Enabling ab initio geometry optimization of strongly correlated systems with transferable deep quantum Monte Carlo
A faithful description of chemical processes requires exploring extended regions of the molecular potential energy surface (PES), which remains challenging for strongly correlated systems. Transferable deep-learning variational Monte Carlo (VMC) offe…
Authors: P. Bernát Szabó, Zeno Schätzle, Frank Noé
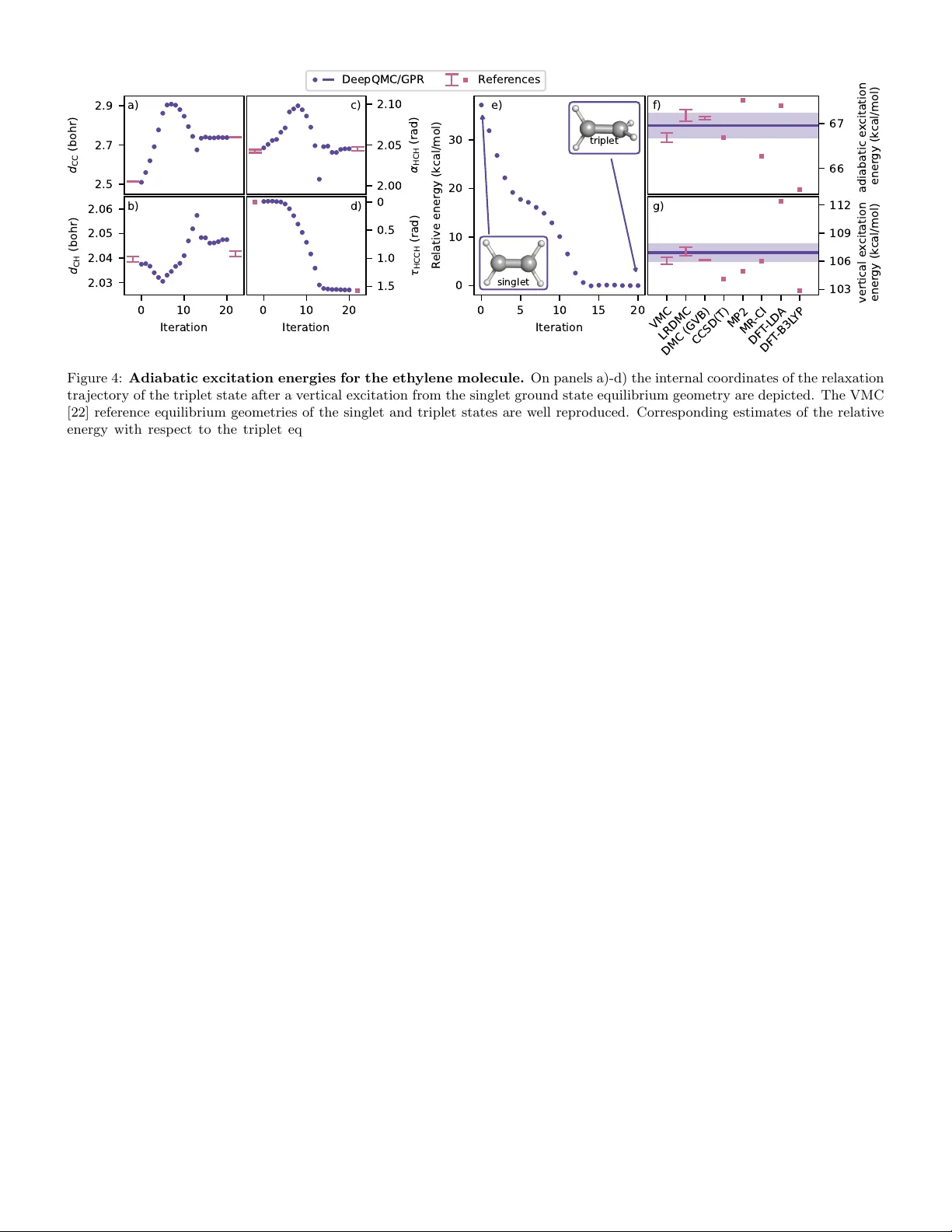
Enabling ab initio geometr y optimiza tion of str ongl y correla ted systems with transferable deep quantum Monte Carlo P . B. Szab´ o 1, † , Z. Sc h¨ atzle 1, † , and F. No ´ e 1,2,3,4,* 1 FU Berlin, Dep artment of Mathematics and Computer Scienc e, Arnimallee 6, 14195 Berlin, Germany 2 Micr osoft R esear ch AI4Scienc e, Karl-Liebkne cht Str. 32, 10178 Berlin, Germany 3 FU Berlin, Dep artment of Physics, Arnimal le e 14, 14195 Berlin, Germany 4 Ric e University, Dep artment of Chemistry, Houston, TX 77005, USA A faithful description of c hemical processes requires exploring extended regions of the molecular potential energy surface (PES), whic h remains c hallenging for strongly correlated systems. T ransferable deep-learning v ariational Mon te Carlo (VMC) offers a promising route by efficien tly solving the electronic Schr¨ odinger equation jointly across molecular geometries at consisten tly high accuracy , yet its stochastic nature renders direct exploration of molecular configuration space nontrivial. Here, we presen t a framework for highly accurate ab initio exploration of PESs that combines transferable deep-learning VMC with a cost-effective estimation of energies, forces, and Hessians. By contin uously sampling nuclear configurations during VMC optimization of electronic wa ve functions, we obtain transferable descriptions that achiev e zero-shot chemical accuracy within chemically relev ant distributions of molecular geometries. Throughout the subsequent c haracterization of molecular configuration space, the PES is ev aluated only sparsely , with lo cal approximations constructed by estimating VMC energies and forces at sampled geometries and aggregating the resulting noisy data using Gaussian process regression. Our metho d enables accurate and efficien t exploration of complex PES landscap es, including structure relaxation, transition-state searches, and minimum-energy path wa ys, for b oth ground and excited states. This op ens the do or to studying b ond breaking, formation, and large structural rearrangemen ts in systems with pronounced m ulti-reference character. The o verarc hing ob jective of computational c hemistry is to c haracterize chemical pro cesses by exploring the molecular potential energy surface (PES). This seemingly uniform prescription masks a great v ariety of problems arising due to the diversit y of c hemical pro cesses them- selv es, comprising among others strongly correlated inter- mediates [1 – 3], excited state pro cesses [4, 5], or ev en light- matter in teraction [6, 7]. The wide range of scenarios in turn necessitated the developmen t of a huge arra y of com- putational metho ds [8], most of which can efficiently solve only a small subset of these problems. Moreov er, to mak e the electronic structure problem computationally tangible, the ma jority of quantum chemistry metho ds are designed to solv e the electronic Sc hr¨ odinger equation for a single molecular configuration at a time. This necessitates inter- mediate steps to arrive from ab-initio electronic structure sim ulation to practical molecular mo deling [9], and renders the cost of directly exploring the PES with accurate quan- tum c hemistry methods prohibitiv e for all but the smallest systems. A common work around is to use computationally less demanding metho ds to generate optimized molecular structures, then supply these geometries to more accurate metho ds to compute single-p oin t energies. While this is considered a work able solution for ground-state, single- reference equilibrium structures, where metho ds suc h as densit y functional theory (DFT) typically yield reasonable geometries, it can fail in sev eral other scenarios suc h as those inv olving excited state s [10, 11] or strong static cor- ∗ Email: frank.no e@fu-b erlin.de † Equal con tribution relation [12]. In suc h cases, the limitations of the cheaper appro ximate methods cast doubt on the quality of the re- sulting geometries, p oten tially rendering the subsequen t energy ev aluations with formally more accurate metho ds unreliable. Another alternative is the optimization of sur- rogate models for predicting the PES from large n umbers of single p oin t simulations [13, 14]. This, how ever, re- quires careful construction datasets emplo ying quan tum c hemistry metho ds tailored to sp ecific regions of molecular configuration space [15], which can b e a costly and lab or in tensive process. F urthermore, working with surrogate mo dels en tails a loss of direct access to the ab initio so- lution, and in tro duces additional sources of errors in form of in termediate computational steps. In this w ork, w e present a metho d that enables the ab initio exploration of molecular PESs via v ariational Mon te Carlo (VMC) optimization of transferable neural net work w av e functions. In particular, w e introduce a no vel tec hnique that exploits the generalization of trans- ferable deep-learning ansatzes across molecular configu- ration space to efficiently ev aluate the prop erties of the PES, including its v alue, gradient, and Hessian with re- sp ect to the nuclear co ordinates. Our electronic structure metho d of c hoice, deep-learning VMC, has been shown to pro vide state-of-the-art accuracy ev en for strongly cor- related systems, while recen t efforts to w ards a transfer- able ansatz hav e enabled the simultaneous mo deling of a range of molecular configurations at a steeply discounted cost [16 – 19]. Emplo ying deep-learning VMC, w e construct transferable wa v e function mo dels that span broad re- gions of molecular configuration space at zero-shot c hem- 1 Figure 1: Sk etch of the DeepQMC/GPR ab initio geometry optimization metho d. The flo wc hart depicts the pro cess of optimizing the minimum energy pathw ay (MEP) of the formaldehyde isomerization. a) A transferable w a ve function is optimized on con tin uously sampled n uclear configurations of a tw o dimensional cut of the formaldeh yde configuration space. Since the training loss consists of exp ectation v alues ov er small electron batches at random nuclear configurations, during transferable training the full PES is never materialized b ey ond noisy estimates. b) T o extract energy and gradien t information from the optimized transferable w av e function, Monte Carlo estimators of the molecular energy and Hellmann-F eynman force are ev aluated. The predictions are further improv ed by ev aluating these exp ectation v alues at a range of molecular configurations in the vicinit y of the geometry under in vestigation and fitting a local approximation of the PES with Gaussian pro cess regression (GPR). F rom the Gaussian pro cess energy , force and Hessian are computed analytically , which suffice to compute second order geometry up dates. c) By a consecutive application of optimization steps, molecular geometries can b e relaxed, transition states can be found and intermediates can b e obtained when employing additional constrains. d) Combining m ultiple geometry optimizations, the full MEP of a c hemical pro cess can b e c haracterized. Notably , for the MEP optimization the PES was only appro ximated sparsely via the samples depicted in gray . ical accuracy ( < 1 k cal/mol), trained at a cost compara- ble to that of a single-p oin t simulation. The universal applicabilit y of deep-learning VMC allows us to account for strong correlation, as w ell as excited states, enabling the use of this single method for the consisten t descrip- tion of entire PESs as well as multiple electronic states [20]. T o enhance sampling efficiency and systematically accoun t for the stochasticit y of Mon te Carlo simulation, w e in tegrate deep-learning VMC with Gaussian process regression (GPR), fitted on the ab initio energy and nu- clear force estimates obtained from the transferable wa ve function. This approach mak es use of the generalization capabilit y of transferable neural net work ansatzes to un- seen geometries, the zero-v ariance property of the energy , as well as the sound statistical form ulation of GPR to de- liv er accurate estimates of the PES along with its first and second deriv ativ es, complete with robust error measures. While direct ab initio geometry optimization with quan- tum Monte Carlo metho ds has b een previously attempted and can yield accurate geometries for small molecules [21– 24], these metho ds are not matured, ha ve not been applied using highly accurate deep-learning-based wa v e functions, and do not leverage the substan tial efficiency gains offered b y transferable wa ve function optimization, whic h we iden- tify as a key innov ation for ac hieving efficient and highly accurate geometry optimization in molecular systems with c hallenging electronic structure. T o demonstrate the re- sulting metho d’s broad applicabilit y , w e p erform a v a- riet y of geometry optimization tasks, such as geometry relaxation, transition state searc h, and minimum energy path wa y (MEP) optimization on ground and excited state PESs for systems ranging from simple one dimensional di- atomic molecules ov er small organic comp ounds to a nine dimensional scan of the HO 2 · + · OH reaction. 1 Resul ts 1.1 Ab initio potential energy sur- f a ces and geometr y optimiza tion with Gaussian pr ocess regression A b initio geometry optimization inv olv es the successiv e so- lution of the electronic Sc hr¨ odinger equation to ev aluate deriv atives of the nuclear p oten tial energy , thereb y guid- ing the system to ward extrema on the Born–Oppenheimer PES. Our metho d pro vides an effectiv e solution to this problem that leverages the excellent accuracy of deep- learning VMC, com bining the transferable sim ulation of electronic states with an appropriate treatmen t of the sto c hasticity inheren t to Monte Carlo estimation of ex- p ectation v alues. The first step of the prop osed algorithm is the transferable optimization of a neural netw ork w av e function ansatz Ψ θ ( r | R ), aiming at zero-shot chemical ac- curacy on large, con tinuous, and c hemically relev ant re- gions of the PES. T o this end, w e con tin uously sample molecular configurations during wa ve function optimiza- tion θ ∗ = argmin θ E R ∼ ρ ( R ) E r ∼| Ψ θ ( r | R ) | 2 " ˆ H ( R )Ψ θ ( r | R ) Ψ θ ( r | R ) # , (1) 2 where ˆ H ( R ) is the electronic Hamiltonian with the ex- ternal p otential of a set of nuclei in configuration R , and ρ ( R ) : Ω → R ≥ 0 is a probability density o ver the relev ant subspace of the molecular configuration space Ω ⊆ R 3 M , i.e. a uniform distribution ov er b ond lengths, angles, and torsions. While nuclear geometries of the outer exp ecta- tion v alues are sampled directly , the exp ectation v alue o ver the electron co ordinates r dep ends on the molecular geom- etry through the transferable w a ve function and is ev alu- ated via Marko v chain Mon te Carlo. T o maintain high sampling efficiency , we emplo y a space-warp tec hnique to up date electron co ordinates in tandem with changes in the molecular geometry [25]. This contin uous transferable optimization yields w av e functions with consisten t ener- gies and forces throughout the range of configurations at an ov erall training cost comparable to an individual single- p oin t optimization. A study that establishes the relev ance of contin uous sampling for consisten t zero-shot accuracy and demonstrates the computational benefits of the trans- ferable simulation is presented in Sec. A.1 of the Supple- men tary Information. With the optimized transferable wa v e function at hand we turn to the exploration of PESs. W orking with the wa ve function directly means that during inference, exp ectation v alues of the energy and force hav e to b e es- timated with Mon te Carlo in tegration. The stochastic- it y of Monte Carlo estimation is a kno wn c hallenge when exploring PESs [18, 23], that we account for b y employ- ing adv anced zero-v ariance force estimators [26, 27], and b y using the ab initio energy and force data to fit sta- tistically optimal local PES models using GPR [28, 29]. GPR for molecular PESs fits non-parametric mo dels to mak e energy predictions for unseen molecular geometries based on observ ations. Gaussian processe s (GPs) pro vide uncertain ty measures for their predictions and facilitate the computation of analytic gradient and Hessian esti- mates. W e acquire a lo cal approximation of the PES, by conditioning a GP on datasets D of energies and forces Y = [ ¯ E 1 , ¯ F 1 , . . . , ¯ E n , ¯ F n ] T at molecular configurations X = [ R 1 , . . . , R n ] T , sampled within a neighborho od of the geometry of in terest R . The estimates ¯ E i and ¯ F i are obtained by ev aluating the appropriate Monte Carlo esti- mators on the optimized transferable wa ve function Ψ θ ∗ ¯ E i , ¯ F i = ˆ H ( R i ) Ψ θ ∗ ( r | R i ) , d ˆ H / d R R i Ψ θ ∗ ( r | R i ) ≈ E ( R i ) , F ( R i ) . (2) Conditioning the GP on the data yields a random v ariable for the energy ˜ E ( R ) | D ∼ N ( m ∗ ( R ) , k ∗ ( R , R )) , (3) where the optimal p osterior mean m ∗ and p osterior v ari- ance k ∗ can b e obtained in closed form and constitute the prediction of the energy and its uncertaint y , resp ectiv ely . An important property of GPs is that their deriv atives are themselv es GPs, enabling the direct prediction of forces and Hessians. Throughout the geometry optimization, at each new molecular configuration we query the GP fitted on the pre- vious energy and force estimates at negligible cost, and c heck whether it has an error estimate below a set ac- curacy threshold. While this is not the case, we sample n uclear configurations in the vicinity of the geometry of in terest to obtain additional data points until sufficient accuracy of the GPR is achiev ed. The energy , n uclear force, and Hessian information obtained from the GP is then used for do wnstream PES exploration tasks, suc h as geometry relaxation or minim um energy pathw ay (MEP) searc h. As demonstrated in Sec. A.2 of the Supplemen- tary Information, our GP force estimates are at least as accurate and efficien t as the best zero-bias zero-v ariance force estimators ev en if only a single geometry is of inter- est, with further efficiency b enefits arising in the geometry optimization setting from reusing the data generated pre- viously for other nearby geometries. Since the dataset for GPR is constructed on-the-fly , materializing the full PES can b e a v oided. With accurate lo cal information ab out the PES at hand, geometry optimization is carried out using standard tec hniques, in particular via the Newton–Raphson metho d in in ternal coordinates [30], employing image functions for transition state searches [31], and the r educed-restricted approac h of Anglada for constrained optimization [32, 33]. 1.2 V alid a ting equilibrium str uctures of dia tomic molecules BH 0 1 d w . r . t . E x p ( b o h r ) 1e 2 Experiment DeepQMC/GPR CCSD(T) MP2 HF 0 1 2 3 1e 3 CO 0 1 1e 2 N2 0 1 2 1e 2 F2 0 1 2 3 1e 2 Figure 2: Optimizing equilibrium bond lengths for di- atomic molecules. DeepQMC/GPR results are compared to exp erimen tal data [34] and to MP2@CBS and CCSD(T)@cc- pV6Z calculations [35]. Before extending to higher dimensional geometry op- timizations, w e v alidate our method b y comparing equilib- rium b ond lengths of diatomic molecules optimized with transferable deep-learning VMC to exp erimental and com- putational references. W e follo w the pro cedure outlined in Sec. 1.1 for a test set of five small molecules, namely BH, HF, CO, N 2 , F 2 . Throughout transferable training, b ond lengths are sampled from a mixture of four Gaussian distri- butions with µ 0 = 1 . 5 bohr , µ 1 = 2 . 5 bohr , µ 2 = 3 . 5 bohr and µ 3 = 4 . 5 b ohr and σ = 1 b ohr. T o a v oid high energy configurations we restrict the interatomic distance to b e larger than 1 bohr for HF and F 2 and 1 . 5 b ohr for BH, CO and N 2 , resp ectively . Using the conv erged transferable w av e functions, geometry optimizations from a stretched 3 1.80 1.85 1.90 1.95 2.00 d N H ( b o h r ) 1.2 1.4 1.6 1.8 2.0 N H ( r a d ) d N H N H a) R eactant guess P r oduct guess DeepQMC/GPR TS DeepQMC/GPR initial nodes DeepQMC/GPR MEP DF T (PBE) CCSD DMC (LS) 1.5 2.0 2.5 3.0 3.5 d C H ( b o h r ) 0.5 1.0 1.5 2.0 2.5 O C H ( r a d ) b) d C H O C H Figure 3: Minimum energy path searc h on t wo dimensional cuts of the ammonia and formaldehyde potential energy surfaces. On panel a) the minim um energy path wa y for the in version of ammonia is shown, while panel b) depicts the isomerization of formaldehyde. Reference and baseline results for the ammonia inv ersion are taken from Ref. [23], CCSD results for formaldehyde are computed in house. The heatmaps visualize PESs extracted from the transferable deep-learning VMC solution and serve as a guide to the eye. b ond length of 3 . 5 bohr are commenced. The relaxations con verge within appro ximately ten iterations to a mean absolute error (MAE) with resp ect to the exp erimen tal reference [34] of 1 . 8 × 10 − 3 b ohr. This is similar to the MAE of 2 . 5 × 10 − 3 b ohr of CCSD(T) calculations in the large cc-pV6Z basis and significantly low er than the MAE of 1 . 6 × 10 − 2 b ohr MP2 calculation extrap olated to the complete basis set limit [35]. The close agreemen t with exp erimen tal references and gold-standard CCSD(T) ge- ometry optimizations for equilibrium structures, whic h are t ypically of single-reference c haracter, supp orts the relia- bilit y of our metho d. 1.3 Exploring reactivity with minimum ener gy p a th sear ches After v alidating the accuracy of our approach on di- atomics, we now turn to show casing the metho dology on the significantly more complex task of MEP optimizations. Being the low est energy pathw a ys on the PES that con- nect tw o local minima (reactants and pro ducts), MEPs are a cen tral concept in the theoretical description of re- activit y . Given their relev ance, several approac hes ha ve b een devised for their computation, such as the growing string [36] and nudged elastic band [37] methods, all of whic h op erate by iterativ ely refining a chain of geometries along the path wa y . As constructing each link of the chain requires several geometry optimization steps, the compu- tation of longer c hains can necessitate dozens or hundreds of PES ev aluations. In this setting, prop erly exploiting the capabilities of transferable deep-learning VMC can result in an enormous computational adv antage, which could be the k ey to unlo c king routinely obtainable, highly accurate, ab initio MEPs for strongly correlated systems. T o test the p erformance of the prop osed transferable PES estimation approac h, we implement a simple version of the double-ended growing string metho d [36]. Describ ed in more detail in Sec. 3.3.2, this algorithm starts b y op- timizing the structures of t wo lo cal minima (reactan t and pro duct), and pro ceeds to connect them via a chain of im- ages. Then, a transition state searc h is carried out starting from the image with the highest energy , after which the original c hain is replaced with new strings connecting the transition state with the reactant and the pro duct. The algorithm is first applied to the inv ersion of the ammonia molecule, where traditional VMC, diffusion Mon te Carlo (DMC), CCSD(T) and DFT references are a v ailable [23]. Figure 3a), displa ys a t wo-dimension cut of the ammonia PES, with all nitrogen-hydrogen bonds constrained to b e the same length, and at the same an- gle to the rotational symmetry axis of the molecule. A transferable neural netw ork ansatz was trained for con- tin uously sampled geometries in the range 1 . 77 bohr ≤ d NH ≤ 2 . 00 bohr and π 3 ≤ α NH − z ≤ 2 π 3 . With a total n umber of 200k training iterations and a total electron batc h size of 4096 the ov erall cost of the transferable op- timization was equiv alent to a small num b er of standard single p oint sim ulations. The trained ansatz was then used to optimize the structures of the t w o local minima, both of whic h are in excellen t agreemen t with the reference CCSD geometries. Lastly , the MEP is constructed as a chain of sev en intermediate images, from which the middle one represen ts the transition state, lying neatly among the ref- erence transition state geometries, and yielding a reaction barrier height of 5.3 k cal/mol, that matches well with the reference CCSD, CCSD(T), and DMC v alues of 5.6, 5.8, and 4 . 4 ± 0 . 7 kcal/mol, resp ectively . Next, an MEP searc h is carried out for the sligh tly more inv olved process of formaldeh yde isomerization, where one of the hydrogen atoms migrates from the car- b on to the o xygen atom, breaking the old and form- ing a new cov alen t b ond along the w ay . As b efore, a 4 2.5 2.7 2.9 d C C ( b o h r ) a) 0 10 20 Iteration 2.03 2.04 2.05 2.06 d C H ( b o h r ) b) c) 0 10 20 Iteration d) 0 5 10 15 20 Iteration 0 10 20 30 R elative ener gy (kcal/mol) singlet triplet e) f) VMC LRDMC DMC (GVB) CCSD(T) MP2 MR -CI DF T -LD A DF T - B3L YP g) DeepQMC/GPR R efer ences 2.00 2.05 2.10 H C H ( r a d ) 0 0.5 1.0 1.5 H C C H ( r a d ) 66 67 adiabatic e x citation ener gy (kcal/mol) 103 106 109 112 vertical e x citation ener gy (kcal/mol) Figure 4: Adiabatic excitation energies for the ethylene molecule. On panels a)-d) the in ternal co ordinates of the relaxation tra jectory of the triplet state after a vertical excitation from the singlet ground state equilibrium geometry are depicted. The VMC [22] reference equilibrium geometries of the singlet and triplet states are well repro duced. Corresp onding estimates of the relative energy with resp ect to the triplet equilibrium geometry are display ed on panel e). The inserts show the optimized equilibrium geometry of the singlet and triplet state. Panel f ) and panel g) compare the adiabatic and the vertical excitation energies to reference calculations [22], where the shaded area indicates chemical accuracy . single neural netw ork ansatz is trained for the tw o di- mensional PES depicted on Figure 3b) and defined b y 1 . 5 b ohr ≤ d CH ≤ 3 . 8 b ohr, and 0 . 4 ≤ α OCH ≤ 2 . 5. The corresp onding MEP comprises a narrow channel near the pro duct and a turn at the transition state. Accordingly , more intermediate images are used here, which further increases the required n um b er of PES ev aluations. The deep-learning VMC MEP is compared with a reference CCSD path computed in house. It is clear that all in ter- mediate geometries, as well as the transition state agree quite w ell b et ween the t wo methods, while the mean abso- lute deviation of relativ e energies across the pathw a y is as lo w as 0.5 k cal/mol. In the pro cess of the full MEP opti- mization a total of 133 geometry up dates were p erformed, and the fact that a single ab initio deep-learning VMC data p oint was used in determining 13.8 updates on av- erage highlights the data efficiency of the GP based force estimator on realistic tasks. 1.4 Geometr y optimiza tion of elec- tr onic excited st a tes The computation of PESs via transferable deep-learning VMC can be straightforw ardly extended to electronic ex- cited states [20]. In this section, w e exploit this gen- eral applicability , to demonstrate that the approac h pre- sen ted here is capable of accurately determining adiabatic excitation energies, as well as studying the reactivity of molecules in their excited states. T o this end, we first study ethylene, as a common mo del for photo c hemistry that undergo es large structural c hanges up on singlet-triplet excitation. W e train a trans- ferable neural net work ansatz for the singlet ground state and first triplet excited state of the ethylene molecule em- plo ying the spin-p enalt y metho d introduced in [39]. The setup of Barb orini et al. [22] is adopted, with the molecule b eing parameterized in terms of the H – C – C – H torsion angle, the H – C – H angle, the C – H b ond length, and the length of the C – C double b ond, forcing the same v alues for all o ccurrences of each. This yields a four dimensional con- figuration space for the transferable ansatz and geometry optimization. Utilizing the transferable wa ve function for the singlet state, we first relax the geometry of the ground state to it’s global minimum. Next, ethylene’s vertical ex- citation energy is computed b y ev aluating the energy of the triplet state at the ground state equilibrium geometry . As display ed on panel g) of Figure 4, our v ertical excita- tion energy estimate is in excellent agreemen t with other QMC derived v alues, as w ell as m ulti-reference CI, while the single reference metho ds CCSD(T) and MP2 exhibit sligh tly larger deviations and DFT estimates v ary signifi- can tly based on the functional b eing used [22]. Lastly , the ground state minimum geometry is relaxed on the triplet surface in order to compute the adiabatic gap. The four dimensional optimization tra jectory is depicted in panels a)-d) of Figure 4, with the relative energy with resp ect to the optimized triplet equilibrium geometry depicted on panel e). Our simulations repro duce the 90 ◦ H – C – C – H torsion and the elongation of the C – C double bond of the triplet minimum, leading to an energy reduction of 36 k cal/mol. The adiabatic excitation energy is display ed on panel f ). Our results are again in excellent agreemen t with the reference QMC v alues b eing within chemical accuracy of our estimate. This indicates that the prop osed geome- try optimization pro cedure successfully translates the ac- curacy of deep-learning VMC to relativ e energies b etw een differen t geometries and electronic states. With applicability established to adiabatic excita- tions, w e mov e to the task of characterizing elemen tary c hemical reactions proceeding entirely on excited surfaces. 5 4 3 2 1 0 1 2 3 4 R e a c t i o n c o o r d i n a t e d H H d N H ( b o h r ) 0 10 20 30 40 50 R elative ener gy (kcal/mol) a 1 A 0 0 F C I / / 6 - 3 1 1 G * o n C A S P T 2 g e o m e t r i e s a 1 A 0 0 D e e p Q M C o n C A S P T 2 g e o m e t r i e s a 1 A 0 0 D e e p Q M C o n D e e p Q M C g e o m e t r i e s b 1 A 0 D e e p Q M C o n a 1 A 0 0 C A S P T 2 g e o m e t r i e s b 1 A 0 D e e p Q M C o n D e e p Q M C g e o m e t r i e s Figure 5: P otential energy surfaces of the H 2 + NH − − − → H · + · NH 2 reaction. The reaction migh t pro ceed either on the a 1 A ′′ or the b 1 A ′ PES. Inset plots sho w the relev ant reactant, transition state, and pro duct structures highligh ting the differences b et ween the t wo electronic states. The FCI results tak en from W u et al. [38] were computed on geometries optimized at the CASPT2 lev el of theory , with the relatively small basis set of 6-311G*. The energies of these geometries were also ev aluated with DeepQMC, shown with solid lines. The radical-radical h ydrogen abstraction reaction of H 2 + NH − − → H · + · NH 2 is selected as our next target. In a recent study [38], the p ossibility of this pro cess pro- ceeding on v arious electronic states was discussed. Com- mencing on the ground state triplet surface, the reaction is w ell describ ed by single reference metho ds. On the other hand, the reaction migh t start from either of the lo w-lying, doubly-degenerate singlet excited states of NH, leading to pro ducts either in the b 1 A ′ or the a 1 A ′′ electronic state, ha ving the unpaired electron of NH 2 either in or out of the molecule’s plane, resp ectiv ely . T o inv estigate reactiv- it y in these states, a transferable neural netw ork ansatz is trained for the b 1 A ′ and a 1 A ′′ surfaces, considering only planar geometries and a collinear attac k of the H 2 molecule (see Sec. B.2 of the Supplemen tary Information for the def- inition of the training geometry distribution). In the work of W u et al. , geometries along the reaction path wa y on the a 1 A ′′ surface are optimized at the CASPT2@6-311G* lev el of theory , while the reaction energy and barrier height of the rev erse reaction are determined to b e 29.9 and 38.6 k cal/mol, respectively , via full configuration interaction (F CI) calculations in the same basis, as sho wn on Figure 5. Ev aluating the energies of the CASPT2 geometries using our neural netw ork ansatz, we obtain estimates of the re- action energy and barrier heigh t that are about 4.0 and 7.3 k cal/mol lo wer than those obtained with F CI, highligh ting the importance of finite basis set errors of second quan- tized metho ds, when using insufficien tly large basis sets. Next, to compare reactivity b etw een the tw o surfaces, the deep-learning VMC energies of the CASPT2 geometries are also ev aluated on the b 1 A ′ surface, as plotted on Fig- ure 5, resulting in a practically barrierless tra jectory that increases in energy to w ards the pro ducts. Finally , p er- forming our full MEP search on the b 1 A ′ surface, we find that the NH 2 radical relaxes by 15.4 kcal/mol into a con- figuration with the H–N–H angle increased to 139 ◦ . As displa yed on Figure 5, on the relaxed b 1 A ′ path wa y we obtain for the rev erse reaction a reaction energy of − 5 . 5 k cal/mol and a barrier height of 3.0 kcal/mol, the latter b eing significantly low er than in the a 1 A ′′ state, indicat- ing that the rev erse reaction migh t tak e place muc h more readily on this excited b 1 A ′ surface. T aken together, the results presented in this section sho w that the ab initio geometry-optimization proto col based on transferable deep-learning VMC facilitates the study of excited-state PESs, offering a p ow erful new to ol for the inv estigation of processes inv olving photoexcita- tions of strongly correlated systems. 1.5 Full potential ener gy surf ace for HO 2 + OH reaction Lastly , we sho w case the scalability of the prop osed metho dology , targeting the practically relev ant problem of characterizing the radical-radical reaction HO 2 · + OH · − − → O 2 + H 2 O. This pro cess is of high imp ortance for b oth atmospheric and com bustion c hemistry , as a ma- jor consumer of OH · and HO 2 · radicals [41, 42], while at the same time b eing difficult to characterize exp erimen- tally o wing largely to the problematic determination of HO 2 · radical concentrations as well as its radical-radical nature [43]. Recen tly , a full PES has b een computed for this process at the explicitly correlated CCSD(T)-F12 lev el of theory , by fitting a p ermutation inv ariant polyno- mial (PIP) neural netw ork to more than h undred thou- sand coupled cluster energies [40]. The authors empha- size the mul ti-reference nature of the system in question, esp ecially in the entrance channel up to the transition state, due to the near-degeneracy of singlet and triplet spin states. Nonetheless, this single-reference metho d yields re- sults that are in agreement with some experiments. Here, w e aim to demonstrate that our proposed metho dology 6 4 3 2 1 0 1 2 3 R e a c t i o n c o o r d i n a t e d O 2 H 1 d H 1 O 3 ( b o h r ) 70 60 50 40 30 20 10 0 R elative ener gy (kcal/mol) CCSD(T)/PIP DeepQMC/GPR DeepQMC on PIP geometries Figure 6: MEP for the HO 2 · + OH · − − − → H 2 O + O 2 reaction. The results of the deep-learning VMC MEP optimization are compared with reference v alues obtained with a p erm utation in v ariant polynomial (PIP) neural netw ork potential trained on 108k CCSD(T)-F12 single-p oint energies [40]. is a useful tool to tackle suc h a realistic c hemical prob- lem, where the application of traditional single-reference electronic structure methods requires significan t computa- tional resources, while not disp elling all doubts regarding strong correlation effects. T o this end, a transferable neural net work ansatz is optimized on a nine dimensional region of the configura- tion space around the MEP iden tified b y Liu et al. [40] (for the pro cedure used to obtain this configuration space see Sec. B.3 of the Supplementary Information). Dur- ing this task, sixteen thousand distinct geometries w ere visited, constituting the largest single molecule configura- tion space, for which a transferable neural netw ork ansatz yields accurate, zero-shot predictions. The pathw a y ob- tained with this transferable ansatz and our MEP finding algorithm is compared with the results rep orted by Liu et al. [40] on Figure 6, with n umerical v alues collected in T able 1. Considering first the reactan t and the pro d- uct, w e observe excellent agreemen t b etw een CCSD(T) and deep-learning VMC, both in terms of molecular ge- ometry (low er half of T able 1), as well as relative energy (upp er half of T able 1). This is in line with the facts that all degrees of freedom within the reactant and pro d- uct fragments are relatively stiff, and that both CCSD(T) and deep-learning VMC are expected to give accurate de- scriptions of such single reference structures. Mo ving to the intermediate geometries, in particular to the lo cal minimum situated in the entrance of the reac- tion channel (CP1), we find larger deviations in molecular structure b et ween coupled cluster and DeepQMC, esp e- cially in angles and torsions. T o disentangle these differ- ences, we also ev aluate our transferable neural netw ork ansatz on the geometries obtained via the CCSD(T)/PIP PESs (second row of T able 1). The t wo sets of deep- learning VMC relativ e energies agree with eac h other within the reported single σ error b ounds, indicating that the differences b etw een the CCSD(T) and deep-learning VMC geometries ha ve limited energetic relev ance. Indeed, the largest con tributors to these geometric differences are the inter-fragmen t (HO 2 – OH) co ordinates, which are ex- p ected to b e the least stiff in the CP1 configuration. F or a sto c hastic method like deep-learning VMC, such a rel- ativ ely flat PES results in a larger v ariance in the opti- mized relative orientation of the t wo fragments. The same, to a lesser extent, is true for the transition state struc- ture (TS), where w e also find non-negligible differences (although noticably smaller than for CP1) in the inter- fragmen t angles and torsions b etw een deep-learning VMC and CCSD(T). T urning now to the relative energies of these intermediate geometries, w e find that deep-learning VMC consistently stabilizes CP1 and destabilizes the tran- sition state compared to CCSD(T), regardless of the exact geometry used. It is therefore conceiv able that this effect is a manifestation of the stronger static correlation to b e exp ected for these geometries, and highlights the b enefits of using black-box metho ds applicable to strongly corre- lated systems suc h as deep-learning VMC when describing PESs of chemical reactions. 2 Summar y and Discussion W e ha ve presented a nov el metho d that enables the ex- ploration of ab initio ground and excited state p otential energy surfaces of strongly correlated systems by in tegrat- ing contin uous transferable deep-learning VMC sim ulation with GPR. Our metho d turns the extrapolation capabil- ities of transferable neural netw ork wa ve functions in to a practical approach for simulating chemical pro cesses, amortizing the cost of transferable neural net work w av e function optimization through rep eated ev aluation across molecular configuration space. This work provides the first demonstration of transferable wa ve functions ac hiev- ing zero-shot chemical accuracy in relative energies across large, con tinuous regions of molecular configuration space, exemplified for systems with up to nine degrees of freedom. 7 T able 1: Comparison of the deep-learning VMC and CCSD(T) c haracterizations of the HO 2 · + OH · − − → H 2 O + O 2 reaction. TS denotes the transition state structure, while CP1 corresponds to the lo cal minimum found near the entrance of the reactiv e channel, first describ ed in Liu et al. [40]. Two sets of geometries are considered: those obtained from the PIP neural- net work potential fitted to CCSD(T)-F12a energies, and those optimized via the presented DeepQMC/GPR metho d. reactan t CP1 TS pro duct relativ e energy (k cal/mol) CCSD(T)/PIP 0 . 0 − 3 . 7 − 1 . 4 − 70 . 6 DeepQMC/GPR 0 . 0 − 4 . 9 ± 0 . 5 0 . 4 ± 0 . 5 − 69 . 8 ± 0 . 4 DeepQMC on PIP geom 0 . 0 − 4 . 3 ± 0 . 6 − 0 . 5 ± 0 . 6 − 69 . 9 ± 1 . 0 DeepQMC v. PIP geometry MAE b onds (a 0 ) 0 . 02 0 . 03 0 . 01 < 0 . 01 angles (deg) 0 . 7 7 . 7 4 . 3 < 0 . 1 torsions (deg) — 12 3 — Utilizing these ansatzes, adv anced Mon te Carlo estimators are employ ed to obtain low v ariance estimates of energy and n uclear force expectation v alues. This data is then used to fit lo cal GPR mo dels of the PES, without sacri- ficing accuracy in the single-p oint setting, while greatly increasing data efficiency during geometry optimization, where the PES m ust b e ev aluated at a n umber of nearby p oin ts. W e illustrate the accuracy and broad usability of our metho d by targeting a range of distinct applications. W e initiate the assessmen t by relaxing five diatomic molecules, reac hing very go od agreemen t with experimental refer- ences. T urning to the more demanding task of char- acterizing elementary c hemical pro cesses, w e obtain re- sults matching reference calculations when simulating the MEPs on the t wo dimensional PESs of the ammonia inv er- sion and formaldehyde isomerization. Next, the feasibility of acc urate geometry optimizations on excited PESs is es- tablished, by computing the adiabatic excitation energy of the singlet-triplet excitation of ethylene, in agreement with computational references on b oth excitation energy , and excited state equilibrium geometry . W e then pro- ceed to characterize the H 2 + NH − − → H · + · NH 2 re- action, qualitatively repro ducing the small-basis FCI en- ergy profile of the reaction pro ceeding in its lo west sin- glet state, and extending the analysis to the first sin- glet excited state, where a new analogous reaction chan- nel is identified. Lastly , we simulate a nine dimensional PES section around the HO 2 · + OH · − − → H 2 O + O 2 reaction pathw ay , demonstrating the scalabilit y of our metho d to unprecedented v olumes of molecular configura- tion space. Using a single transferable ansatz for the en tire PES, quantitativ e agreemen t is reac hed with a reference neural-net work potential fitted on more than one hundred thousand CCSD(T)-F12a single-p oint energy calculations. Small deviations around the in termediate geometries in- dicate that the use of multi-reference algorithms such as deep-learning VMC might b e warran ted. Com bining transferable QMC and GPR for the opti- mization of molecular geometries is a nov el paradigm that lev erages the interpolation capabilities of neural netw orks while retaining the computation’s ab initio character. An- c hored in deep-learning VMC, the metho d inherits all its b eneficial attributes, suc h as its applicability to strongly correlated systems, a consisten t description of all molec - ular configurations, i.e. the intermediates of chemical re- actions, the straight forw ard extension to the simulation of excited states, and its ov erall black-box nature. F ur- thermore, the metho d provides direct access to the wa ve function, and hence all electronic prop erties, at any point in configuration space and will benefit from any improv e- men ts made in the rapidly developing field of transferable deep-learning VMC. While not strictly necessary , GPR further impro ves efficiency of our method b y reusing in- formation from previous force and energy ev aluations of the PES and enables the automation of geometry opti- mizations by querying the costly deep-learning VMC ora- cle only when necessary . Ov erall our metho d is a to ol for the exploration of in tricate PESs of small molecules that streamlines the pro cess of quan tum chemistry computa- tion, fitting the PES with surrogate models and the op- timization of chemical pro cesses and significantly reduces the cost compared to the alternative of consecutiv e, inde- p enden t ab initio sim ulations. While the presented results are promising, imp ortant c hallenges remain. W e estimate the simulation of the nine dimensional PES of H 2 O + O 2 to be at the limit of our curren t capabilities. T o increase system sizes in terms of the n umber of electrons as well as the dimensionality of the molecular configuration space tec hnical innov ations, suc h as the integration with finite range embeddings [44] or sparse w av e function arc hitectures [45], will b e required. F urthermore, the metho d w ould greatly b enefit from more mature and adv anced geometry optimization protocols and could b e employ ed as a to ol for ab initio molecular dynamics simulation. W e hop e that our method will b e further developed, and can b ecome the default scheme for the PES exploration of strongly correlated, medium size systems. 3 Methods The method presented here is concerned with solving tw o main tasks: obtaining neural netw ork ansatzes that ac- curately describ e the electronic structure for a large and con tinuous range of molecular geometries, and the subse- quen t extraction of all information necessary for geometry optimization from these ansatzes. The rest of this section details the tec hniques used to accomplish these tw o ob- jectiv es, and is organized as follows. In Section 3.1, we 8 describ e solving the Schr¨ odinger equation for a contin u- ous range of molecular geometries with transferable deep- learning VMC. Then, in Section 3.2, the tec hniques of obtaining energies, nuclear forces and Hessians from con- v erged deep-learning VMC solutions are discussed, along with some asp ects of utilizing this information in do wn- stream tasks such as geometry or MEP optimizations in Section 3.3. 3.1 Transferable deep-learning v aria- tional Monte Carlo for continuous molecular configura tion sp aces This section discusses the core concepts of deep-learning v ariational Monte Carlo as w ell as its extension to the geometrically transferable setting. The generalization to con tinuous configuration spaces is comprised three main con tributions, the transferable optimization across molec- ular geometries, the extended w av efunction ansatz and the sampling of electronic and nuclear degrees of freedom. 3.1.1 Deep-learning v aria tional Monte Carlo The time-indep enden t, non-relativistic Schr¨ odinger equa- tion takes the form of an eigen v alue equation ˆ H Ψ = E Ψ Ψ ∈ H − , (4) where ˆ H is the Hamiltonian of the physical system un- der in vestigation, Ψ is the wa ve function which, describ- ing fermions, m ust b elong to the anti-symmetric subspace of the many-bo dy Hilb er-space ( H − ) and E is its as- so ciated energy . Here, w e are concerned with the b e- ha vior of electrons in molecular systems sub ject to the Born–Opp enheimer appro ximation, and therefore with the clamp ed n ucleus Coulom b Hamiltonian (in atomic units) ˆ H = − 1 2 X i ∇ 2 i + X i
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment