w , (2) where ¯ w is a reverse Wiener pro cess, and the reverse time τ ∈ [ T , 0] [ 55 ]. The mo del learns s θ ( x , t ) ≈ ∇ x log p τ ( x ) so that b y initializing the reverse SDE with noise x τ ∼ N (0 , I ) and solving it numerically , w e can sample from the data distribution after training. The training is generally self-sup ervised and requires only nuclear p ositions. How ever, we require the diffusion mo del to additionally be conditioned on each metastable state to sample from the transition region b etw een each state. Hence, w e annotate eac h data p oin t with its metastable state class c and train a denoising score matching mo del with Classifier-F ree Guidance (CFG) [ 50 ]. The labeling is trivial as the training data is generated from t wo separate molecular dynamics simulations for systems without any pre-existing tra jectory data. The scores are then parametrized b y the CFG-based score s c θ ( x , t ) ≈ ∇ log p t ( x | c ) , giv en b y the extrap olation s c θ ( x , t ) = s θ ( x , t, ∅ ) + γ ( s θ ( x , t, c ) − s θ ( x , t, ∅ )) , (3) where c is the condition, replaced by a null token ∅ for the unconditional score, and γ is the h yp erpa- rameter of the guidance scale that controls the conditioning strength. W e trained the diffusion mo dels with the Denoising Diffusion Probabilistic Mo dels[ 68 ] (DDPM) framework. Recen t metho ds hav e prop osed v arious approac hes to steer the diffusion sampling pro cess at infer- ence time[ 69 , 70 ]. W e are particularly in terested in sampling an in termediate region b et ween tw o metastable states while pro ducing structures lying on the data manifold. The Sup erposition of Diffu- sion mo dels [ 52 ] (Sup erDiff ) prop osed a principled wa y to sample the surface of equal density under 9 t wo conditional mo dels denoted A and B , where the equal densit y surface is sampled by follo wing the rev erse SDE while resp ecting d log p A t ( x ) = d log p B t ( x ) . (4) Our k ey intuition i s that the TS region ov erlaps with this equal density region of tw o conditional generativ e mo dels trained on the resp ectiv e metastable states MD sampling. W e form ulate this infer- ence steering follo wing the Sup erDiff AND op erator. The rev erse diffusion pro cess is biased to follow along the target equal probability surface by in terp olating b et ween the tw o conditional scores s SBI ( x , t ) = s B θ ( x , t ) + κ ( s A θ ( x , t ) − s B θ ( x , t )) , (5) where the interpolation weigh t κ controls the contribution of each state’s score. T o obtain the κ that follo ws the path dictated by equation 4 , the change of probabilit y densities can b e estimated with an Itô density estimator. One can then obtain the v alue of κ at each time step by solving a simple system of linear equations. W e refer to the original Sup erDiff pap er for details on the deriv ation and implemen tation [ 52 ]. W e get our initial guesses following the rev erse diffusion pro cess of equation 2 with the interpolated score of equation 5 . W e ev aluate this metho d against Score A veraging (SA) that provides a simpler baseline that directly av erages the tw o conditional scores, which corresponds to setting κ = 0 . 5 [ 71 ]. 4.2 Score-Aligned Ascent for Saddle Poin t Search TSs are first-order saddle points on a m ulti-dimensional PES. Lo cating a TS corresp onds to an optimization problem, where the energy needs to b e maximized along the given transformation, often referred to as the reaction co ordinate r , and minimized in all orthogonal directions. This is achiev ed b y decomp osing the negativ e gradient of the energy with resp ect to all nuclear co ordinates −∇ U ( x ) into comp onen ts parallel and p erpendicular to the reaction co ordinate. The parallel comp onen t is inv erted to create an ascen t force that pushes the system energetically up ward along r , while preserving the original comp onen ts that minimize the energy in all orthogonal directions. This op eration yields the ideal ascent force F = −∇ U ( x ) + 2( ∇ U ( x ) · r ) r / ∥ r ∥ 2 2 . (6) The key challenge of this metho d lies in the definition of r . F or example, existing approaches such as the Dimer algorithm [ 72 – 76 ], find lo cal appro ximations to r by lo cating the direction of least curv ature with gradient calculations of t wo close-lying configurations on the PES. W e detail existing metho ds repro ducing this kind of dynamics in section 4.4.1 that serve as baselines for our approac h. W e propose to leverage the ability of diffusion mo dels to em ulate these dynamics with the Score- Aligned Ascent (SAA). In practice, tow ards the end of the reverse diffusion pro cess steered with SBI, w e pause the denoising and optimize with SAA. W e find that, at this low noise level, an appro ximate reaction co ordinate r SAA can b e dynamically form ulated at each step by taking the difference of the scores conditioned on each state A and B r SAA = s B θ ( x , t ) − s A θ ( x , t ) . (7) This vector p oin ts from one metastable state to the other (see App endix E ) and provides an efficien t ascen t direction. Then, by calculating the ph ysical force −∇ U ( x ) that produced the training d a ta, one computes the SAA force F SAA = −∇ U ( x ) + 2( ∇ U ( x ) · r SAA ) r SAA / ∥ r SAA ∥ 2 2 . (8) There are t wo main ob jectives when p erforming the SAA dynamics at a low laten t lev el. First, b ecause of its sensitivity to small top ological changes, the force field must b e ev aluated on clean structures, i.e. configurations on the data manifold. Therefore, we estimate the denoised data p oin t ˆ x t for a configuration x t from the p osterior mean ˆ x t ≡ E p ( x 0 | x t ) [ x 0 ] with ˆ x t = 1 √ ¯ α t x t − √ 1 − ¯ α t ϵ θ ( x t , t ) , (9) where we follo w the standard DDPM notations and write ˆ x t to emphasize the dep endency of that estimate on time. Ho wev er, this estimate deteriorates rapidly as the diffusion timestep increases, 10 particularly for molecular systems. W e address this limitation by p erforming SAA at a low noise lev el. Moreo ver, the score difference used as an approximation for the ascen t direction is computed at the selected laten t lev el. Therefore, there is a discrepancy b et ween r SAA ( x t ) and the forces −∇ U ( ˆ x t ) , as these tw o di rections are not computed at the same laten t level. T o effectiv ely minimize the impact of this structural mismatch in equation 8 , we c hose a lo wer noise lev el for SAA dynamics. A second, equally imp ortan t reason for pausing the denoising and not operating SAA directly on the data manifold is cen tral to the success of the metho d. The ascen t direction deriv ed from equation 7 can lead to non-physical structures, b ecause the score difference provides no guarantee of mo vemen t along the true physical structure manifold. Nonetheless, this direction offers an efficient and meaningful path wa y to connect t wo states, which can subsequently b e pro jected back onto the manifold by denoising to τ = 0 . By p erforming the dynamics at an intermediate latent level, we can correct such non-ph ysical structures after SAA optimization by completing the denoising reverse pro cess. 4.3 ASTRA Summarizing the w orkflow, we first solv e the reverse-time SDE with in terp olated score up to a chosen fixed timestep close to the SDE endp oin t to ensure b oth reliable force and score calculations. W e then pause the reverse SDE pro cess, and optim ize with ˆ F SAA = −∇ U ( x ) + 2( ∇ U ( x ) · r SAA ) r SAA / ∥ r SAA ∥ 2 2 to steer the configuration to wards a TS. W e then resume the reverse SDE sampling, whic h transfers the sample on to the data manifold and corrects non-physical configurations stemming from the non- ph ysical ascent direction. W e refer to Algorithm 1 for the complete details of the metho d and its key h yp erparameters. 4.4 Ev aluation and baselines Except for coarse-grained chignolin, all baseline and b enc hmark methods were ev aluated with the same classical force field or electronic structure metho d that w as used to generate the dataset and to perform SAA. A common force field reference ensures that the transition states and other reference structures targeted b y eac h algorithm are consisten t with the underlying PES, so that observed differences reflect metho dological p erformance rather than differences b et w een p oten tials. In that regard, we emphasize that the notion of ’true’ TS is relativ e to the giv en force field used. 4.4.1 T ransition state characterization Our primary metric for assessing the quality of the sampled TS configurations is the ev aluation of the committor v alue q ( x ) . It corresp onds to the probability that a tra jectory starting from a given structure x will reach one metastable state b efore reac hing another metastable state. Thus, a TS is c haracterized b y q ( x ) = 0 . 5 as a simulation launched from a TS has the same probabilit y of ending in either state. This definition requires us to define the outlines of the metastable states. W e define the outlines within a dimensionality-reduced space, based on the observed MD distributions in the training sets that sampled these basins. Then, after sampling 100 structures with our metho d, we run a 2 ps long MD simulation and filter out all the configurations leading to instabilit y that are lab eled as non-ph ysical. F rom the v alidated TS structures w e then run multiple replicas of a Langevin Dynamics tra jectory with velocities initialized randomly from the Maxw ell–Boltzmann distribution. The committor v alue is calculated b y coun ting the num b er of simulations that reac hed metastable state A b efore reaching state B and dividing it b y the n umber of total sim ulations. T o further assess the quality of our TS samples, w e demonstrate that established reference TS optimization methods conv erge rapidly when initialized from these samples. Such metho ds typically require accurate initial guesses to ensure conv ergence. Therefore, we carried out short single-ended TS optimizations implemented in SCINE ReaDuct [ 77 , 78 ]. W e report (i) the conv ergence rate, (ii) the a verage num b er of steps required for conv ergence, (iii) the av erage energy difference b et ween the ASTRA samples and the final conv erged structures, and (iv) the av erage ro ot mean square deviation (RMSD) of nuclear p ositions b et ween the tw o. Our TS optimization method of choice in this w ork is the dimer metho d [ 72 ], a Hessian-free, minim um-mo de-follo wing technique that requires only energy gradients and can b e initialized from a single configuration. The metho d represents the search direction using a “dimer” (t wo nearby images separated by a small displacemen t) and iterates cycles of (i) dimer rotation to iden tify the lo west- curv ature (minim um) mo de based solely on forces, and (ii) configuration translation (climbing) along 11 this mo de while sim ultaneously relaxing all orthogonal degrees of freedom. Iterations contin ue until the curv ature is minimal and the gradient con verges to zero. This approac h is particularly well suited to large systems where explicit Hessians are impractical, and sev eral impro vemen ts, such as shrinking/optimization-based v ariants, L-BFGS acceleration, and reduced gradient-ev aluation sched- ules, hav e b een prop osed to enhance con vergence and robustness [ 73 – 76 ]. In our implemen tation, w e initialize the dimer direction from a Hessian calculation rather than from a random guess, as supported b y SCINE ReaDuct [ 78 ]. 4.4.2 TS sampling baselines W e b enc hmarked the quality of our TS guesses against traditional approac hes. As a primary baseline, w e emplo yed the NEB[ 79 , 80 ] method, a standard approach for lo cating transition states and minim um energy paths (MEPs) b et ween kno wn reactant and pro duct states. NEB represents the path wa y as a c hain of discrete images connected by virtual springs. The springs preven t all images from con verging to one of the tw o minima while the energy of all images is minimized. This “n udging” drives the images tow ards the MEP , enabling accurate saddle p oin t lo calization without explicit kno w ledge of the reaction co ordinate. NEB has b ecome widely applied in computational c hemistry and materials science for exploring energy barriers and reaction mec hanisms. A ckno wledgemen ts. H.L. ackno wledges the Korea Institute for Adv ancemen t of T ec hnology (KIA T). This pap er was supp orted by a KIA T gran t funded by the K orea Go vernmen t (Ministry of Education)(P0025681-G02P22450002201-10054408, Semiconductor-Sp ecialized Universit y). M.S. gratefully ac knowledges the Mobilit y fello wship P500PN_225736 from the Swiss National Science F oundation. Y.D. ackno wledges the support from Cornell Univ ersity . The authors ackno wledge MIT SuperCloud and Lincoln Laboratory Supercomputing Center for pro viding (HPC, database, consultation) resources that hav e contributed to the research results rep orted within this pap er. App endix A Related W orks A.1 Mac hine Learning for T ransition State Searc h Numerous machine learning approaches ha ve b een dev elop ed to study rare even ts and in particular transition search. Early w orks leveraged a dataset of accumulated transition states to train generative mo dels [ 37 ] with diffusion mo dels [ 38 , 39 ] and flow matc hing [ 40 ]. How ev er, to alleviate the require- men t of creating a transition state dataset, later work aimed to leverage geometric optimization and energy ev aluations during training to find transition paths and extract transition states from it [ 45 , 46 ]. Ref. [ 47 ] also leveraged the metric induced by lo cal molecular dynamics sim ulation to learn a gener- alized flow matching mo del, while Ref. [ 48 ] relied on the learned force field from pre-trained diffusion mo dels with the Onsager–Machlup functional to appro ximate the transition path. A dditionally , a large bo dy of literature tac kled the committor function estimation problem and esti- mate reaction rates directly . When transition path data are av ailable, maximum-lik eliho od metho ds are straightforw ard to learn the committor function [ 23 – 25 ]. Ref. [ 26 , 27 ] directly solved the v aria- tional formulation of the K olmogorov backw ard equation (KBE) with equilibrium data. Ref. [ 35 , 36 ] prop osed active approac hes to use the current estimate of committor function to improv e sampling of transition state regions when solving the KBE equation. Ref. [ 28 – 30 ] instead considered the F eynman— Kac formulation of the KBE and used self-consistency ob jectives to match the committor v alues across the domain and their exp ected v alues after time ev olution. A.2 Inference-time Control for Diffusion Mo dels Diffusion models excel at generating high-quality data, spanning images, texts to molecular structures. Bey ond sampling from the learned distribution, they can b e guided to generate from mo dified target distributions that enco de design ob jectiv es, constraints, or rewards. The early and well-kno wn exam- ples are classifier guidance and classifier-free guidance to sample from conditional distributions [ 50 ]. Later on, inference-time control has been extended to comp ositional, reward-tilted, annealed, equal densit y distributions and more [ 51 – 53 ]. One p opular branch of metho ds dev elop up on the heuristics and approximate guidance [ 81 ]. Another branch of metho ds study exact guidance where exp ensiv e Mon te Carlo (MC) estimations are required [ 82 ]. Recen tly , sequential Monte Carlo methods ha ve b ecome relev an t to reweigh t on path space to reduce the v ariance of inference-time control [ 53 , 83 , 84 ]. 12 App endix B Bac kground B.1 Score-Based Diffusion Mo dels Giv en a forward process that transforms a data distribution p 0 ( x 0 ) ov er a con tinuous time v ariable t ∈ [0 , T ] into a known prior distribution p T ( x T ) , typically the standard Normal distribution N (0 , I ) , a diffusion mo del s θ ( x , t ) can b e trained suc h that a reverse pro cess transforms samples from the prior distribution to the data distribution [ 85 ]. The mo del can learn the added noise in each time step t [ 68 ] or the gradient of the log-probability of the data distribution ∇ x log p t ( x ) , the so-called score function [ 86 ]. Both training strategies can b e related as discretizing the same con tinuous reverse pro cess describ ed by the sto c hastic differential equation (SDE) d x = [ f ( x , τ ) − g ( τ ) 2 ∇ x log p τ ( x )] dτ + g ( τ ) d ¯ w , (B1) where f ( x , t ) is the drift co efficien t, g ( t ) is the diffusion co efficien t, ¯ w is a reverse Wiener pro cess, and the rev erse time τ ∈ [ T , 0] [ 55 ]. Once the mo del s θ ( x , t ) is trained, it can b e applied as a generativ e mo del by initializing the reverse SDE with noise x τ ∼ N (0 , I ) and solving it n umerically . B.2 Classifier-F ree Guidance Classifier-F ree Guidance (CFG) [ 50 ] is a tec hnique for conditional generation that trains a single diffusion mo del to handle b oth conditional and unconditional generation scenarios. The mo del is trained to predict the score of the data distribution, conditioned on the label c ∈ { C 1 , C 2 , ..., ∅} . During training, the conditioning information is randomly dropp ed with a given probability p drop . This technique enables the single mo del to learn b oth the conditional score functions, ∇ log p t ( x | c ) , for each lab el, and the unconditional score function, ∇ log p t ( x ) , ov er the entire data manifold. During inference, the CFG-based score s c θ ( x , t ) is given by interpolation s c θ ( x , t ) = s θ ( x , t, ∅ ) + γ ( s θ ( x , t, c ) − s θ ( x , t, ∅ )) , (B2) where s θ ( x , t, c ) is the conditional score, s θ ( x , t, ∅ ) is the unconditional score, where the condition c is replaced by a n ull tok en ∅ , and γ is the guidance scale that controls the strength of conditioning. This approach provides fine-grained control o ver conditioning strength without requiring separately trained conditional mo dels. B.3 Sup erposition of Diffusion Mo dels The Sup erp osition of Diffusion Mo dels (Sup erDiff ) [ 52 ] provides a principled wa y to combine the score functions from m ultiple mo dels, each trained on a different data distribution. Given tw o score mo dels, s A and s B , trained on distributions p A and p B resp ectiv ely , a comp osite score function can b e constructed by a weigh ted av erage: s SBI ( x , t ) = s B θ ( x , t ) + κ ( s A θ ( x , t ) − s B θ ( x , t )) . (B3) The interpolation factor κ ( t ) determines the nature of the comp osition, enabling the generation of samples from distributions that represent logical combinations of the base distributions. F or example, setting κ ( t ) based on the relative likelihoo ds of a sample under each mo del can pro duce samples from the union (OR) of the distributions, while setting κ based on equal likelihoo ds can produce samples from the in tersection (AND). In the context of transition state sampling, we are interested in the latter and a κ ( t ) that satisfies the equal densit y: d log p A t ( x ) = d log p B t ( x ) (B4) can b e found by solving a set of three linear equations [ 52 ]. As a comparison, we also include in our ablations Simple A v eraging of the scores to interpolate and steer the diffusion process [ 71 ]. B.4 Lo cal mo de maximization T ransition states are first-order saddle points on a multi-dimensional p oten tial energy surface (PES). Lo calizing a transition state corresponds to an optimization problem, where the energy needs to b e 13 maximized along the given transformation and minimized in all orthogonal directions. If the mo de of transformation r , commonly referred to as reaction co ordinate, is known, a gradient-based optimiza- tion to the transition state can b e achiev ed with a sufficiently close start condition. The optimization is driven by the step ∆x i that is determined b y the gradient of the p oten tial energy with resp ect to n uclear coordinates g ( x ) ∆x i = − g ( x ) + 2 g ( x ) T r r . (B5) Ho wev er, the definition of r is non-trivial for a giv en chemical sy stem. Existing approaches, such as the Dimer algorithm [ 72 – 76 ], find lo cal appro ximations to r by lo cating the direction of least curv ature with gradient calculations for multiple close-lying configurations on the PES. App endix C A dditional Details on Exp erimen ts C.1 Double W ell P oten tial The double w ell potential is a simple p otential with tw o distinct states. The analytical formula for this p oten tial energy surface is given by: V ( x ) = 0 . 1 x 4 − x 2 + 0 . 1 x + 1 . 0 (C6) C.2 Müller-Bro wn P oten tial F or the Müller-Bro wn potential, our metho dology adheres to the training procedure detailed in Ref. 48 . A score-based diffusion mo del w as trained on the ‘tiny’ subset of the pro vided dataset, which consists of 4,000 samples. The tw o metastable states, A and B, are defined b y the co ordinate criteria y < 20 and y > 20, resp ectiv ely . The analytical surface of this system is defined as: V ( x, y ) = 4 X i =1 A i exp α i ( x − a i ) 2 + β i ( x − a i )( y − b i ) + γ i ( y − b i ) 2 (C7) with the parameters taken directly from the sim ulation co de: A = 10 × barrier × [ − 1 . 73 , − 0 . 87 , − 1 . 47 , 0 . 13] α = 10 − 2 × [ − 0 . 39 , − 0 . 39 , − 2 . 54 , 0 . 273] a = [48 , 32 , 24 , 16] β = 10 − 2 × [0 , 0 , 4 . 30 , 0 . 23] b = [8 , 16 , 32 , 24] γ = 10 − 2 × [ − 3 . 91 , − 3 . 91 , − 2 . 54 , 0 . 273] where ‘barrier‘ is a scaling factor for the heigh t of the p oten tial barrier, for which we used 1.0. C.3 Double Path P otential F or the double path p oten tial, the dataset was generated with Langevin Dynamics simulations, fol- lo wing the pro cedure outlined in Ref. 48 . A score-based diffusion mo del w as subsequently trained on a curated subset of these simulation data, with 4,000 samples. State A and B are defined b y the regions x < 0 and x > 0 , resp ectiv ely . 14 The analytical formula for the p oten tial energy surface is given by: V ( x, y ) = 10 2 + 4 3 x 4 − 2 y 2 + y 4 + 10 3 x 2 ( y 2 − 1) + 7 exp − ( x + 0 . 7) 2 + ( y − 0 . 8) 2 0 . 4 2 + exp − ( x − 1 . 0) 2 + ( y + 0 . 3) 2 0 . 4 2 − 6 exp − ( x + 1 . 0) 2 + ( y + 0 . 6) 2 0 . 4 2 (C8) (a) Alanine dip eptide. (b) Chignolin. Fig. C1 : P otential Energy Surface (PES) of alanine dip eptide and chignolin. C.4 Alanine Dip eptide (a) Conditioned on C5. (b) Conditioned on C7ax. (c) Unconditional. Fig. C2 : Poten tial Energy Surface (PES) of alanine dipeptide sample from the trained diffusion mo del. W e generated the dataset by running Langevin Dynamics from each state (C5 and C7ax) for 500 ps with a 1 fs time step saving ev ery 10 steps and a temp erature of 300 K. W e then sliced parts of the tra jectory that co vers the widest region in Ramachandran space ( ϕ, ψ ) and subsample randomly 15 2,500 configurations for each of the t wo states. In Figure C1a , we show the PES of alanine dip eptide. W e propagated the dynamics with a custom force field taken from Ref. 48 . W e then trained a diffusion mo del based on the EquiformerV2 arc hitecture [ 87 ] with up to L = 2 representations, four attention heads and 64 channels. The radius graph is computed with r cutof f = 5 . 0 Å. W e use the A dam W optimizer with a constant learning rate 6 . 10 − 4 , 0 . 001 w eight decay . W e use an 0.999 Exp onen tial Mo ving A verage (EMA) deca y with an effective batc h size of 128 , and train for 500 epo c hs. F or diffusion, w e lev erage the Denoising Diffusion Probabilistic Mo del framew ork (DDPM) with 1000 time steps and a cosine schedule [ 68 ]. F or SAA, we use 1000 optimization steps by Adam optimizer with a learning rate of 0.01, β 1 = 0 . 0 , and β 2 = 0 . 999 . The committor probabilities are computed from 100 configurations sampled from the metho d. W e first run a 2 ps long sim ulation and filter out all the configurations leading to instability that are lab elled as non ph ysical. F or all rep orted results, more than 98% of samples were k ept and used for the committor calculations. W e compute the committor probabilities by running 100 replicas sto c hastic Langevin Dynamics with randomly initialized velocities using the same force field that generated the dataset to sta y consistent with the PES distribution. The simulations are run for 1 ps with 1 fs steps at T=300 K. W e define eac h state limits and record the region first reach for each replica to compute the probability of reaching one state or the other. C.5 Chignolin F or our analysis of Chignolin, whic h is a small fast-folding protein, we employ ed the D. E. Shaw Researc h coarse-grained (CG) dataset [ 3 ]. In Figure C1b , we show the PES of the chignolin training dataset. W e remov ed all the conformations with a ML committor v alue b et ween 0.0001 and 0.9999 in the training dataset. Our conditional diffusion mo del is an adaptation of the pre-trained architecture from Ref. 62 . W e finetuned this base mo del to enable conditional generation by incorp orating a group em b edding that sp ecifies the state, i.e., folded or unfolded, at the b eginning of the forw ard pro cess. Sampling was then p erformed using SAA h yp erparameters with 100 optimization steps, a learning rate of 0.0005, and a pause ratio of 0.05. Finally , the generated configurations were v alidated by computing their committor probabilities with the machine-learned committor predictor mo del from Ref. 36 . C.6 Electro cyclical reaction W e generated the dataset by running Langevin Dynamics from the reactant and pro duct structure eac h for 500 ps with a 0.5 fs time step saving every 10 steps and a temperature of 300 K. W e propagated the dynamics with the semi-empirical PM6 metho d [ 88 ] calculated with Scine Sp arr o w [ 89 , 90 ]. W e then trained a diffusion mo del with the exact same pro cedure as b efore with alanine dip eptide. 16 App endix D Conformational diversit y of the transition state ensem ble for the electro cyclical reaction T o v alidate the structural diversit y of ASTRA-generated samples for the DASA system (Figure D3 ), w e generated a large set of reference structures using multiple transition-state optimization meth- o ds with v arying initializations. By comparing the ASTRA samples directly against these resulting reference clusters, we characterize the cov erage and v alidity of our generativ e approach. Notably , the only cluster missed by ASTRA is 2). It contains structures that contin ue a structural trend of an increasing dihedral angle and hence structural distortion going from the top right to the b ottom left of cluster 1), hence, cluster 2) is an extreme version of cluster 1). The lac k of cluster 2) structures indicates that the distorted structure is disfav ored in the diffusion pro cess. Due to this distortion, the chemical relev ance of this cluster might b e low as all structures in this cluster are higher in energy than the ASTRA generated structures in cluster 1). Beyond the cov erage of each cluster, ASTRA can also capture a finer grained diversit y within a certain cluster. Cluster 5) mixes different meth yl group orientations, with the righ t side of the cluster including structures with down ward methyl orien tation, while the left side of the cluster includes upw ard orien tations. W e observe that ASTRA successfully captures this in ternal div ersity of structures. Similarly , cluster 3) has a large extent of p ossible carb on-carbon d istances while preserving the other structural features, which is cov ered by ASTRA samples. This demonstrates that our algorithm can reco ver ph ysically v alid v ariabilit y of transitory configurations without prior knowledge or sampling of such ensembles due to our SBI and SAA sampling process. Our ASTRA algorithm pro duced an additional structure outside of cluster 3) that exhibits similar structural features, but includes an asymmetrical p osition of the transferred proton. This is missed b y the other methods, but it also includes a strong distortion of the 6-membered ring and is high in energy . The ASTRA algorithm also pro duced three out of four structures in cluster 4). The structures of this cluster show an upw ard tilted left oxygen atom while the righ t 6-mem b ered ring stays flat. Structures within all clusters generally sho w different amine eth yl group orien tations that aim at reducing steric hindrance. Based on these findings, we are confident that the structural div ersity of ASTRA samples observed in the smaller test systems b efore also extends to more complex c hemical reactions. F or examples, the structures in cluster 1) exhibit a small dihedral angle of the transferred proton and the 6-mem b ered ring and the geminal meth yl groups are p ositioned do wnw ard. In comparison, cluster 5), the largest set of p oin ts, shows a strong tilt of the o xygen atoms transferring the proton with the left one tilted upw ards and the right one down wards. Sampling with ASTRA encompasses this diversit y of structural features typically encountered in traditional approac hes with a div erse initialization. 17 Fig. D3 : T wo dimensional represen tation of the reactive p oten tial energy surface (PES) spanned by t wo imp ortan t intern uclear distances. The MD simulation data p oin ts are plotted as densities. The v arious p oin ts represent structures optimized with the Dimer algorithm where the initial structure w as generated with different TS guess structure generation algorithms. The v arious baseline metho ds are detailed in section G.2.2 . The zo omed-in version of the PES features representativ e structures for eac h cluster of p oin ts that are num b ered from 1 to 5. App endix E V alidation of Score Difference Appro ximation to Reaction Co ordinate T o v alidate the claim that the score difference approximates the reaction co ordinate, i.e., s A ( x , t ) − s B ( x , t ) ≈ r , we compare the tw o vectors in Figure E4 . The visualization sho ws strong alignment b et w een our appro ximation (blue arro ws) and the true reaction coordinate (red arro ws). This is quan tified by the high av erage cosine similarity: 0.9275 on the Müller-Brown p oten tial and 0.9584 on the double path p oten tial. 18 (a) Müller-Brown p oten tial. (b) Double path p oten tial. Fig. E4 : V alidation of Score Difference Approximation. The score difference v ector (red) closely aligns with the true reaction co ordinate (blue) on the (a) Müller-Bro wn p oten tial and (b) double path p oten tial. App endix F ASTRA Sampling Algorithm 19 Algorithm 1 Sampling with Score-Based In terp olation and Score-Aligned Ascent Require: Denoising model ϵ θ ( x t , t, c ) with classifier-free guidance; Energy-based force field F ( x ) ; T otal num b er of time steps T ; Guidance pause time step T pause ; Number of optimization steps N opt ; Optimizer hyperparameters Θ opt . Output: Sampled molecular conformation x 0 . 1: function SBI_Combine ( x t , t ) 2: Compute conditional noise estimates: ϵ A t ← ϵ θ ( x t , t, A ) , ϵ B t ← ϵ θ ( x t , t, B ) . 3: Compute the interpolation factor: κ t ← SBI ( ϵ A t , ϵ B t , x t , t ) . 4: Com bine noise estimates: ϵ comb ← ϵ B t + κ t ( ϵ A t − ϵ B t ) . 5: return ϵ comb , ϵ A t , ϵ B t 6: end function 7: Initialize p ositions x T ∼ N (0 , I ) . // Phase 1: Denoising with Sc or e-Base d Interp olation 8: for t ← T , T pause + 1 do 9: ϵ comb , _ , _ ← SBI_Combine ( x t , t ) 10: P erform one rev erse diffusion step to obtain x t − 1 from x t and ϵ comb . 11: end for // Phase 2: Sc or e-Aligne d Asc ent Optimization at latent time T p ause 12: Let z ← x T pause . Initialize an optimizer O with Θ opt . 13: for k ← 1 , N opt do 14: ϵ comb , ϵ A T pause , ϵ B T pause ← SBI_Combine ( x T pause , T pause ) 15: Appro ximate the reaction co ordinate: τ ← ϵ B T pause − ϵ A T pause . 16: Predict the clean sample: ˆ x 0 ← predict_x 0 ( z , T pause , ϵ comb ) . 17: Ev aluate the force from the external field: f ← F ( ˆ x 0 ) . 18: Compute the Score-Aligned Ascent force: f SAA ← f − 2 f · τ ∥ τ ∥ 2 2 τ . 19: Up date p ositions with gradient descen t: z ← O ( z , ∇ z L ) , where ∇ z L = − f SAA . 20: end for 21: x T pause ← z . // Phase 3: R esume d Denoising 22: for t ← T pause , 1 do 23: ϵ comb , _ , _ ← SBI_Combine ( x t , t ) 24: P erform one rev erse diffusion step to obtain x t − 1 from x t and ϵ comb . 25: end for return x 0 . App endix G Extended analysis of ASTRA dynamics and robustness G.1 Mo dules c haracterization In this section we attempt to characterize and interpret the b eha vior of ASTRA. W e base our discus- sions on the alanine dip eptide systems as its multiple and geometrically diverse transition path wa ys pro vide a c hallenging task. All ablation studies results can b e found in Section H . The ASTRA frame- w ork is comp osed of tw o fundamental mo dules, Score-Based In terp olation (SBI) for generating an initial guess of the transition ensemble and Score-Aligned Ascen t (SAA) for its subsequent refinemen t. Therefore, w e ev aluate the efficacy and contribution of eac h individual comp onen ts through ablation studies that are discussed using the committor and T ransition State optimization metrics. G.1.1 Score-Based In terp olation W e examine the effectiveness of the SBI mo dule as an initialization strategy for SAA by replacing it with established transition state (TS) guess methods, and b y comparing t wo distinct SBI-based approac hes. Sp ecifically , we b enc hmark our primary metho d—Isodensity Interpolation (I I)—against 20 alternativ e initialization techniques including Simple A veraging (SA), geo desic in terp olation [ 91 ], and linear interpolation in Cartesian space. F rom SBI-based approaches, ASTRA successfully recov ers at least tw o out three TSs as sho wn in Figure H8 . These results are in agreemen t with prior results obtained using our force field [ 48 ]. While SAA shines in its con vergence capabilities, SBI - as an informed initialization prior to SAA optimization - is the main feature allowing a priori sampling of a diverse TS ensemble. In particular, b y sampling across a broad region of the (approximate) iso-probabilit y surface connecting the t wo metastable states—under their resp ectiv e conditional generativ e mo dels, I I provides a diverse set of initial guesses that helps av oid premature conv ergence to a single dominant pathw a y . This allo ws to sample all three transition states. In terestingly , this comprehensive sampling of the TS is only observed when employing a guidance scale of 1.0. This phenomenon is illustrated in Figure H19 and Fig. H20 , where increasing the guidance scale from 1.0 to 3.0 progressively concentrates the TS guesses into the direct region separating the high-probabilit y metastable states. This b eha vior is consistent with a stronger guidance driving the sampling tow ard more lo calized, higher-densit y regions: a reduction in directional diversit y constrains the sampling tra jectories to few er dominant mo des. Notably , SA fails to generate initial guesses that con verge to all three TSs under SAA, whereas II succeeds, alb eit with the third TS (b ottom-righ t region of the Ramac handran plot) b eing sampled less frequently , as sho wn in Figure H19 . While the difference in sampling densit y for this third TS is substantial, it highligh ts the potential of II to recov er underrepresen ted transition mo des. Lo oking ahead, further improving the diversit y and breadth of PES cov erage during initialization could enhance the robustness of the ov erall method, particularly in systems exhibiting complex, multi-path wa y reaction dynamics. T o ev aluate the adv an tage of SBI ov er traditional TS guess methods for initializing SAA, we com- pare it against linear interpolation and geo desic interpolation. In b oth cases, w e construct a path of 25 intermediate structures and extract the five central images, which are then p erturbed with latent noise at a PR = 0.05 lev el b efore being used as inputs to SAA (see Figure H18 . T o ensure fair compar- ison and assess the ability of each metho d to conv erge tow ard distinct TSs, we av oid random starting configurations and instead ensure that the interpolated paths pass sufficiently close to the true TS region. This allows us to sample all TSs. Results presented in T able H4 demonstrate that SAA initial- ized with both linear and geo desic in terp olation can yield reasonable TS candidates, as supp orted by committor v alues and dimer conv ergence metrics. Ho wev er, SBI consisten tly produces sligh tly more accurate TS guesses, as measured by low er RMSD and energy difference to the reference TS. W e high- ligh t a key limitation of classical in terp olation-based methods: they require prior structural kno wledge of the TS region or the mechanism, to construct meaningful and diverse paths. In contra s t, SBI op er- ates without any such prior, generating diverse and physically plausible TS guesses directly from the conditional generative mo del. The broad cov erage of the intermediate region betw een the tw o basins, facilitates the discov ery of multiple v alid transition states without prior access to the PES. G.1.2 Score-Aligned Ascen t W e pro ceed to analyzing the b eha vior of SAA by sweeping the hyperparameter space. The results are rep orted T able H2 and H3 . W e start with the P ause Ratio (PR) that defines at which step is the rev erse diffusion process halted to p erform SAA b efore resuming the last few denoising steps. The PR is critical for the o verall p erformance of the method. W e define PR as Pause time step T otal n umber of time steps . The PR intrinsically manages the quality of the force field prediction, and the level of discrepancy b et ween the SAA ascen t direction and the structure it is optimized on, in terms of noise lev el. In fact, the force field should b e ev aluated on a clean structure and therefore we should use the p osterior expectation as input. Ho wev er the score difference defining the ascen t direction should b e ev aluated at the laten t lev el. Consequently , whether the optimization is carried out in latent space or on the denoised structure introduces an inheren t trade-off: in either case, a discrepancy arises b et w een the domain where the ascent direction or the forces are computed and where they are applied. In fact, we observ e in Figure H16 nearly iden tical p erformances whether optimization is p erformed directly in laten t space or on the denoised structure. In our implem en tation, we choose to p erform the optimization on the denoised structure and subsequently pro ject it bac k to the laten t space corresp onding to the PR level, using the same predicted score used to compute the p osterior exp ectation. Then, the next score ev aluation can b e computed, and so on. 21 In addition, w e find that pausing the rev erse diffusion at a nonzero latent noise lev el plays a critical role in correcting for the partially non-physical nature of the ascent direction in SAA, whic h can otherwise driv e samples a wa y from the true data manifold. This is a key feature of the method. Sp ecifically , the difference b et ween conditional scores yields a highly informative ascen t direction, but one that is more reliable in latent space - consistent with prior observ ations that denoising score matc hing models tend to struggle at v ery low noise levels [ 92 ]. The strength of the method is that the outcome of this laten t ascen t can be effectiv ely mapped back to the data manifold by simply resuming the reverse SDE from the pause time step to completion. In fact, we observ e in T able F2 that computing SAA at τ = 0 is not numerically stable. Increasing the pausing ratio generally increases the num b er of samples that hav e physical structures. How ev er, when pausing to o far from the SDE endp oin t, the distribution of committer v alues spreads out of the TS region range. W e attribute this degradation partially to the aforementioned discrepancy b et ween the structure on which the forces are computed, and the structure on which the ascent direction is computed. W e also hypothesize that the I I scores ma y actually drive the samples out of the n arro w TS region Therefore, w e find that there exists an optimal pause ratio around 0.05 that ac hieves the b est balance b et ween these constrain ts. When increasing the Optimization Step (OS), w e observe a clear conv ergence, across all metrics, demonstrating the stability and reliability of SAA. W e note that the trends of b oth the committor analysis, and dimer optimization are consisten t with one another. Our optimization algorithm reac hes a plateau around 750 OS ac hieving about a 85% conv ergence rate, 20 steps needed for con vergence and guess TS structures as close as a 0.01Å and a -0.5kcal.mol − 1 energy difference a wa y from the optimized ground truth TS. Changing the n umber of optimization steps, w e also identify a characteristic of SAA. In fact, we observ e that SAA can drive samples out of the data manifold. As OS increases, the n umber of stable samples decreases b efore reac hing a plateau of ab out 75% of ph ysical structures. As non-ph ysical structures can b e easily filtered out by running a short MD simulation from them, we rep ort the % of Committor v alue b et ween 0.4 and 0.6, with resp ect to the n umber of stable (filtered) structures. W e ma y in part in terpret harsher SAA conditions as a filter here. As the distribution narro ws around the TS regions, non promising TS guesses from SBI are driven to o far aw ay from the manifold for them to b e corrected by the subsequen t denoising. They can then b e easily filtered out, ac hieving in the end an excellent quality of the TS guesses, of almost all structures. The Step Size (SS) presen ts an optimum v alue of around 0.01 Å corresp onding to a standard tradeoff b et w een a slow con vergence, and an ov ersho oting leading to non-ph ysical structures. W e emphasize that the filtered ASTRA samples still remain close to the true TS in harsh SS conditions, although there are logically very few of them. G.2 Baselines G.2.1 Alanine dip eptide Our main baseline for alanine dip eptide is NEB, a well established metho d to optimize a MEP and find the saddle p oin t. W e provide in T able H4 a comparison of SAA against NEB to optimize an initial guess structure (in the case of SAA) and path (in the case of NEB) to the true TS. T o dra w a fair comparison, we use identical initialization algorithms (linear and geo desic interpolation) as input to SAA and NEB. F or NEB, we take the whole paths obtained from energy minimized structures (cen tral to obtaining the TS as the highest energy point). F or SAA, we retain the middle points as inputs. The initial structure from which the paths are interpolated are the same as in section G.1.2 and the results for the optimized paths are shown Figure H17 . The principal observ ation is that, when a structure from NEB can b e conv erged by dimer, it is extremely close to the true TS structure b oth in terms of p osition RMSD and energy difference. W e highlight the closeness of SAA generated samples in terms of RMSD when initialized from geodesic interpolation. This is the most relev ant case as we are able to recov er the tw o main TS in contrast to linear interpolation where the upp er path cannot b e conv erged by NEB. This is actually one of the main adv antage of using SAA here. NEB is not able to recov er tw o TS from the linear interpolated paths. This is probably due to non-physical structures that the reference metho d struggles to handle (see Figure H14 ). In contrast, SAA noises the structure bac k to the latent space and has the ability to correct unphysical structures back to the manifold. Our metho d is then able to sample the t wo main TSs from the linear interpolated paths. Therefore, SAA is more robust compared to NEB with resp ect to lesser qualit y interpolation metho d. This emphasizes once against the strength of such an approac h. Where no knowledge of the PES is 22 needed and where the o verall obtaining of the TS is straightforw ard, with little user input needed to obtain a go od TS guess. Finally , our method do es not need minimized structures which allows a broader cov erage of tran- sition states samples with conv entio n a l interpolation technics as illustrated Figure H18 . Indeed, we are not constrained to initiate paths b et ween the main minimum energy structures. This broad co ver- age of the TS ensem ble is less straightforw ard for NEB dep ending on the system and its PES which significan tly hinders systematic approach to T ransition state search. In contrast, ASTRA allo ws a generalized exploration without system-sp ecific considerations. G.2.2 Electro cyclic reaction W e to ok multiple established TS generation metho ds as baselines for the D ASA system to ensure that ASTRA co vers all relev ant transition states. W e ran a CREST simulation [ 93 , 94 ] with the default settings for b oth the reactant and pro duct. This resulted in 20 reactant and 162 pro duct conformers. W e then carried out a Newton T ra jectory 2 scan [ 10 ] for each reactant and pro duct conformer with the asso ciations and disso ciations defined by the tw o oxygen-h ydrogen b onds and the carb on-carbon b ond within the formed 5-membered ring. Additionally , we carried out double-ended searc hes. W e ran one double-ended search p er conformer, where we s elected the conformer of the other side to b e the one with the low est RMSD. The double-ended search was then carried out with b oth the Direct Max Flux (DMF) metho d [ 95 ] and with a B-Spline-based optimization [ 96 ]. The initial tra jectory w as determined by geo desic in terp olation [ 91 ] for DMF and with linear interpolation in Cartesian space for the B-Spline-based optimization. All generated guess structures were sub jected to the identical Dimer optimization algorithm with a maximum of 300 optimization steps. App endix H Ablation Studies H.1 Effect of Score-Based In terp olation and Score-Aligned Ascent W e inv estigate the effects of tw o interpolation metho ds, Iso densit y In terp olation (I I) and Simple A v eraging (SA) , both with and without the application of Score-Aligned Ascen t (SAA) , on the double w ell potential, as sho wn in Figure H5 . When used alone, SA fails to capture the true transition state, instead generating samples scattered betw een the tw o basins. In contrast, II pro duces samples closer to the true transition state. The subsequen t application of SAA further refines these outcomes. F or b oth interpolation metho ds, SAA guides the diffusion pro cess tow ard the transition state (TS) region, correcting deviations and ensuring the final configurations closely align with the true TS. Notably , the com bination of I I and SAA yields the most accurate and physically meaningful transition path. 23 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H5 : Impact of Score-Based Interpolation and Score-Aligned Ascent on double w ell p otential. W e extended this ablation study to the tw o-dimensional p oten tial energy surfaces of the Müller- Bro wn and double path p oten tial. On b oth systems, as shown in Figures H6 and H7 , II dra ws a dividing line b et ween the tw o states, while SA is less effective, producing a scattered distribution of samples, sev eral of which are located in non-physical high-energy regions. Neither in terp olation metho d alone is sufficient for precise TS lo calization. The application of SAA prov es crucial, refining the samples b y collapsing the broad distributions onto the precise lo cations of the low- and high- energy transition states. As quantified by the L 2 distance statistics in T able H1 , b oth interpolation metho ds alone yield substan tial errors, whereas subsequently applying SAA dramatically reduces the distance to the nearest transition state, confirming its critical role. 24 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H6 : Impact of Score-Based Interpolation and Score-Aligned Ascent on Müller-Brown p oten tial. T able H1 : L2 Distance statistics for analytical p oten tials from the closer transition state. The b est results are b olded , and the second-b est results are underline d . P otential I I I I+SAA SA SA+SAA Double W ell 0.1174 ± 0.0101 0.0002 ± 0.0000 0.5870 ± 0.5000 0.0002 ± 0.0000 Müller-Bro wn 7.3632 ± 1.0707 0.0376 ± 0.0044 6.8723 ± 3.1354 0.0449 ± 0.0107 Double P ath 0.6810 ± 0.1008 0.0511 ± 0.0377 0.6998 ± 0.1511 0.0384 ± 0.0358 25 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H7 : Impact of Score-Based Interpolation and Score-Aligned Ascent on double path p oten tial. W e further extend our ablation study to alanine dip eptide to assess the metho d’s p erformance in a high-dimensional system. The results, presented in Figure H8 , highlight the critical role of SAA. When used alone, b oth I I and SA incorrectly identify the transition state region by generating samples lo cated b et ween the tw o basins. Up on introducing SAA, the p erformance of both metho ds improv es notably . SAA successfully guides the generated samples to probable TS regions, causing them to con verge precisely onto the kno wn, physically meaningful transition states on the potential energy surface. 26 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H8 : Impact of Score-Based Interpolation and Score-Aligned Ascent on alanine dip eptide. A similar trend is observed for the folding of chignolin, depicted in Figure H9 . While b oth inter- p olation metho ds can generate a coarse path b et w een the folded and unfolded states, the resulting samples are diffuse and fail to define the transition pathw a y clearly . The addition of SAA is essential for refining this pathw ay , guiding the scattered p oin ts to ward the transition state region. This result demonstrates that the combination of I I and SAA is a robust and effective metho d for iden tifying transition states in complex biomolecular systems. 27 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H9 : Impact of Score-Based Interpolation and Score-Aligned Ascent on Chignolin. F or the electro cyclical reaction, as illustrated in Figure H10 , b oth I I and SA fail to precisely lo cate the TS of the reaction. Instead, the resulting samples are broadly distributed in the region b et w een the tw o energy minima corresponding to the op en (colored) and closed (colorless) isomers. The application of SAA is critical, as it refines this scattered distribution, driving the system to the more probable TS structures. This result underscores the metho d’s ability to na vigate the complex PES of a chemical reaction. 28 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H10 : Impact of Score-Based Interpolation and Score-Aligned Ascent on D ASA reaction. H.2 Committor Analysis of Chemical Systems In Figure H11 , we report histograms of the distributions of committor v alues calculated based on the samples in Figure H8 . W e observe that giv en the partial cov erage of the metastable states by our MD sim ulations at 300 K, simply using Score-Based In terp olation allows sampling an intermediate region but not the TSs themselv es. The SAA algorithm successfully directs the samples tow ards the true TSs as demonstrated b y a p eak ed distribution of the committor v alues around 0.5. Sup erDiff AND surpasses SA in our exp erimen ts. 29 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H11 : Histogram of calculated committors on alanine dip eptide. W e also rep ort ML committor v alues computed with the trained mo dels from [ 36 ] to compare the effects of SBI and SAA algorithm. W e observ e that SAA consistently increases the num b er of samples close to the q = 0 . 5 region. W e note that the ML committor model draws muc h sharp er iso committor surfaces compared to our MD ev aluation for alanine dipeptide. 30 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H12 : Histogram of ML predicted committors on Chignolin. 31 (a) I I. (b) SA. (c) I I + SAA. (d) SA + SAA. Fig. H13 : Histogram of ML predicted committors on DASA reaction. 32 H.3 Sensitivit y to Hyp erparameters W e present a detailed hyperparameter sw eep for ASTRA, and a comparison of SAA against NEB. These results are the basis for the discussion in Sections G.1 and G.2 . T able H2 : Hyp erparameter Ablation Study ev aluated by running the Dimer metho d on ASTRA generated samples. The main result configuration is shown first. Eac h subsequen t ro w ablates a single hyperparameter, with the mo dified v alue in b old . No v alue means that the SAA w as unstable and the force field even tually returns NaN. W e rep ort median v alues for all metrics. Method GS PR OS SS Conv. Rate ↑ Con v. Step ↓ P os. RMSD ↓ Energy Diff. ↓ (Å) (kcal · mol − 1 ) Main 1.0 0.05 1000 0.0100 0.84 14 0.010 -0.50 Ablation on the Guidanc e Sc ale (GS) GS = 2.0 2.0 0.05 1000 0.0100 0.86 34 0.011 -0.51 GS = 3.0 3.0 0.05 1000 0.0100 0.87 11 0.011 -0.46 Ablation on the Pause R atio (PR) PR = 0.00 1.0 0.00 1000 0.0100 – – – – PR = 0.25 1.0 0.25 1000 0.0100 0.78 70 0.05 -0.12 PR = 0.75 1.0 0.75 1000 0.0100 0.41 129 0.018 -1.1 PR = 0.10 1.0 0.10 1000 0.0100 0.13 294 0.06 -1.2 Ablation on the Optimization Steps (OS) Steps = 100 1.0 0.05 100 0.0100 0.27 193 0.38 -1.8 Steps = 250 1.0 0.05 250 0.0100 0.55 161 0.17 -0.83 Steps = 500 1.0 0.05 500 0.0100 0.79 84 0.03 -0.53 Steps = 750 1.0 0.05 750 0.0100 0.84 17 0.01 -0.50 Steps = 1250 1.0 0.05 1250 0.0100 0.84 20 0.01 -0.49 Steps = 1500 1.0 0.05 1500 0.0100 0.88 14 0.01 -0.48 Ablation on the Step Size (SS) SS = 0.0005 1.0 0.05 1000 0.0005 0.25 209 0.47 -2.8 SS = 0.001 1.0 0.05 1000 0.0010 0.31 – 0.36 -1.6 SS = 0.005 1.0 0.05 1000 0.005 0.80 18 0.018 -0.51 SS = 0.02 1.0 0.05 1000 0.02 0.81 76 0.018 -0.49 SS = 0.05 1.0 0.05 1000 0.05 0.50 108 0.07 -0.54 33 T able H3 : Hyp erparameter Ablation Study ev aluated b y computing the MD committor. The main result configuration is shown first. Each subsequen t ro w ablates a single h yp erparameter, with the modified v alue in b old . No v alue means that the SAA w as unstable and the force field even tually returns NaN. The num b er of stable samples corresp onds to the num b er of structure for which running MD for 2 ps starting, from these configurations, is stable. The metrics w ere ev aluated by sampling 100 configurations from ASTRA. Metho d GS PR OS SS % of Comm. ↑ Num. stable samples in [0.4,0.6] (out of 100) Main 1.0 0.05 1000 0.0100 85 76 Ablation on the Guidanc e Sc ale (GS) GS = 2.0 2.0 0.05 1000 0.0100 81 88 GS = 3.0 3.0 0.05 1000 0.0100 80 78 Ablation on the Pause R atio (PR) PR = 0.00 1.0 0.00 1000 0.0100 – – PR = 0.25 1.0 0.25 1000 0.0100 42 44 PR = 0.75 1.0 0.75 1000 0.0100 61 79 PR = 0.10 1.0 0.10 1000 0.0100 21 98 Ablation on the Optimization Steps (OS) Steps = 100 1.0 0.05 100 0.0100 19 96 Steps = 250 1.0 0.05 250 0.0100 40 82 Steps = 500 1.0 0.05 500 0.0100 75 76 Steps = 750 1.0 0.05 750 0.0100 88 75 Steps = 1250 1.0 0.05 1250 0.0100 86 76 Steps = 1500 1.0 0.05 1500 0.0100 84 74 Ablation on the Step Size (SS) SS = 0.0005 1.0 0.05 1000 0.0005 12 100 SS = 0.001 1.0 0.05 1000 0.0010 15 100 SS = 0.005 1.0 0.05 1000 0.005 80 78 SS = 0.02 1.0 0.05 1000 0.02 68 77 SS = 0.05 1.0 0.05 1000 0.05 11 14 34 H.3.1 Qualit y of samples from classic interpolation approaches W e ev aluate tw o baseline methods for generating conformational pathw ays: linear in terp olation, p er- formed after applying the Kabsch algorithm [ 97 ], and a geodesic in terp olation approac h based on in ternal co ordinates, as proposed b y Ref. 91 , in Figure H14 and H15 Sp ecifically for the alanine dip ep- tide, b oth methods fail to pro duce physically realistic pathw a ys, as they tra verse significant energy barriers on the PES. Linear interpolation, in particular, is prone to generating intermediate confor- mations with sev ere steric clashes, leading to geometrically in v alid structures. While the geo desic metho d a voids such direct structural inconsistencies, the path wa y it defines remains energetically pro- hibitiv e and is, therefore, an unviable represen tation of the transition. Therefore, further optimization is needed, using NEB for example. F or suc h flexible systems with structurally very differen t pathw ays, these classical interpolation metho ds might struggle to provide a broad and v alid set of initial guesses for optimization algorithms. (a) Linear interpolation. (b) Geo desic interpolation. (c) Structure of linear interpolation. (d) Structure of geo desic interpolation. Fig. H14 : Comparison of linear and geo desic in terp olation for alanine dip eptide. The top row visu- alizes the interpolated pathw ays on the potential energy surface (PES), while the bottom ro w shows represen tative in termediate structures. 35 (a) Linear interpolation. (b) Geo desic interpolation. (c) Structure of linear interpolation. (d) Structure of geo desic interpolation. Fig. H15 : Comparison of linear and geo desic interpolation for the folding of c hignolin. The top row displa ys the path wa ys on the PES, and the b ottom row shows in termediate molecular structures. 36 H.4 Noise level for SAA optimization (a) Latent-lev el optimization. (b) Clean structure-level optimization. Fig. H16 : Committor histograms comparison of a latent-lev el optimization and a clean structure- lev el optimization. This is for PR=0.05. In Figure H16 , we provide the comparison b et ween optimizing at the latent level or at the clean structure lev el. The results show that the p erformance is almost iden tical. This stems from the fact that the force predictions are computed at the clean structure level while the SAA direction is computed at the latent level inducing a discrepancy in b oth cases. 37 H.5 SAA and NEB from standard in terp olation metho ds Figure H18 shows the optimized minimum energy paths obtained by running NEB starting from linear and geo desic-in terp olated paths. The paths are made of 25 in termediate images. W e clearly observe that NEB fails to conv erge the second TS from the linearly-interpolated path. This probably stems from the p o or quality of these samples as highlighted in Section H.3.1 . W e also pro vide in Figure H18 the details of the linear and geo desic interpolations. W e show the different interpolated paths and the 5 selected middle frames that are used to initialize SAA for the results in T able H4 . A dditionally , we highligh t that when energy minimization is not necessary (as is the case for ASTRA, unlik e NEB), w e can sample a broader ensemble of guesses, allowing for b etter cov erage of the TSE. (a) Linear Interpolation. (b) Geo desic Interpolation. Fig. H17 : NEB transition state guesses and minimum energy paths computed from linear and geo desic interpolation guess paths. The yello w empty circles corresp ond to the initial guesses computed b y interpolating b et ween the start (red) and end (blue) configurations using the giv en interpolation tec hnique. The optimized paths are colored with the viridis color sc heme. T able H4 : Ev aluation of TS samples generated b y running SAA or NEB after initializing guess structures from classical interpolation algorithms (linear and geodesic in terp olation). W e report metrics asso ciated with running the Dimer metho d from these structures. The "minimized" or "non-minimized" initialization corresp onds to minimizing the energy of the structure b efore interpolating the initial guess path. Metho d % of Comm. ↑ Num. stable samples Conv. Rate ↑ Conv. Step ↑ P os . RMSD ↓ Energy Diff. ↓ [0.4,0.6] (out of 56) (Å) (kcal · mol − 1 ) Geo d e sic Interpolation (minimized) SAA 0.83 54 0.93 20 0.013 -0.82 NEB (baseline) 56 0.98 0.07 -0.009 Geo d e sic Interpolation (non-minimized) SAA 0.9 40 0.85 23 0.012 -0.69 NEB (baseline) 5 0.0 – – – Linear Interpolation (minimized) SAA 0.3 53 0.52 137 0.17 -1.01 NEB (baseline) 50 0.98 0.008 -0.025 38 (a) Minimized. (b) Non-minimized. Fig. H18 : In terp olation paths using geo desic interpolation with the start and end structures used to ev aluate SAA p erformance and the NEB baseline. W e highlight in orange the 5 middle points for eac h path that were taken as input for SAA. The tw o graphs (a) and (b) corresp ond to whether the initial configurations are optimized with energy minimization b efore computing the interpolated path. As required by NEB, the minimized configuration (b) where used for NEB calculations. 39 H.6 Score-Based Interpolation TS region co vering Figure H19 shows the distribution of samples obtained from Iso densit y Interpolation when v arying the guidance scale. Figure H20 shows this same distribution when using Simple A veraging of scores instead of I I. (a) Guidance 1.0. (b) Guidance 2.0. (c) Guidance 3.0. Fig. H19 : Effect of the guidance scale when sampling 100 structures using Isodensity Interpolation to guide the reverse diffusion pro cess. 40 (a) Guidance 1.0. (b) Guidance 2.0. (c) Guidance 3.0. Fig. H20 : Effect of the guidance scale when sampling 100 structures using Simple A v eraging to guide the reverse diffusion pro cess. 41 References [1] Eyring, H.: The activ ated complex in chemical reactions. The Journal of c hemical physics 3 (2), 107–115 (1935) [2] Wigner, E.: The transition state metho d. T ransactions of the F arada y So ciet y 34 , 29–41 (1938) [3] Lindorff-Larsen, K., Piana, S., Dror, R.O., Shaw, D.E.: How fast-folding proteins fold. Science 334 (6055), 517–520 (2011) [4] Tiw ary , P ., W alle, A.: A review of enhanced sampling approac hes for accelerated molecular dynamics. Multiscale materials mo deling for nanomec hanics, 195–221 (2016) [5] Y ang, Y.I., Shao, Q., Zhang, J., Y ang, L., Gao, Y.Q.: Enhanced sampling in molecular dynamics. J. Chem. Phys. 151 (7) (2019) [6] Hénin, J., Lelièvre, T., Shirts, M.R., V a l sson, O., Delemotte, L.: Enhanced sampling metho ds for molecular dynamics simulations. arXiv preprint arXiv:2202.04164 (2022) [7] Shen, W., Zhou, T., Shi, X.: Enhanced sampling in molecular dynamics simulations and their latest applications – A review. Nano Research 16 (12), 13474–13497 (2023) [8] Dewy er, A.L., Zimmerman, P .M.: Finding Reaction Mec hanisms, Intuitiv e or Otherwise. Org. Biomol. Chem. 15 (3), 501–504 (2017) [9] Simm, G.N., V aucher, A.C., Reiher, M.: Exploration of Reaction Path wa ys and Chemical T ransformation Netw orks. J. Phys. Chem. A (2018) [10] Unsleb er, J.P ., Reiher, M.: The Exploration of Chemical Reaction Netw orks. Annu. Rev. Phys. Chem. 71 (1), 121–142 (2020) [11] Baiardi, A., Grimmel, S.A., Steiner, M., Türtscher, P .L., Unsleb er, J.P ., W eymuth, T., Reiher, M.: Expansiv e Quantum Mechanical Exploration of Chemical Reaction Paths. Acc. Chem. Res. 55 (1), 35–43 (2022) [12] Ismail, I., Ma jerus, R.C., Habershon, S.: Graph-Driven Re action Discov ery: Progress, Challenges, and F uture Opp ortunities. J. Phys. Chem. A 126 (40), 7051–7069 (2022) [13] Steiner, M., Reiher, M.: Autonomous reaction netw ork exploration in homogeneous and hetero- geneous catalysis. T op. Catal. 65 (1), 6–39 (2022) [14] Steiner, M., Reiher, M.: A human-mac hine interface for automatic exploration of chemical reaction netw orks. Nat. Commun. 15 (1), 3680 (2024) [15] Mehdi, S., Smith, Z., Herron, L., Zou, Z., Tiwary , P .: Enhanced sampling with mac hine learning. Ann u. Rev. Ph ys. Chem. 75 (2024), 347–370 (2024) [16] Mendels, D., Piccini, G., Parrinello, M.: Collective v ariables from lo cal fluctuations. J. Chem. Ph ys. Lett. 9 (11), 2776–2781 (2018) [17] W ang, Y., Rib eiro, J.M.L., Tiw ary , P .: P ast–future information b ottlenec k for sampling molecular reaction co ordinate sim ultaneously with thermo dynamics and kinetics. Nat. Comm un. 10 (1), 3573 (2019) [18] Kang, P ., Zhang, J., T rizio, E., Hou, T., Parrinello, M.: Committors without descriptors. arXiv preprin t arXiv:2510.18018 (2025) [19] T rizio, E., Kang, P ., P arrinello, M.: Everything ev erywhere all at once: a probability-based enhanced sampling approach to rare even ts. Nat. Comput. Sci., 1–10 (2025) [20] Das, S., Raucci, U., T rizio, E., Kang, P ., Neves, R.P ., Ramos, M.J., P arrinello, M.: A machine 42 learning-driv en, probabilit y-based approach to enzyme catalysis. ACS Catal. 15 , 9785–9792 (2025) [21] Zhang, J., Bonati, L., T rizio, E., Zhang, O., Kang, Y., Hou, T., P arrinello, M.: Descriptor-free collectiv e v ariables from geometric graph neural netw orks. J. Chem. Theory Comput. 20 (24), 10787–10797 (2024) [22] T an, A.R., Dietsc hreit, J.C., Gómez-Bom barelli, R.: Enhanced sampling of robust molecular datasets with uncertaint y-based collective v ariables. J. Chem. Phys. 162 (3) (2025) [23] Jung, H., Covino, R., Hummer, G.: Artificial intelligence assists disco very of reaction co ordinates and mec hanisms from molecular dynamics simulations. arXiv preprint arXiv:1901.04595 (2019) [24] Sun, L., V andermause, J., Batzner, S., Xie, Y., Clark, D., Chen, W., K ozinsky , B.: Multitask mac hine learning of collective v ariables for enhanced sampling of rare even ts. J. Chem. Theory Comput. 18 (4), 2341–2353 (2022) [25] Jung, H., Co vino, R., Arjun, A., Leitold, C., Dellago, C., Bolhuis, P .G., Hummer, G.: Mac hine-guided path sampling to disco ver mechanisms of molecular self-organization. Nature Computational Science 3 (4), 334–345 (2023) [26] Kho o, Y., Lu, J., Ying, L.: Solving for high-dimensional committor functions using artificial neural netw orks. Research in the Mathematical Sciences 6 , 1–13 (2019) [27] Li, Q., Lin, B., Ren, W.: Computing committor functions for the study of rare even ts using deep learning. The Journal of Chemical Physics 151 (5) (2019) [28] Li, H., Kho o, Y., Ren, Y., Ying, L.: A semigroup metho d for high dimensional committor func- tions based on neural netw ork. In: Mathematical and Scien tific Machine Learning, pp. 598–618 (2022). PMLR [29] Strahan, J., Finkel, J., Dinner, A.R., W eare, J.: Predicting rare even ts using neural netw orks and short-tra jectory data. Journal of computational ph ysics 488 , 112152 (2023) [30] Mitc hell, A.R., Rotskoff, G.M.: Committor guided estimates of molecular transition rates. Journal of Chemical Theory and Computation 20 (21), 9378–9393 (2024) [31] Megías, A., Con treras Arredondo, S., Chen, C.G., T ang, C., Roux, B., Chipot, C.: Iterativ e v ariational learning of committor-consisten t transition pathw a ys using artificial ne ural net works. Nat. Comput. Sci., 1–11 (2025) [32] Arredondo, S.C., T ang, C., T almazan, R.A., Megías, A., Chen, C.G., Chip ot, C.: F rom atoms to dynamics: Learning the committor without collective v ariables. arXiv preprint (2025) [33] T almazan, R.A., Chip ot, C.: F rom static path wa ys to dynamic mec hanisms: A committor-based data-driv en approac h to chemical reactions. J. Chem. Inf. Mo del. (2025) [34] Giusepp e Chen, C., T ang, C., Megías, A., T almazan, R.A., Con treras Arredondo, S., Roux, B., Chip ot, C.: F ollowing the committor flow: A data-driv en discov ery of transition pathw ays. J. Chem. Theory Comput. (2026) [35] Rotsk off, G.M., Mitchell, A.R., V anden-Eijnden, E.: A ctive imp ortance sampling for v ariational ob jectives dominated by rare ev ents: Consequences for optimization and generalization. In: Mathematical and Scientific Machine Learning, pp. 757–780 (2022). PMLR [36] Kang, P ., T rizio, E., Parrinello, M.: Computing the committor with the committor to study the transition state ensemble. Nat. Comput. Sci. 4 (6), 451–460 (2024) [37] P attanaik, L., Ingraham, J.B., Grambow, C.A., Green, W.H.: Generating transition states of 43 isomerization reactions with deep learning. Physical Chemistry Chemical Physics 22 (41), 23618– 23626 (2020) [38] Duan, C., Du, Y., Jia, H., Kulik, H.J.: Accurate transition state generation with an ob ject-aw are equiv arian t elementary reaction diffusion mo del. Nature computational science 3 (12), 1045–1055 (2023) [39] Kim, S., W oo, J., Kim, W.Y.: Diffusion-based generative ai for exploring transition states from 2d molecular graphs. Nature Communications 15 (1), 341 (2024) [40] Duan, C., Liu, G.-H., Du, Y., Chen, T., Zhao, Q., Jia, H., Gomes, C.P ., Theo dorou, E.A., Kulik, H.J.: Optimal transp ort for generating transition states in chemical reactions. Nature Machine In telligence 7 (4), 615–626 (2025) [41] Lewis, S., Hemp el, T., Jiménez-Luna, J., Gastegger, M., Xie, Y., F oong, A.Y.K., Satorras, V.G., Ab din, O., V eeling, B.S., Zap orozhets, I., Chen, Y., Y ang, S., F oster, A.E., Sc hneuing, A., Nigam, J., Barbero, F., Stimp er, V., Campb ell, A., Yim, J., Lienen, M., Shi, Y., Zheng, S., Sc hulz, H., Munir, U., Sordillo, R., T omiok a, R., Clementi, C., Noé, F.: Scalable emulation of protein equilibrium ensembles with generative deep learning. Science 389 (6761), 9817 (2025) [42] Jing, B., Stärk, H., Jaakk ola, T., Berger, B.: Generativ e modeling of molecular dynamics tra jectories. Adv. Neural. Inf. Pro cess. Syst. 37 , 40534–40564 (2024) [43] Costa, A.d.S., P onnapati, M., Rubin, D., Smidt, T., Jacobson, J.: A ccelerating Protein Molecular Dynamics Simulation with DeepJump. arXiv preprint arXiv:2509.13294 (2025) [44] Thiemann, F.L., Resch ützegger, T., Esp osito, M., T addese, T., Olarte-Plata, J.D., Martelli, F.: F orce-free molecular dynamics through autoregressiv e equiv arian t netw orks. arXiv preprint arXiv:2503.23794 (2025) [45] Nam, J., Steiner, M., Misterk a, M., Y ang, S., Singhal, A., Gómez-Bombarelli, R.: T ransferable learning of reaction pathw ays from geometric priors. J. Phys. Chem. Lett. 16 (45), 11690–11699 (2025) [46] Hait, D., Estrada Pabon, J.D., Stohr, M., Martínez, T.J.: Lo cating ab initio transition states via geo desic construction on machine-learned p oten tial energy surfaces. Journal of Chemical Theory and Computation 21 (22), 11632–11644 (2025) [47] W ang, H., Qiu, Y., W ang, Y., Brek elmans, R., Du, Y.: Generalized flow matching for transition dynamics mo deling. arXiv preprint arXiv:2410.15128 (2024) [48] Ra ja, S., Šípk a, M., Psenk a, M., Kreiman, T., Pa velk a, M., Krishnapriyan, A.S.: Action- minimization meets generativ e mo deling: Efficien t transition path sampling with the onsager- mac hlup functional. arXiv preprin t arXiv:2504.18506 (2025) [49] T uo, P ., Chen, J., Li, J.: Flow matc hing for reaction pathw ay generation. arXiv preprin t arXiv:2507.10530 (2025) [50] Ho, J., Salimans, T.: Classifier-free diffusion guidance. arXiv preprin t arXiv:2207.12598 (2022) [51] Du, Y., Durk an, C., Strudel, R., T enenbaum, J.B., Dieleman, S., F ergus, R., Sohl-Dic kstein, J., Doucet, A., Grathw ohl, W.S.: Reduce, reuse, recycle: Comp ositional generation with energy- based diffusion mo dels and mcmc. In: In ternational Conference on Machine Learning, pp. 8489– 8510 (2023). PMLR [52] Skreta, M., A tanack ovic, L., Bose, J., T ong, A., Neklyudov, K.: The sup erposition of diffusion mo dels using the itô density estimator. In: The Thirteen th International Conference on Learning Represen tations (2024) [53] Skreta, M., Akhound-Sadegh, T., Ohanesian, V., Bondesan, R., Aspuru-Guzik, A., Doucet, A., 44 Brek elmans, R., T ong, A., N eklyudo v, K.: F eynman-k ac correctors in diffusion: Annealing, guid- ance, and pro duct of exp erts. In: F ort y-second In ternational Conference on Machine Learning (2025) [54] Nam, J., Máté, B., T oshev, A.P ., Kaniselv an, M., Gómez-Bombarelli, R., Chen, R.T., W o o d, B., Liu, G.-H., Miller, B.K.: Enhancing diffusion-based sampling with molecular collective v ariables. arXiv preprint arXiv:2510.11923 (2025) [55] Song, Y., Sohl-Dickstein, J., Kingma, D.P ., Kumar, A., Ermon, S., Poole, B.: Score-based generativ e modeling through sto c hastic differential equations. arXiv preprin t (2020) [56] Del Camp o, J.M., Köster, A.M.: A hierarc hical transition state search algorithm. The Journal of c hemical ph ysics 129 (2) (2008) [57] P eters, B., Heyden, A., Bell, A.T., Chakrab ort y , A.: A gro wing string metho d for determining transition states: Comparison to the nudged elastic band and string metho ds. The Journal of c hemical ph ysics 120 (17), 7877–7886 (2004) [58] Zimmerman, P .: Reliable transition state searches in tegrated with the growing string metho d. Journal of chemical theory and computation 9 (7), 3043–3050 (2013) [59] Maiti, S.R., Buttar, D., Duarte, F.: Benchmark of double-ended transition state search metho ds for metal-catalysed reactions (2025) [60] Müller, K., Brown, L.D.: Lo cation of saddle p oin ts and minim um energy paths by a constrained simplex optimization pro cedure. Theor. Chim. A cta 53 (1), 75–93 (1979) [61] Chekmarev, D.S., Ishida, T., Levy , R.M.: Long-time conformational transitions of alanine dip ep- tide in aqueous solution: Contin uous and discrete-state kinetic mo dels. The Journal of Physical Chemistry B 108 (50), 19487–19495 (2004) [62] Arts, M., Garcia Satorras, V., Huang, C.-W., Zugner, D., F ederici, M., Clementi, C., Noé, F., Pinsler, R., Berg, R.: T w o for one: Diffusion mo dels and force fields for coarse-grained molecular dynamics. Journal of Chemical Theory and Computation 19 (18), 6151–6159 (2023) [63] Molgedey , L., Sch uster, H.G.: Separation of a mixture of indep enden t signals using time delay ed correlations. Physical review letters 72 (23), 3634 (1994) [64] Pérez-Hernández, G., P aul, F., Giorgino, T., De F abritiis, G., Noé, F.: Iden tification of slo w molecular order parameters for marko v mo del construction. J. Chem. Phys. 139 (1) (2013) [65] Rey es, C.A., Karr, A., Ramsp erger, C.A., K, A.T.G., Lee, H.J., Picazo, E.: Compartmentalizing donor–acceptor stenhouse adducts for structure–prop ert y relationship analysis. J. Am. Che m. So c. 147 (1), 10–26 (2024) [66] T ang, C., Pandey , M.P ., Chen, C.G., Megías, A., Dehez, F., Chip ot, C.: Breaking the timescale barrier: Generative discov ery of conformational free-energy landscap es and transition pathw ays. arXiv preprint arXiv:2510.24979 (2025) [67] K oulischer, F., Handke, F., Deleu, J., Demeester, T., Am brogioni, L.: F eedback guidance of diffusion mo dels. arXiv preprint arXiv:2506.06085 (2025) [68] Ho, J., Jain, A., Abbeel, P .: Denoising diffusion probabilistic mo dels. Adv ances in neural information pro cessing systems 33 , 6840–6851 (2020) [69] Skreta, M., Akhound-Sadegh, T., Ohanesian, V., Bondesan, R., Aspuru-Guzik, A., Doucet, A., Brek elmans, R., T ong, A., Neklyudov, K.: F eynman-k ac correctors in diffusion: Annealing, guidance, and pro duct of exp erts. arXiv preprint arXiv:2503.02819 (2025) 45 [70] Singhal, R., Horvitz, Z., T eehan, R., Ren, M., Y u, Z., McKeo wn, K., Ranganath, R.: A gen- eral framew ork for inference-time scaling and steering of diffusion mo dels. arXiv preprin t arXiv:2501.06848 (2025) [71] Liu, N., Li, S., Du, Y., T orralba, A., T enen baum, J.B.: Comp ositional visual generation with comp osable diffusion models. In: Europ ean Conference on Computer Vision, pp. 423–439 (2022). Springer [72] Henk elman, G., Jónsson, H.: A dimer method for finding saddle p oin ts on high dimensional p oten tial surfaces using only first deriv atives. J. Chem. Ph ys. 111 (15), 7010–7022 (1999) [73] Olsen, R., Kroes, G., Henkelman, G., Arnaldsson, A., Jónsson, H.: Comparison of metho ds for finding saddle p oin ts without knowledge of the final states. J. Chem. Phys. 121 (20), 9776–9792 (2004) [74] Heyden, A., Bell, A.T., Keil, F.J.: Efficien t metho ds for finding transition states in chemical reactions: Comparison of improv ed dimer metho d and partitioned rational function optimization metho d. The Journal of chemical ph ysics 123 (22) (2005) [75] Kästner, J., Sherw o od, P .: Sup erlinearly conv erging dimer method for transition state searc h. The Journal of chemical physics 128 (1) (2008) [76] Shang, C., Liu, Z.-P .: Constrained bro yden minimization combined with the dimer metho d for lo cating transition state of complex reactions. Journal of Chemical Theory and Computation 6 (4), 1136–1144 (2010) [77] W eymuth, T., Unsleber, J.P ., Türtscher, P .L., Steiner, M., Sob ez, J.-G., Müller, C.H., Mörchen, M., Klasovita, V., Grimmel, S.A., Ec khoff, M., Csizi, K.-S., Bosia, F., Bensb erg, M., Reiher, M.: SCINE – Softw are for chemical interaction netw orks. J. Chem. Ph ys. 160 (22) (2024) [78] Bensb erg, M., Brunk en, C., Csizi, K.-S., Grimmel, S.A., Gugler, S., Sob ez, J.-G., Steiner, M., Türtsc her, P .L., Unsleb er, J.P ., V aucher, A., W eymuth, T., Reiher, M.: qcscine/readuct: Release 6.0.0. Zeno do (2024). https://doi.org/10.5281/zenodo.13372944 . https://doi.org/10. 5281/zeno do.13372944 [79] Henk elman, G., Ub eruaga, B.P ., Jónsson, H.: A Climbing Image Nudged Elastic Band Metho d for Finding Saddle P oints and Minimum Energy Paths. J. Chem. Phys. 113 (22), 9901–9904 (2000) [80] Henk elman, G., Jónsson, H.: Improv ed tangent estimate in the nudged elastic band metho d for finding minimum energy paths and saddle p oints. J. Chem. Phys. 113 (22), 9978–9985 (2000) [81] Ch ung, H., Kim, J., Mccann, M.T., Klasky , M.L., Y e, J.C.: Diffusion p osterior sampling for general noisy inv erse problems. arXiv preprint arXiv:2209.14687 (2022) [82] Lu, C., Chen, H., Chen, J., Su, H., Li, C., Zh u, J.: Con trastive energy prediction for exact energy-guided diffusion sampling in offline reinforcemen t learning. In: In ternational Conference on Machine Learning, pp. 22825–22855 (2023). PMLR [83] W u, L., T rippe, B., Naesseth, C., Blei, D., Cunningham, J.P .: Practical and asymptotically exact conditional sampling in diffusion models. Adv ances in Neural Information Pro cessing Systems 36 , 31372–31403 (2023) [84] He, J., Hernández-Lobato, J.M., Du, Y., V argas, F.: Rne: a plug-and-pla y framew ork for diffusion densit y estimation and inference-time control. arXiv preprint arXiv:2506.05668 (2025) [85] Sohl-Dic kstein, J., W eiss, E., Mahesw aranathan, N., Ganguli, S.: Deep unsupervised learning using nonequilibrium thermodynamics. In: International Conference on Machine Learning, pp. 2256–2265 (2015). pmlr [86] Song, Y., Ermon, S.: Generative modeling by estimating gradients of the data distribution. 46 A dv ances in neural information pro cessing systems 32 (2019) [87] Liao, Y.-L., W o o d, B., Das, A., Smidt, T.: Equiformerv2: Impro ved equiv ariant transformer for scaling to higher-degree representations. arXiv preprint arXiv:2306.12059 (2023) [88] Stew art, J.J.: Optimization of parameters for semiempirical metho ds V: Mo dification of NDDO appro ximations and application to 70 elements. J. Mol. Mo d. 13 (12), 1173–1213 (2007) [89] Bosia, F., Husch, T., Müller, C., Polonius, S., Sobez, J.-G., Steiner, M., Unsleber, J.P ., Alain, V., W eymuth, T., Reiher, M.: qcscine/sparrow: Release 5.1.0. Zeno do (2024). h ttps://doi.org/10. 5281/zeno do.13372942 [90] Bosia, F., Zheng, P ., V aucher, A., W eymuth, T., Dral, P .O., Reiher, M.: Ultra-fast semi-empirical quan tum chemistry for high-throughput computational campaigns with Sparrow. J. Chem. Phys. 158 (5), 054118 (2023) [91] Zh u, X., Thompson, K.C., Martínez, T.J.: Geo desic in terp olation for reaction path wa ys. The Journal of Chemical Physics 150 (16) (2019) [92] De Bortoli, V., Hutchinson, M., Wirnsb erger, P ., Doucet, A.: T arget score matching. arXiv preprin t arXiv:2402.08667 (2024) [93] Prac ht, P ., Bohle, F., Grimme, S.: Automated exploration of the low-energy chemical space with fast quantum chemical metho ds. Phys. Chem. Chem. Phys. 22 , 7169–7192 (2020) [94] Prac ht, P ., Grimme, S., Bannw arth, C., Bohle, F., Ehlert, S., F eldmann, G., Gorges, J., Müller, M., Neudeck er, T., Plett, C., Spic her, S., Steinbac h, P ., W esoło wski, P .A., Zeller, F.: CREST—A program for the exploration of low-energy molecular c hemical space. J. Chem. Ph ys. 160 (11), 114110 (2024) [95] K o da, S.-i., Saito, S.: Lo cating T ransition States b y V ariational Reaction P ath Optimization with an Energy-Deriv ative-F ree Ob jective F unction. J. Chem. Theory Comput. 20 (7), 2798–2811 (2024). PMID: 38513192 [96] V aucher, A.C., Reiher, M.: Minimum Energy P aths and T ransition States by Curve Optimization. J. Chem. Theory Comput. 14 (6), 3091–3099 (2018). PMID: 29648812 [97] La wrence, J., Bernal, J., Witzgall, C.: A purely algebraic justification of the k absc h-umeyama algorithm. Journal of researc h of the National Institute of Standards and T echnology 124 , 1 (2019) 47
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment