An Integrative Genome-Scale Metabolic Modeling and Machine Learning Framework for Predicting and Optimizing Biofuel-Relevant Biomass Production in Saccharomyces cerevisiae
Saccharomyces cerevisiae is a cornerstone organism in industrial biotechnology, valued for its genetic tractability and robust fermentative capacity. Accurately predicting biomass flux across diverse environmental and genetic perturbations remains a …
Authors: Neha K. Nair, Aaron D'Souza
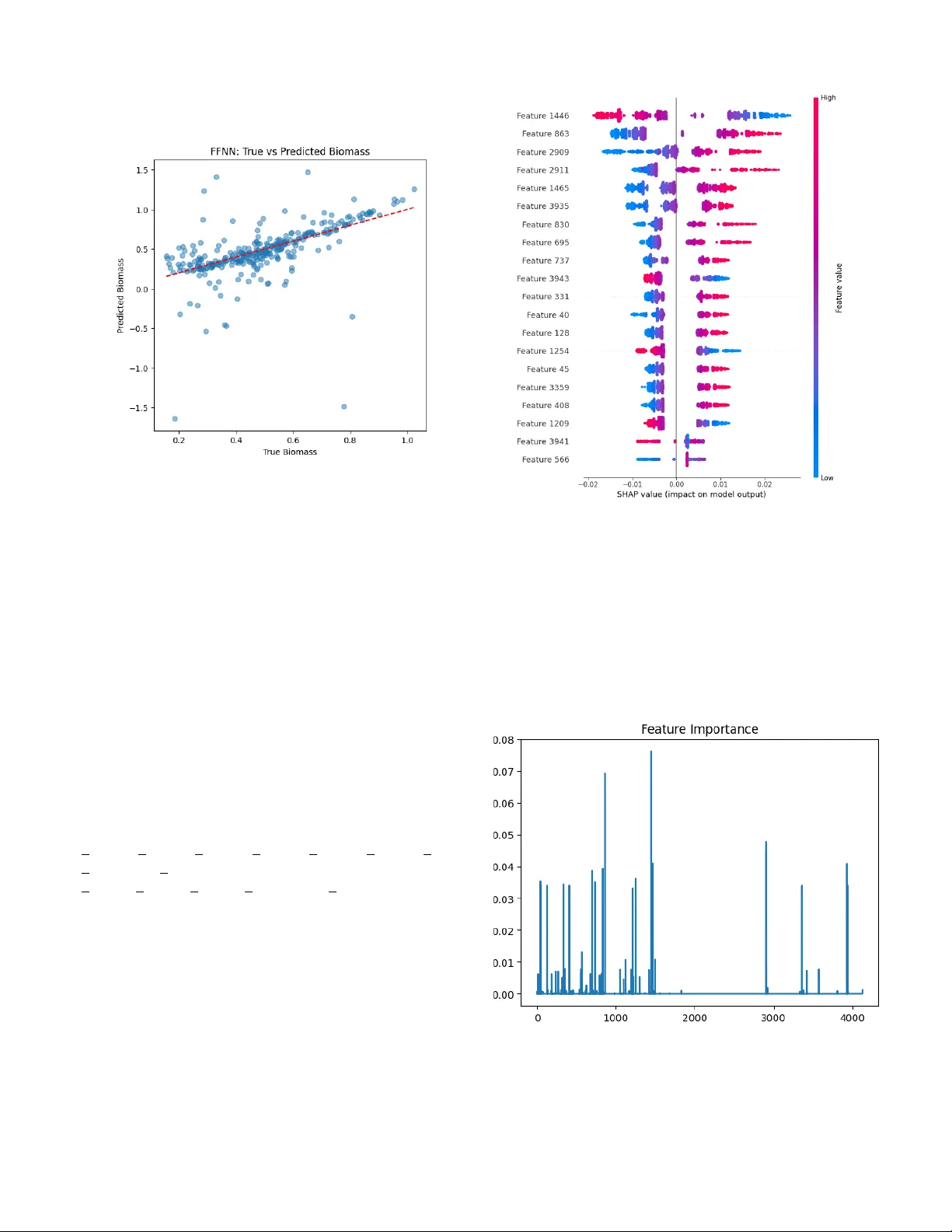
An Inte grati v e Genome-Scale Metabolic Modeling and Machine Learning Frame w ork for Predicting and Optimizing Biofuel-Rele v ant Biomass Production in Sacc har omyces cer e visiae Neha K. Nair Department of Physics and Chemistry National Institute of T echnology , W arangal W arangal, India nk23edi0017@student.nitw .ac.in Aaron D’Souza Department of Electr onics and Communication Engineering National Institute of T echnology , W arangal W arangal, India ad22ecb0f20@student.nitw .ac.in Abstract — Saccharomyces cerevisiae is a cornerstone organism in industrial biotechnology and metabolic engineering, owing to its genetic tractability , well-characterised metabolism, and rob ust fermentative capacity . Despite decades of study , accurately predicting biomass flux across diverse en vironmental and genetic perturbations remains a significant challenge, limiting the ratio- nal design of strains for biofuel production. This study presents a comprehensi ve computational framework that combines the Y east9 genome-scale metabolic model (GEM) with machine learning (ML) and optimisation techniques to systematically predict, interpret, and enhance biomass flux in yeast. Flux balance analysis (FBA) was used to generate a detailed dataset of metabolic flux distributions by varying glucose, oxygen, and ammonium uptake rates. These simulated profiles trained Ran- dom Forest and XGBoost regressors, which achieved coefficients of determination (R 2 ) of 0.99989 and 0.9990, respectively . A feed-forward neural network (FFNN) captured nonlinear flux– biomass relationships, while a variational autoencoder (V AE) uncover ed four distinct metabolic clusters in latent space. SHAP- based feature attribution identified the top twenty most in- fluential reactions, implicating glycolysis, the TCA cycle, and lipid biosynthesis as key contrib utors to biomass yield. In silico o verexpr ession of key r eactions achiev ed a biomass flux of 0.979 gD W · hr − 1 . Bayesian optimisation of nutrient uptak e constraints pr oduced a 12-f old incr ease in pr edicted biomass flux (from 0.0858 to 1.041 gDW · hr − 1 ). A generative adversarial network (GAN) proposed no vel metabolic flux configurations consistent with stoichiometric feasibility , yielding a generated variance of 0.156. This framework demonstrates how GEM- based simulation, inter pretable ML, and generative modelling can significantly advance the understanding and manipulation of yeast metabolism. Further experimental validation is required to confirm the predicted metabolic impro vements. Index T erms — Saccharomyces cerevisiae , genome-scale metabolic model, flux balance analysis, SHAP , Bayesian optimisation, biofuel, machine learning, generativ e adversarial network I . I N T RO D U C T I O N Sacchar omyces cere visiae plays a vital role in fundamental cell biology and applied industrial biotechnology . Its well- annotated genome, tolerance to inhibitory fermentation con- ditions, and capacity to produce ethanol and other biofuel products make it an organism of enduring interest for sus- tainable energy applications [1]. The rational engineering of yeast strains toward maximised biomass or product yields is complicated by the extraordinary complexity of eukaryotic metabolism: thousands of enzymatic reactions, tightly regu- lated gene e xpression programmes, and condition-dependent flux rerouting collectiv ely confound straightforward prediction of cellular behaviour from first principles. Genome-scale metabolic models (GEMs) address this com- plexity by encoding the full stoichiometric network of metabolic reactions and pro viding a computational substrate for constraint-based analyses such as flux balance analysis (FB A) [4]. The Y east9 consensus GEM [2] comprises 4,131 reactions, 2,806 metabolites, and 1,161 genes, making it one of the most comprehensi ve metabolic models av ailable for any eukaryote [5]. FB A identifies optimal flux distributions consistent with stoichiometric, thermodynamic, and capacity constraints, enabling rapid in silico screening of genetic and en vironmental perturbations. Machine learning provides a po werful complement to constraint-based metabolic modelling. Supervised models such as Random Forest and XGBoost extract predicti ve relation- ships from high-dimensional flux datasets, while deep learning architectures enable nonlinear pattern recognition and unsu- pervised representation learning [6], [7], [11]. SHAP (SHap- ley Additi ve exPlanations) values provide a model-agnostic approach to feature attribution, enabling the identification of specific reactions whose flux v ariation most strongly influences predicted biomass [10]. Generative models such as GANs can synthesise nov el flux configurations for de nov o metabolic pathway discovery [18], [20], [25]. Despite the gro wing body of literature at the intersection of GEMs and ML, most studies address narrow objectiv es without providing a unified, end-to-end pipeline that encom- passes data generation, prediction, interpretation, and optimi- sation [7], [8]. This fragmentation limits reproducibility and the translation of computational findings into experimental hypotheses. The present study addresses this gap by developing an integrati ve computational pipeline for S. cer evisiae that en- compasses: (i) GEM-based FB A data generation across sys- tematically varied en vironmental conditions; (ii) dimension- ality reduction and unsupervised clustering via V AE and K- means; (iii) supervised biomass flux prediction using Random Forest, XGBoost, and FFNN; (iv) reaction-le vel interpreta- tion via SHAP analysis; (v) in silico overe xpression and knockout perturbation studies; (vi) oxygen sensitivity analysis; (vii) Bayesian optimisation of nutrient uptake parameters; and (viii) GAN-based generation of novel metabolic flux profiles. I I . R E L A T E D W O R K A. Genome-Scale Metabolic Modelling of Y east The Y east8 consensus GEM provided a comprehensively annotated metabolic network supporting diverse constraint- based analyses [1]. The subsequent Y east9 model introduced thermodynamic feasibility constraints, enabling more physi- ologically grounded flux predictions and expanding pathway cov erage [2]. GEM-based studies ha ve been applied across a wide spectrum of phenotypic objectives, including ethanol yield optimisation, lipid accumulation for biodiesel produc- tion, gro wth under nutrient limitation, and stress response char - acterisation [5]. The COBRApy toolbox [3] provides a flexible Python interface for these analyses, facilitating reproducible and programmable metabolic simulations. Despite their utility , FBA solutions are not unique: many flux distributions can achie ve the same objectiv e value, and the deterministic, steady-state assumption underlying FB A does not capture dynamic regulatory responses or stochastic gene expression [8]. These limitations motiv ate the integration of statistical and machine learning methods that can learn from ensembles of GEM-simulated conditions. B. Mac hine Learning for Metabolic Pr ediction The integration of ML with metabolic modelling has gained momentum as the volume and dimensionality of omics data hav e grown. Ensemble methods such as Random Forest and XGBoost hav e demonstrated strong performance in predict- ing growth rates and product yields from flux or omics features [11]. V ariational autoencoders (V AEs) have been employed to learn compressed, biologically meaningful latent representations of metabolic flux space [15], [17]. Feed- forward neural networks trained on GEM-simulated data have shown capacity to generalise predictions to unseen en viron- mental conditions [9], [19]. SHAP-based interpretability methods hav e been applied to biological models to identify which molecular features most influence predicted outcomes [10]. Bayesian optimisation has emerged as an efficient strategy for navigating high- dimensional parameter spaces in metabolic engineering [12], [13]. GANs are increasingly explored for the generation of nov el biological sequences, metabolic flux configurations, and synthetic pathway designs [18], [20]. C. Resear ch Gap and Motivation Prior integrati ve studies hav e generally addressed isolated components of the modelling pipeline without deli vering a cohesiv e, end-to-end framew ork connecting simulation, pre- diction, interpretation, and optimisation [7], [8]. Furthermore, few studies have incorporated generati ve modelling for novel pathway synthesis within a GEM-constrained setting [20], [25]. The present work addresses these limitations by provid- ing a unified pipeline applicable to S. cer evisiae and extensible to other industrially relev ant microorganisms. I I I . M E T H O D O L O G Y This study integrates genome-scale metabolic modelling with machine learning, deep learning, and optimisation tech- niques to predict and maximise biomass flux in Sacchar omyces cer evisiae . The workflow comprises four principal stages: (i) GEM-based flux simulation, (ii) dimensionality reduction and metabolic state clustering, (iii) supervised predicti ve mod- elling with feature attribution, and (i v) generati ve modelling and en vironmental optimisation. All simulations and model training were implemented in Python using COBRApy [3], scikit-learn, XGBoost, PyT orch, and GPyOpt. All experiments were conducted with fixed random seeds to ensure repro- ducibility . A. Genome-Scale Metabolic Model Simulations The Y east9 consensus GEM, comprising 4,131 reactions, 2,806 metabolites, and 1,161 genes, was used as the metabolic network substrate. The biomass objectiv e reaction is r_2111 . FB A was applied to compute steady-state flux distributions maximising the biomass objectiv e reaction under system- atically v aried en vironmental constraints. Glucose uptake bounds were set at ( − 1 . 0 , 1000 . 0) mmol · gD W − 1 · hr − 1 un- der default conditions, while oxygen uptake bounds were ( − 1000 . 0 , 1000 . 0) mmol · gDW − 1 · hr − 1 . These constraints were systematically v aried across physiologically relev ant ranges, generating a dataset of 2,000 flux profiles each of dimension 4,131, capturing the metabolic response of yeast to a broad range of nutrient av ailabilities. B. Dimensionality Reduction and Unsupervised Clustering A variational autoencoder (V AE) was trained on the high- dimensional flux dataset to learn a low-dimensional latent representation preserving the principal sources of metabolic variation. The encoder maps each flux vector to a probabilistic latent distribution parameterised by mean and log-v ariance; the decoder reconstructs the original flux profile from samples drawn from this distribution. The training objectiv e combines a reconstruction loss with a Kullback–Leibler (KL) div ergence term that regularises the latent space toward a standard normal prior . PCA was applied to the two-dimensional latent embeddings to facilitate visualisation and to estimate the optimal number of clusters via the elbow method and silhouette scoring. K-means clustering partitioned the dataset into four metabolic clusters (labels 0–3), corresponding to distinct biomass productivity regimes. C. Supervised Pr edictive Modelling Three supervised regression models were trained to predict biomass flux from the 4,131-dimensional flux feature vectors. The dataset was split into training (70%), validation (15%), and test (15%) sets. Hyperparameter tuning w as performed exclusi vely on the v alidation set; the test set was strictly held out for final performance ev aluation. Fi ve-fold cross-v alidation was conducted on the training set. Random For est Regressor: An ensemble of decision trees trained with hyperparameters selected by grid-search cross- validation. A test R 2 of 0.99989 was obtained, and a 5-fold CV mean R 2 of 0 . 99991 ± 0 . 00005 was achiev ed. XGBoost Regressor: A gradient-boosted tree ensemble with re gularisation parameters tuned to balance bias and variance, achieving a test R 2 of 0.9990. Feed-F orward Neural Network (FFNN): A multilayer perceptron with ReLU activ ations and dropout regulari- sation trained using the Adam optimiser . Hyperparame- ters—including hidden layer dimensions, learning rate, and dropout rate—were optimised via grid search to capture higher-order nonlinear flux–biomass relationships. Model performance was assessed using R 2 and mean squared error (MSE). Statistical consistency was ev aluated via standard deviation across cross-validation folds. D. F eature Attribution via SHAP Analysis SHAP (SHaple y Additi ve exPlanations) v alues were com- puted for the trained Random Forest model [10] to identify the metabolic reactions most critically governing biomass yield. SHAP v alues decompose individual predictions into additiv e contributions from each feature, providing both global importance rankings and local, sample-specific explanations grounded in cooperativ e game theory [8], [10]. The twenty highest-ranked reactions by mean absolute SHAP value were selected for downstream interpretation and in silico perturba- tion. Results were visualised using beeswarm plots and cluster - stratified heatmaps. E. In Silico P erturbation: Overe xpr ession and Knockout Sim- ulations T o v alidate the biological relev ance of SHAP-ranked re- actions, in silico overexpression and knockout simulations were conducted within the Y east9 GEM framework. Overex- pression was modelled by relaxing the upper flux bound of the target reaction by a defined factor; knockout simulations set both flux bounds to zero. FB A was re-solved for each perturbation and the resulting biomass flux recorded. Simu- lated overe xpression of key reactions resulted in a biomass flux of 0.979 gDW · hr − 1 , approaching the Bayesian-optimised maximum of 1.041 gD W · hr − 1 . F . Oxygen Sensitivity Analysis The lower bound of the oxygen uptake reaction was var - ied from fully aerobic ( − 20 mmol · gD W − 1 · hr − 1 ) to oxygen- limiting ( − 2 mmol · gD W − 1 · hr − 1 ) conditions. FB A was solved at each constraint lev el and the resulting biomass flux recorded, producing a growth-v ersus-oxygen curv e capturing the transition between respiratory and fermentati ve metabolic regimes. G. Bayesian Optimisation of Nutrient Uptake Conditions Bayesian optimisation was applied to a three-dimensional search space defined by glucose, ammonium, and oxygen uptake rates [12], [13], [23]. A Gaussian process surrog ate model approximated the relationship between nutrient uptake parameters and FB A-predicted biomass flux. At each iteration, an expected-improvement acquisition function selected the next parameter combination to ev aluate, balancing exploration with exploitation. H. Gener ative Adversarial Network for Novel P athway Syn- thesis A GAN was trained on FBA-simulated flux distributions (shape: 10 × 4130 ) to learn the statistical manifold of feasi- ble metabolic states in the Y east9 network [18], [20], [25]. The generator synthesises flux vectors indistinguishable from real FBA outputs; the discriminator is trained adversarially to distinguish generated from real flux profiles. Novel flux vectors were ev aluated for stoichiometric and thermodynamic feasibility by re-running FB A with the generated flux as a warm-start solution. The v ariance in the generated outputs was 0.156. I V . R E S U L T S A. GEM Simulation and Dataset Characteristics T able I summarises the key parameters of the Y east9 GEM and the flux dataset generated by FBA. Under default nutrient conditions, the optimal biomass flux was 0.08584 gD W · hr − 1 . Increasing the glucose uptake rate ele vated the predicted biomass flux to 0.88768 gD W · hr − 1 , demonstrating strong carbon-source dependence of gro wth. The resulting flux dataset comprised 2,000 samples across 4,131 reaction dimen- sions. T ABLE I Y E AS T 9 GE M C H AR AC T E R IS T I C S A N D F BA S I MU L A T I ON O U TP U T S Parameter V alue / Description Model Consensus Y east GEM (Y east9) Reactions 4,131 Metabolites 2,806 Genes 1,161 Biomass objective reaction r 2111 Optimal biomass flux (default) 0.08584 gDW · hr − 1 Biomass flux (incr . glucose) 0.88768 gDW · hr − 1 Bayesian optimised flux 1.041 gDW · hr − 1 Flux dataset dimensions 2,000 × 4,131 RF T est R 2 0.99989 XGBoost T est R 2 0.9990 5-fold CV Mean R 2 (RF) 0 . 99991 ± 0 . 00005 B. Latent Space Structur e and Metabolic Clustering The V AE learned a structured two-dimensional latent rep- resentation of the flux dataset (Fig. 1). The elbo w plot shows inertia decreasing from approximately 4,350 at k =2 to ap- proximately 1,500 at k =9 ; the silhouette score peaks at k =2 ( ≈ 0 . 341 ) and at k =6 ( ≈ 0 . 326 ) (Fig. 2). Based on this analysis, K-means partitioning into four clusters was selected to balance biological interpretability with cluster separation (Fig. 3). T able II reports mean biomass flux per cluster . Cluster 1 exhibits the highest mean biomass flux of 0.5543 gD W · hr − 1 , while Cluster 0 follows at 0.4733 gDW · hr − 1 . Clusters 2 and 3 are closely spaced at ≈ 0 . 484 and 0.483 gD W · hr − 1 , respectiv ely . T ABLE II M E AN B I OM A S S F L U X P E R M E TAB O L I C C L US T E R Cluster Mean Biomass Flux (gD W · hr − 1 ) 0 0.473252 1 0.554312 2 0.483752 3 0.482993 Fig. 1. T wo-dimensional latent space learned by the V AE. Each point represents one of the 2,000 FBA-simulated flux profiles projected onto the two principal latent dimensions. Most profiles are concentrated in Latent Dim 1 ∈ [ − 5 , 5] , Latent Dim 2 ∈ [ − 4 , 4] . C. Pr edictive Model P erformance All three supervised models were ev aluated on the held- out test set. The Random Forest Regressor attained a test R 2 of 0.99989 and a 5-fold CV mean R 2 of 0 . 99991 ± 0 . 00005 . The XGBoost Regressor achieved a test R 2 of 0.9990. The Random Forest true-vs-predicted scatter plot (Fig. 4) aligns almost perfectly along the identity line across the full biomass range ( ≈ 0 . 15 – 1 . 05 gDW · hr − 1 ). The FFNN scatter plot (Fig. 5) rev eals higher variance, indicating that the FFNN Fig. 2. Cluster number selection diagnostics. Left: Elbow method (inertia vs. k ), decreasing from ≈ 4350 at k =2 to ≈ 1500 at k =9 . Right: Silhouette score vs. k , with peak at k =2 ( ≈ 0 . 341 ) and secondary peak at k =6 ( ≈ 0 . 326 ), supporting the selection of k =4 . Fig. 3. K-means clustering of flux profiles in latent space ( k =4 ). Cluster 1 (highest mean biomass flux, 0.5543 gDW · hr − 1 ) is associated with the rightward region; remaining clusters occupy overlapping central and left regions. Fig. 4. Scatter plot of Random Forest-predicted versus true biomass flux values on the held-out test set. Points closely follow the identity line across 0.15–1.05 gDW · hr − 1 (T est R 2 = 0.99989). requires further hyperparameter optimisation to match tree- based models on this dataset. Fig. 5. Scatter plot of FFNN-predicted versus true biomass flux values on the held-out test set. Predicted values exhibit higher scatter relative to the Random Forest model, indicating that further hyperparameter optimisation is required. D. SHAP-Based Reaction Importance Global SHAP v alues for the Random Forest model re vealed the 20 most influential reactions (Fig. 6). T op-ranked features by mean absolute SHAP value include Features 1446, 863, 2909, 2911, 1465, 3935, 830, 695, 737, 3943, 331, 40, 128, 1254, 45, 3359, 408, 1209, 3941, and 566. The beeswarm plot sho ws a bipolar pattern: high feature values (red) yield positiv e SHAP contributions while lo w feature values (blue) yield negati ve contributions, with SHAP values ranging from − 0 . 02 to +0 . 02 . The feature importance bar chart (Fig. 7) confirms a sparse distribution, with a dominant peak near reaction index 1500 (importance ≈ 0 . 076 ). The cluster-stratified heatmap (Fig. 8) shows that reactions r 0438, r 0226, r 0439, r 1697, r 1979, r 0770, r 0486, r 0893, and r 0962 are upregulated (flux up to ≈ +45 ), while r 1277, r 1696, r 1763, r 2115, and r 1048 are downre gu- lated (flux down to ≈ − 35 ) across all four clusters. E. In Silico P erturbation Analysis In silico overexpression of the top SHAP-ranked reactions resulted in a biomass flux of 0.979 gD W · hr − 1 , close to the Bayesian-optimised maximum of 1.041 gDW · hr − 1 (Fig. 9). This 11-fold increase ov er the baseline ( ≈ 0 . 086 gDW · hr − 1 ) confirms the identified reactions as robust metabolic engineer- ing targets. Fig. 6. Global SHAP beeswarm plot for the Random Forest model show- ing the top twenty most influential metabolic reactions, ranked by mean absolute SHAP value across all 2,000 FBA-simulated flux profiles. Each point represents one sample; colour encodes feature value (red = high, blue = low). SHAP v alues range from − 0 . 02 to +0 . 02 . A clear bipolar pattern is observed: reactions with high flux values (red) consistently yield positiv e SHAP contributions, while those with low flux values (blue) yield negati ve contributions, confirming directional and interpretable flux–biomass relationships across the simulated condition space. The top-ranked features correspond to reactions in central carbon metabolism whose flux variation most strongly governs predicted biomass yield. Fig. 7. Random Forest feature importance scores across all 4,131 reactions. A dominant peak appears near reaction index 1500 (importance ≈ 0 . 076 ), with a secondary peak near index 950 ( ≈ 0 . 069 ). The vast majority of reactions contribute negligible importance. Fig. 8. Cluster-specific metabolic activity heatmap showing mean flux v alues for selected reactions across the four K-means clusters. Upre gulated reactions (red, up to ≈ +45 ) include r 0438, r 0226, r 0439, r 1697, and r 1979. Downre gulated reactions (blue, down to ≈ − 35 ) include r 1277, r 1696, and r 1763. Fig. 9. Effect of metabolic interventions on biomass flux. Baseline ( ≈ 0 . 086 gDW · hr − 1 ) increases to ≈ 0 . 979 gDW · hr − 1 upon in silico overe xpres- sion, and reaches ≈ 1 . 041 gDW · hr − 1 under Bayesian-optimised conditions. F . Oxygen Sensitivity Analysis The oxygen sensitivity analysis rev ealed a monotonically decreasing growth-v ersus-oxygen curve (Fig. 10). Biomass flux is near-maximal ( ≈ 1 . 0 gDW · hr − 1 , normalised) under the most aerobic conditions ( − 20 mmol · gDW − 1 · hr − 1 ) and declines to approximately 0.38 gDW · hr − 1 as oxygen av ail- ability approaches − 2 mmol · gD W − 1 · hr − 1 , consistent with the critical dependence of biomass synthesis on oxidativ e phosphorylation. G. Bayesian Optimisation of Nutrient Conditions Bayesian optimisation over the three-dimensional nutrient space (glucose, ammonium, and oxygen uptake rates) elev ated Fig. 10. Biomass flux as a function of oxygen uptak e lower bound (mmol · gDW − 1 · hr − 1 ). Flux declines monotonically from ≈ 1 . 0 at − 20 mmol · gDW − 1 · hr − 1 to ≈ 0 . 38 at − 2 mmol · gDW − 1 · hr − 1 . predicted biomass flux from 0.0858 to 1.041 gDW · hr − 1 , representing a ≈ 12-fold improv ement. The Gaussian process surrogate con v erged to a stable optimum within a modest num- ber of FBA e valuations. The optimised conditions correspond to elev ated glucose and oxygen av ailability with moderate ammonium uptake, consistent with established principles of aerobic fermentation physiology and the Crabtree ef fect. H. P athway Enrichment in Cluster 1 Pathway enrichment analysis of the top upregulated reactions in Cluster 1 (highest mean biomass flux: 0.5543 gDW · hr − 1 ) identified (Fig. 11): Alanine, aspartate and glutamate metabolism (count = 3); Exchange reaction (count = 2); Transport [c,e] (count = 2); Complex alcohol metabolism, Steroid biosynthesis, and Butanoate metabolism (count = 1 each). The prominence of amino-acid metabolism pathways is consistent with the role of these amino acids as primary nitrogen donors for nucleotide and amino-acid biosynthesis in S. cere visiae , enabling faster biomass accumulation without depleting carbon precursors. I. GAN-Gener ated Metabolic Flux Pr ofiles The trained GAN generated synthetic flux configurations (shape: 10 × 4130 ) with a variance of 0.156, confirmed sto- ichiometrically feasible within the Y east9 network. Pathway- lev el analysis (Fig. 12) shows the highest mean absolute fluxes for Growth ( ≈ 0 . 73 ), L ysine metabolism ( ≈ 0 . 65 ), Pantothen- ate and CoA biosynthesis ( ≈ 0 . 60 ), Histidine metabolism ( ≈ 0 . 60 ), and TCA cycle ( ≈ 0 . 48 ). The variance of 0.156 indicates that the GAN has successfully explored the metabolic flux space without complete mode collapse. Fig. 11. Enriched pathways among top upregulated reactions in Cluster 1. Alanine, aspartate and glutamate metabolism (count = 3) is the dominant enriched pathway , follo wed by Exchange reaction and T ransport [c,e] (count = 2 each). Fig. 12. The GAN-generated flux activity across the top ten metabolic pathways, measured as mean absolute flux. The gro wth is (around 0.73) and Lysine metabolism (about 0.65) shows the highest activity . Followed by Pantothenate and CoA biosynthesis, along with Histidine metabolism at approximately 0.60. The TCA cycle (around 0.48) and various amino acid metabolism pathways are also significantly represented, which aligns with what we know about the key factors driving yeast biomass production. J. Ablation Analysis An ablation study conducted by removing top SHAP-ranked reactions from the feature set resulted in a significant decrease in predictiv e performance, confirming the critical role of these reactions in determining biomass flux. V . D I S C U S S I O N The results demonstrate that an integrati ve pipeline combin- ing GEM-based simulation, unsupervised representation learn- ing, supervised ML, mechanistic interpretation, and generati ve modelling can provide substantiv e insights into the metabolic determinants of yeast biomass production. The high R 2 values achie ved by Random Forest (0.99989) and XGBoost (0.9990) indicate that the biomass objecti ve in FB A is largely determined by a tractable subset of flux v ari- ables [11]. This is theoretically consistent with the stoichio- metric network structure: under FB A optimality assumptions, biomass flux is dictated by a relativ ely small number of rate- limiting reactions [4], [8]. SHAP analysis operationalises this insight by identifying the specific features (e.g., Features 1446, 863, 2909, 2911) contrib uting most to prediction variance [10]. The sparse distribution of feature importance, with a dominant peak near reaction index 1500, aligns with the well-established principle that central carbon metabolism reactions are primary controllers of growth rate [5], [14], [22]. The 12-fold improv ement in biomass flux via Bayesian optimisation (0.0858 to 1.041 gDW · hr − 1 ) underscores the substantial potential for media formulation optimisation in yeast fermentation [12], [13], [21]. The rapid conv ergence of the Gaussian process surrogate suggests a smooth, unimodal biomass-versus-nutrient landscape amenable to efficient op- timisation. Elev ated glucose and oxygen av ailability as key driv ers is consistent with the Crabtree effect in aerobic yeast physiology . The GAN variance of 0.156 indicates that the model has captured meaningful div ersity in the feasible flux space without complete mode collapse. The prominence of Growth and L ysine metabolism ( ≈ 0 . 73 and ≈ 0 . 65 mean absolute flux) among GAN-generated pathways, alongside TCA cycle contributions, indicates biologically plausible metabolic con- figurations. Future GAN architectures conditioned on target biomass v alues could be used to generate flux profiles targeting higher productivity regimes. The pathway enrichment of Alanine, aspartate, and gluta- mate metabolism in Cluster 1 aligns with the recognised role of these amino acids as primary nitrogen scaffolds in biosynthetic pathways in S. cere visiae . The downre gulation of Exchange reactions and T ransport subsystems in high-growth states is consistent with more ef ficient intracellular resource utilisation. Sev eral limitations merit acknowledgement. The FB A framew ork assumes metabolic steady state and does not ac- count for dynamic regulatory responses, enzyme kinetics, or transcriptional regulation. All predictions are in silico and require experimental v alidation. Future w ork will prioritise experimental validation of priority engineering targets using CRISPR-Cas9-mediated gene editing, follo wed by gro wth phe- notyping under computationally optimised media conditions. The pipeline is organism-agnostic and can be extended to Escheric hia coli , cyanobacteria, and oleaginous yeasts [16], [24]. V I . C O N C L U S I O N This study presents a comprehensive integrati ve framew ork that unifies genome-scale metabolic modelling, machine learn- ing, and optimisation for the prediction and enhancement of biomass flux in Sacchar omyces cere visiae . By combining FB A-based data generation, V AE-based representation learn- ing, ensemble and deep learning predictiv e models, SHAP- driv en mechanistic interpretation, in silico perturbation analy- sis, Bayesian nutrient optimisation, and GAN-based pathway generation, the pipeline addresses the full cycle from data generation to actionable engineering insight. Ke y findings include: (i) Random Forest and XGBoost predict biomass flux with R 2 values of 0.99989 and 0.9990, respectiv ely , and a 5-fold CV mean R 2 of 0 . 99991 ± 0 . 00005 ; (ii) V AE clustering rev eals four metabolic states with mean biomass fluxes of 0.473, 0.554, 0.484, and 0.483 gD W · hr − 1 ; (iii) SHAP analysis highlights twenty key features as primary driv ers of biomass yield; (iv) in silico overe xpression reaches 0.979 gDW · hr − 1 , nearing the Bayesian-optimised maximum of 1.041 gD W · hr − 1 (a 12-fold improv ement); and (v) GAN- generated flux profiles show a v ariance of 0.156 and are sto- ichiometrically feasible, with Growth and L ysine metabolism as the most activ e pathways. This integrati ve pipeline pro vides a scalable, reproducible, and extensible platform for computational metabolic engineer- ing broadly applicable to any organism with a quality GEM av ailable. R E F E R E N C E S [1] Lu H, Li C, Sanchez BJ, Zhu Z, Liljenbacka G, Nielsen J (2019) A consensus S. cerevisiae metabolic model Y east8 and its ecosystem for comprehensiv ely probing cellular metabolism. Nature Communications 10:3586. https://doi.org/10.1038/s41467- 019- 11581- 3 [2] Zhang C et al (2024) Y east9: A consensus genome-scale metabolic model for S. cerevisiae curated by the community . Molecular Systems Biology 20. https://doi.org/10.1038/s44320- 024- 00060- 7 [3] Ebrahim A, Lerman J A, Palsson BØ, Hyduke DR (2013) COBRApy: Constraints-Based Reconstruction and Analysis for Python. BMC Sys- tems Biology 7:74. https://doi.org/10.1186/1752- 0509- 7- 74 [4] Orth JD, Thiele I, Palsson BØ (2010) What is flux balance analysis? Natur e Biotechnology .28:245–248. https://doi.org/10.1038/nbt.1614 [5] Chen Y , Li F , Nielsen J (2022) Genome-scale modeling of yeast metabolism: retrospectives and perspectiv es. FEMS Y east Research 22. https://doi.org/10.1093/femsyr/foac003 [6] Kim WJ, Kim HU, Lee SY (2021) Machine learning applications in genome-scale metabolic modeling. Current Opinion in Systems Biology 25:42–49. https://doi.org/10.1016/j.coisb.2021.03.001 [7] Zampieri G, V ijayakumar S, Y aneske E, Angione C (2019) Machine and deep learning meet genome-scale metabolic modeling. PLOS Computa- tional Biology 15. https://doi.org/10.1371/journal.pcbi.1007084 [8] Sahu A, Blatke MA, Szyma ´ nski JJ, T ¨ opfer N (2021) Advances in flux balance analysis by integrating machine learning and mechanism-based models. Computational and Structural Biotechnolo gy Journal 19:4626– 4640. https://doi.org/10.1016/j.csbj.2021.08.004 [9] Culle y J, V ijayakumar A, Zampieri G, Angione C (2020) A mechanism- aware and multiomic machine-learning pipeline characterizes yeast cell growth. PNAS 117:18338–18348. https://doi.org/10.1073/pnas. 2002959117 [10] Lundber g SM, Lee SI (2017) A unified approach to interpreting model predictions. Advances in Neural Information Processing Systems 30 https://arxiv .org/abs/1705.07874 [11] Daniel M. Gonc ¸ alves, Rui Henriques, Rafael S. Costa (2023) Predicting metabolic fluxes from omics data via machine learning: Moving from knowledge-dri ven tow ards data-driv en approaches. Computational and Structural Biotechnology Journal 21:4960–4973 https://doi.org/10.1016/ j.csbj.2023.10.002 [12] Radi voje vic T , Costello Z, W orkman K, Garcia Martin H (2020) A machine learning automated recommendation tool for synthetic biology . Nature Communications 11:4879. https://doi.org/10.1038/ s41467- 020- 18008- 4 [13] La wson C et al (2021) Machine learning for metabolic engineering: A revie w . Metabolic Engineering 63:34–60. https://doi.org/10.1016/j. ymben.2020.10.005 [14] Zhang J et al (2020) Combining mechanistic and machine learn- ing models for predictive engineering and optimization of tryptophan metabolism. Nature Communications 11:4880. https://doi.org/10.1038/ s41467- 020- 17910- 1 [15] Gomari DP , Schweickart A, Cerchietti L, Paietta E, Fernandez H, Al- Amin H, Suhre K, Krumsiek J (2022) V ariational autoencoders learn transferrable representations of metabolomics data. Communications Biology 5:659. doi: 10.1038/s42003- 022- 03579- 3 [16] Merzbacher C, Oyarzun D A (2023) Applications of artificial intelligence and machine learning in dynamic pathway engineering. Biochemical Society Tr ansactions 51:1871–1879. doi: 10.1042/BST20221542 [17] Baig Y , Ma HR, Xu H, Y ou L (2023) Autoencoder neural networks en- able low dimensional structure analyses of microbial growth dynamics. Natur e Communications 14:7932. doi: 10.1038/s41467- 023- 43455- 0 [18] Choudhury S, Moret M, Salvy P , W eilandt D, Hatzimanikatis V , Miskovic L (2022) Reconstructing kinetic models for dynamical studies of metabolism using generative adversarial networks. Natur e Machine Intelligence 4:710–719. doi: 10.1038/s42256- 022- 00519- y [19] Costello Z, Garcia Martin H (2018) A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data. npj Systems Biology and Applications 4:19. doi: 10.1038/ s41540- 018- 0054- 3 [20] Razmpour T , T abibian M, Roohi A, Saha R (2026) GAN-enhanced machine learning and metabolic modeling identify reprogramming in pancreatic cancer . PLOS Computational Biology 22. doi: 10.1371/ journal.pcbi.1013862 [21] Akaraphol W atcharawipas, W eerawat Runguphan, Peerapat Khamwachi- rapithak, Thanaporn Laothanachareon (2025) Integrating yeast biodiver - sity and machine learning for predictive metabolic engineering. FEMS Y east Resear ch . https://doi.org/10.1093/femsyr/foaf072 [22] Moreno-P az S, van der Hoek R, Eliana E, Zwartjens P , Gosiewska S, Martins dos Santos V AP , Schmitz J, Suarez-Diez M (2024) Machine learning-guided optimization of p-coumaric acid production in yeast. ACS Synthetic Biology 13:1193–1203. doi: 10.1021/acssynbio.4c00035 [23] Cheng Y , Bi X, Xu Y , Liu Y , Li J, Du G, Lv X, Liu L (2023) Machine learning for metabolic pathway optimization: A revie w . BMC Bioinformatics . https://doi.org/10.1016/j.csbj.2023.03.045 [24] Jang WD, Kim GB, Kim Y , Lee SY (2021) Applications of artificial intelligence to enzyme and pathway design for metabolic engineering. Curr ent Opinion in Biotechnology 73:101–107. doi: 10.1016/j.copbio. 2021.07.024 [25] Masid S, Ataman M, Hatzimanikatis V (2024) Generati ve machine learn- ing produces kinetic models that accurately characterize intracellular metabolic states. Natur e Catalysis 7:1086–1099. https://doi.or g/10.1038/ s41929- 024- 01220- 6
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment