RAFT-UP: Robust Alignment for Spatial Transcriptomics with Explicit Control of Spatial Distortion
Spatial transcriptomics (ST) profiles gene expression across a tissue section while preserving the spatial coordinates. Because current ST technologies typically profile two-dimensional tissue slices, integrating and aligning slices from different re…
Authors: Yaqi Wu, Jingfeng Wang, Xin Maizie Zhou
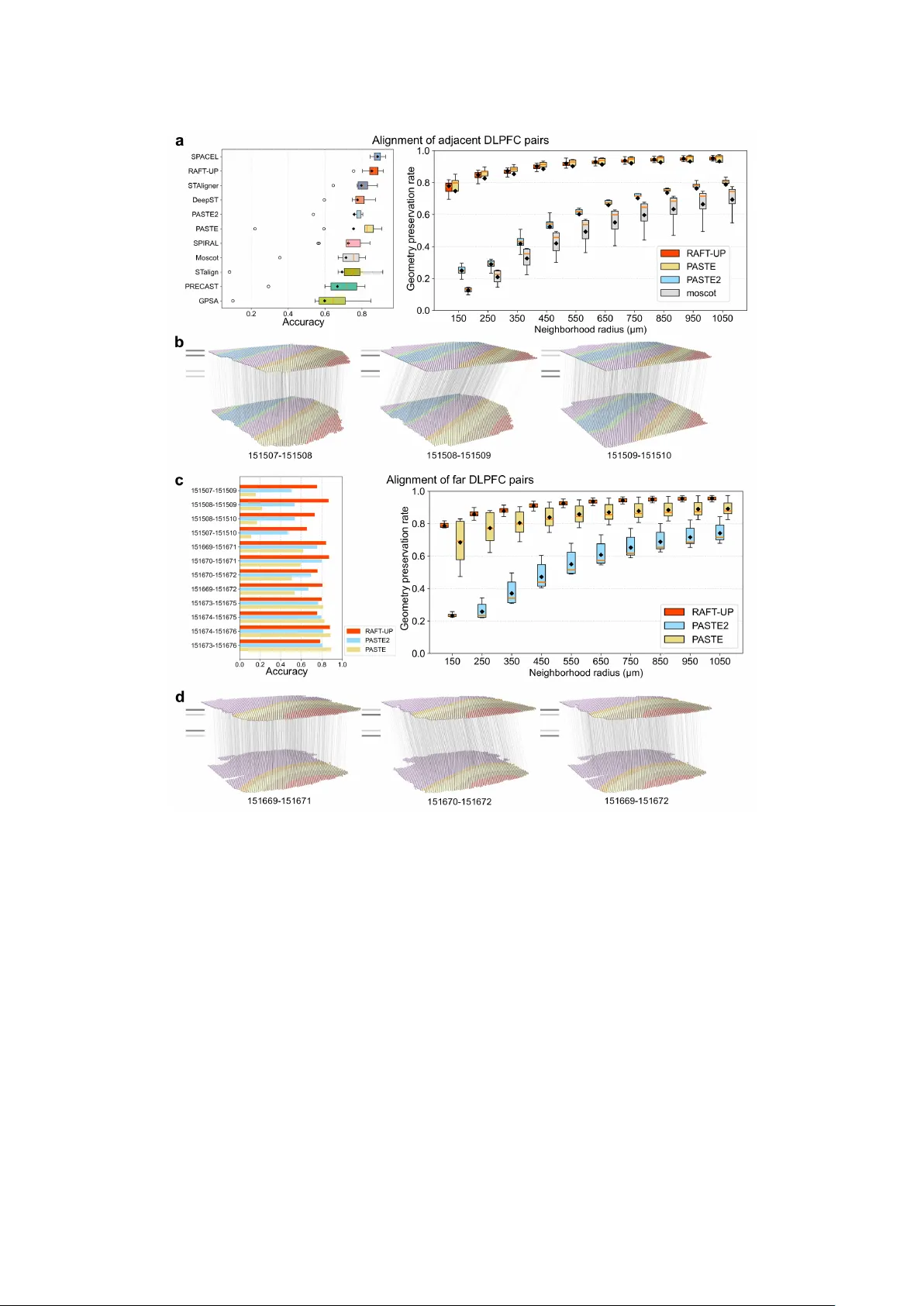
RAFT-UP: Robust Alignmen t for Spatial T ranscriptomics with Explicit Con trol of Spatial Distortion Y aqi W u 1 , Jingfeng W ang 2 , Xin Maizie Zhou 3 , Y anxiang Zhao 1* , Zixuan Cang 2,4* 1 Departmen t of Mathematics, George W ashington Univ ersit y , W ashington D.C, USA. 2 Departmen t of Mathematics, North Carolina State Univ ersity , Raleigh, NC, USA. 3 Departmen t of Biomedical Engineering, V anderbilt Univ ersit y , Nash ville, TN, USA. 4 Cen ter for Research in Scientific Computation, North Carolina State Univ ersity , Raleigh, NC, USA. *Corresp onding author(s). E-mail(s): yxzhao@email.gwu.edu ; zcang@ncsu.edu ; Abstract Spatial transcriptomics (ST) profiles gene expression across a tissue section while preserving the spatial co ordinates. Because curren t ST tec hnologies typically profile tw o- dimensional tissue slices, in tegrating and aligning slices from differen t regions of the same three-dimensional tissue or from samples under differen t conditions enables analyses that rev eal 3D organization and condition-asso ciated spatial patterns. Two ma jor challenges remain. First, interpretable and flexible con trol ov er spatial distortion is needed b ecause rigid transformations can be o verly restrictiv e, whereas highly deformable mappings ma y arbitrarily distort spatial proximit y . Second, biologically plausible matching is also needed, esp ecially when the slices ov erlap partially . Here, we in tro duce RAFT-UP , a to ol for robust ST alignment that pro vides explicit control o ver spatial distance preserv ation through a fused supervised Gromov-W asserstein (FsGW) optimal transport framework. FsGW com bines expression and spatial information, incorp orates sp ot-wise constrain ts to discourage biologically implausible matc hes, and enforces a pairwise distance-consistency constrain t that prev ents mapping tw o pairs of spots when their spatial distances differ b ey ond a specified tolerance. W e demonstrate that RAFT-UP accurately aligns slices from differen t regions of the same tissue and slices from different samples. Benc hmark- ing sho ws that RAFT-UP improv es spatial distance preserv ation while ac hieving sp ot lab el matc hing accuracy comparable to state-of-the-art methods. Finally , w e demonstrate RAFT-UP on t w o spatially constrained do wnstream applications, including spatiotem- p oral mapping of dev eloping mouse midbrain and comparativ e cross-slice analysis of cell-cell comm unication. RAFT-UP is av ailable as op en-source softw are. Keyw ords: F used Sup ervised Gromo v-W asserstein, P artial Alignmen t, Spatial T ranscriptomics 1 In tro duction Spatial transcriptomics (ST) tec hnologies [ 1 , 2 ] measure gene expression across spatially resolved lo cations within tissue sections. There are several ST technologies, including sequencing-based approac hes such as 10x Visium [ 3 ], Slide-seq [ 4 ], and Stereo-seq [ 5 ], which provide transcriptome- wide co verage at differen t spatial resolutions, and imaging-based approac hes such as seqFISH [ 6 ], osmFISH [ 7 ], and MERFISH [ 8 ], which ac hieve subcellular resolution for selected genes. ST data has provided critical insigh ts into cellular organization and tissue architecture in their nativ e spatial con text, imp ortan t information that is lost in single-cell RNA sequencing data (scRNA-seq). V arious computational approaches ha ve b een dev elop ed sp ecifically for extracting biological kno wledge from ST data. These include metho ds for iden tifying spatially v ariable genes [ 9 ], inferring cell-cell com- m unication pro cesses [ 10 ], and segmen ting spatial tissue domains [ 11 ]. These analyses often focus on individual ST slices. T o gain a comprehensive understanding of a system and to identify key differences across condi- tions, it is crucial to p erform joint analyses of m ultiple ST slices from different regions of the same three-dimensional tissue or from differen t samples representing different conditions or stages. Sev- eral computational metho ds hav e b een developed for aligning ST slices. GPSA [ 12 ] is a probabilistic metho d that uses a tw o-lay er Gaussian pro cess where a w arping function maps spatial co ordinates to a common co ordinate system (CCS) and a second function maps CCS to phenotype, that is, gene expression profile. It provides alignment in t wo mo des, mapping both slices to a de novo CCS or fixing one slice as the CCS and mapping the other one on to it. PRECAST [ 13 ] is another prob- abilistic approach that uses spatial factor analysis to obtain join t laten t em b eddings of multiple slices. ST align [ 14 ] is a geometric approac h based on large deformation diffeomorphic metric map- ping (LDDMM), and represents the alignment in the form of a diffeomorphism b etw een the spatial domains of the slices. SP ACEL [ 15 ] is a comprehensive tool with three mo dules including Sp oint for cell type deconv olution using a m ulti-la yer perception model and a probabilistic model, Splane for domain segmen tation across multiple slices using a graph con v olution neural netw ork, and subse- quen tly Scub e that p erforms a rigid-b o dy transformation to stack the slices. There are also sev eral deep learning-based approac hes including DeepST, SPIRAL, and ST Aligner, whic h emplo y differ- en t v ariants of graph neural netw ork architectures to deriv e shared laten t embeddings of sp ots. The shared latent embeddings obtained in DeepST [ 16 ], SPIRAL [ 17 ], ST Aligner [ 18 ], and PRE- CAST provide an implicit form of alignmen t, which can b e conv erted into explicit spot-to-sp ot 2 corresp ondences through postpro cessing suc h as k-nearest neighbor matc hing. Additionally , ST Al- igner and SPIRAL also pro vide dedicated functionality that extends the shared latent embeddings in to three-dimensional reconstructions, where ST Aligner p erforms rigid transformation using the iterativ e closest p oint algorithm and SPIRAL first p erforms a fused Gromo v-W asserstein optimal transp ort to obtain a transp ort matrix connecting the t wo slices and maps each sp ot to the a verage co ordinates of its top corresponding sp ots in the other slice. The problem of aligning multiple slices of ST data can b e viewed as a sp ecial case of in tegrating and aligning high-dimensional and high-resolution omics datasets. Optimal T ransp ort (OT) [ 19 , 20 ] is naturally suited for such tasks, as it provides a general framework for finding corresp ondence b et ween complex distributions with the flexibility to accoun t for structural similarity . OT has b een widely applied to finding v arious corresp ondences among single-cell or spatial omics data. F or exam- ple, W addingtonOT [ 21 ] and SpaT rac k [ 22 ] use OT to infer developmen tal tra jectories from time series of scRNA-seq datasets and ST datasets, resp ectively . SCOT [ 23 ] uses Gromov-W asserstein (GW) [ 24 ] OT to align scRNA-seq data and single-cell A T A C sequencing data, while no voSpaRc [ 25 ], SpaOTsc [ 26 ], and T ACCO [ 27 ] use different v ariants of the fused Gromo v-W asserstein (FGW) OT [ 28 ] to in tegrate scRNA-seq data with spatial gene expression data. Recen tly , several approac hes ha ve utilized OT for aligning ST slices including P ASTE [ 29 ] using F GW, P ASTE2 [ 30 ] using partial F GW, and moscot [ 31 ] using an FGW algorithm with impro ved efficiency . The F GW approaches used in these to ols utilize gene expression dissimilarit y as in ter-dataset costs and promote the preser- v ation of in tra-dataset spatial distances. In these OT-base d approac hes, the alignmen t is represen ted b y the transp ort plans in the form of probability matrices. P ASTE and P ASTE2 further generate a consensus slice to which every slice is mapp ed, by computing the FGW barycenter. Existing metho ds either rely on rigid transformations, whic h lack the flexibility to capture lo cal spatial v ariation, or imp ose only soft p enalties, which could lead to substantial distortions in spatial distances. Precise con trol of spatial distance distortion across aligned ST slices is critical for biolog- ically meaningful downstream analyses, particularly in applications with strong spatial constrain ts suc h as cell-cell communication analysis [ 10 ]. T o address these limitations, we present RAFT-UP , a Robust Alignmen t Framew ork for spatial Transcriptomics with User-Prescrib ed spatial distortion con trol. RAFT-UP also a voids biologically implausible mapping and simultaneously infers the partial o verlap portion b etw een slices. The core algorithm of RAFT-UP is a new F used sup ervised Gromo v- W asserstein (FsGW) optimal transp ort metho d. FsGW extends our previous works, sup ervised OT 3 [ 32 ] and sup ervised GW [ 33 ], to simultaneously incorp orate element-wise constraints on the trans- p ort matrix to prohibit corresp ondences b etw een dissimilar sp ots and higher-order constrain ts on pairs of elemen ts in the transp ort matrix to enforce distance preserv ation within a tolerance. In the FsGW framew ork, the cross-dataset cost is deriv ed from a join t em b edding of the slices, obtained b y extending our earlier graph neural netw ork-based spatially aw are em b edding method [ 34 ] to train in an alternating scheme across differen t slices. T o handle large datasets, we first do wnsample the sp ots to obtain a uniformly spaced set of p oints. The FsGW is then computed on the downsampled data, after which the full transport matrix is reco vered by solving a supervised OT problem. W e show that RAFT-UP accurately aligns ST slices from differen t regions of the same tissue and from samples across different developmen tal stages, using real datasets generated by v arious tec h- nologies including 10X Visium, Stereo-seq, and MERFISH. The b enchmarking results demonstrate that RAFT-UP effectively manages the trade-off betw een spatial preserv ation and accuracy on cell t yp e matc hing. It outp erforms rigid transformation based approaches in accuracy and achiev es b etter spatial proximit y preserv ation than the approac hes allowing spatial flexibilit y while also achieving top accuracy . W e further demonstrate the utility of RAFT-UP in comparative analysis of cell-cell comm unication, a pro cess with strong spatial constrain t, across m ultiple slices. Results Ov erview of RAFT-UP Across spatially adjacent or temporally consecutiv e ST slices, there is often mo derate spatial distor- tion, v ariation in gene expression, and partial ov erlap. W e therefore seek an alignmen t metho d that b oth allo ws these differences in the mapping and regulates their exten t. T o address these challenges, RAFT-UP uses a F used supervised Gromo v-W asserstein (FsGW) optimal transport framew ork. FsGW integrates supervised OT [ 32 ] and sup ervised GW [ 33 ], extending FGW to incorp orate elemen t-wise and higher-order constraints on the transp ort plan, for controlling gene expression similarit y and spatial distance preserv ation, respectively . Unlik e the partial OT framew ork, whic h requires a predetermined transp orted mass, FsGW naturally infers the prop ortion of partial ov erlap from these constraints. An ov erview of the core algorithm of RAFT-UP is shown in Fig. 1 . Giv en t wo ST slices of n 1 and n 2 sp ots, an in ter-dataset cost C ∈ R n 1 × n 2 + is computed from a join t em b edding of the sp ots in the tw o slices, which describ es the gene expression profile of each 4 Fig. 1 : Overview of RAFT-UP . a , An inter-dataset cost matrix that capture gene expression dissimilarit y b etw een sp ots and their spatial neigh b orho o ds is obtained from a Deep Graph Infomax mo del trained alternately on the tw o datasets. The t wo intra-dataset cost matrices are obtained from the spatial distances b et ween the sp ots within each slice. b , An optimal probabilistic mapping b et ween the t wo slices is obtained b y solving a F used supervised Gromo v-W asserstein optimal trans- p ort, whic h takes the costs defined in (a) and enforces element -wise and second-order constraints on the transp ort plan. c , T o handle large ST datasets, RAFT-UP uses a downsampling and lifting strategy . An initial mapping is first obtained across geometrically uniform downsampling of the tw o slices. A full alignmen t is then reco vered by solving a sup ervised optimal transp ort problem with a cost based on the distance of each sp ots to the anchor p oints in the initial mapping. sp ot and its spatial neighborho o d. W e train a Deep Graph Infomax model [ 34 , 35 ] alternately on the t w o datasets to obtain the join t embedding, and later compute C ij as the Euclidean distance of spot i of slice A and sp ot j of slice B in the embedding space. Two intra-dataset cost matrices D 1 ∈ R n 1 × n 1 + and D 2 ∈ R n 2 × n 2 + are computed from the spatial distances betw een the sp ots within eac h slice (Fig. 1 a). RAFT-UP then finds an optimal coupling matrix P ∗ ∈ R n 1 × n 2 b et ween the slices b y solving a FsGW problem that has the following prop erties. (1) P ∗ ij has a high weigh t indicating corresp ondence betw een spot i in slice A and sp ot j in slice B if the t wo sp ots’ spatial gene expression profiles are similar. (2) RAFT-UP can also explicitly forbid matc hes whose expression dissimilarity exceeds a user-con trolled threshold ρ f , which helps preven t mismatches such as aligning different cell t yp es. (3) When b oth P ∗ ij and P ∗ kl ha ve high v alues, the in tra-dataset distances D 1 ik and D 2 j l are similar. (4) RAFT-UP further explicitly con trols the spatial structure preserv ation by enforcing 5 that any pair of corresp ondences can only co exist when the distance distortion is within a threshold ρ s . As a result, RAFT-UP naturally obtains a partial coupling using the in terpretable parameters ρ f and ρ s that con trol feature consistency and geometry consistency , resp ectiv ely (Fig. 1 b). In practice, RAFT-UP uses a do wnsampling, FsGW coupling, and lifting to full data w orkflow to impro ve the computational efficiency (Fig. 1 c). Sp ecifically , a geometrically uniform downsampling is first p erformed on the tw o slices. F or 10X Visium data, we exploit the near-hexagonal grid of sp ots. F or other ST data, we use a max-min sampling pro cedure [ 36 ] to promote even spreading of sampled points across the tissue domain. W e then compute an FsGW coupling b et ween the tw o do wnsampled slices. Finally , we lift this corresp ondence to the full datasets by constructing a cross- slice spatial cost for all spot pairs, which is deriv ed from their spatial proximit y to the sampled p oints within each slice and the precomputed cross-slice corresp ondence of the sampled p oints. Using this cross-slice spatial cost for full data, we solve a sup ervised optimal transp ort problem [ 32 ] to obtain a coupling ov er all spots. Details of the RAFT-UP model and algorithm are pro vided in Metho ds and Supplemen tary Note . Ac hieving high accuracy while preserving spatial structure W e first ev aluate the p erformance of RAFT-UP on mapping accuracy and preserv ation of spatial structure using the human dorsolateral prefrontal cortex (DLPFC) dataset generated with the 10x Visium platform [ 38 ]. The DLPFC dataset con tains 12 cortical sections with exp ert annotations of cortical lay ers 1 to 6 and white matter (WM). These 12 slices are organized into three groups, eac h con taining four spatially consecutive sections (A, B, C, and D) from the same tissue sample. The pairs A-B and C-D are 10 µ m apart and the pair B-C is 300 µ m apart. W e use tw o metrics to ev aluate the alignmen t p erformance, including (1) alignment accuracy of spatial domains and (2) geometric neigh b orho o d preserv ation. An alignmen t b etw een tw o slices is represented b y a mapping matrix P ∈ R n 1 × n 2 , where P ij is the matching w eight b et ween sp ot i in slice A and sp ot j in slice B. First, w e rep ort alignment accuracy , defined as the fraction of sp ots whose highest-w eight matching in the other slice has the same lay er lab el. Second, w e rep ort the Geometric Preserv ation Rate (GPR), whic h quantifies ho w well local spatial neigh b orho o ds, defined within a fixed radius r , are preserved under the alignmen t P , b y measuring how m uch transported mass from a sp ot’s neigh b orho o d maps in to the neighborho o ds of its matched sp ots. Both metrics take v alues in [0 , 1], with higher v alues indicating better alignment. See Metho ds for detailed definition of these ev aluation m etrics. 6 Fig. 2 : Benchmarking alignment on human DLPF C Visium slices. a , Performance on adjacen t DLPFC section pairs. Left: alignment accuracy for RAFT-UP and other metho ds. Righ t: geometric preserv ation rate (GPR) as a function of neighborho o d radius, comparing RAFT-UP , P ASTE2, P ASTE, and moscot. SP ACEL is ev aluated under its native, lab el-informed configuration and the remaining metho ds are ev aluated without lab el information. The accuracy of PRECAST, ST align, DeepST, SPIRAL, and GPSA are taken from a recent b enc hmark [ 37 ]. b , Represen tative RAFT-UP alignments of three adjacen t pairs. Sp ots are colored by annotated cortical lay er and line segmen ts indicate the computed sp ot-level corresp ondence b et ween slices. c , Performance on non adjacen t DLPFC pairs. Left: Accuracy for eac h pair comparing RAFT-UP , P ASTE2, and P ASTE. Righ t GPR v ersus neighborho o d radius for far pairs. d , Represen tative RAFT-UP alignments for three far pairs. In the b oxplots, b oxes indicate the in terquartile range (25th-75th p ercentiles) with the median as the center line, whisk ers extend to the most extreme v alues within 1.5 × IQR, and blac k diamonds denote the mean. 7 Fig. 3 : Benchmarking alignment on MERFISH sections. a , Lab el matching accuracy for MERFISH data for adjacent pairs (left) and far pairs (righ t). SP ACEL is ev aluated under its native lab el-informed configuration and other metho ds are ev aluated without access to lab el information. b , Representativ e RAFT-UP alignments for an adjacent pair (top) and a far pair (b ottom) showing b oth alignment of all cells (left) and alignment of ep endymal cells (right) highlighting its p erformance on aligning fine structures. c , Geometric preserv ation rate (GPR) as a function of neigh b orho o d radius comparing RAFT-UP , P ASTE2, P ASTE, and moscot on adjacent pairs (top) and far pairs (b ottom). The alignmen t p erformance of RAFT-UP on DLPF C sections is summarized in Fig. 2 . Across all adjacen t alignmen ts, RAFT-UP achiev es consisten tly high alignment accuracy (Fig. 2 a, b). Among the compared metho ds, the alignment mo dule of the comprehensiv e SP ACEL pack age utilizes lab el information under its native setup, and therefore SP ACEL achiev es the highest lab el accuracy . The other metho ds op erate under the same setup without access to lab el information, and among these metho ds, RAFT-UP ac hieves the strongest o verall accuracy . RAFT-UP also sho ws strong preser- v ation of lo cal spatial geometry on adjacen t pairs when compared to other OT-based approaches (Fig. 2 b, Supplemen tary Fig. 1), where RAFT-UP p erforms comparably to P ASTE and outp erform P ASTE2 and moscot across neighborho o d radii. T o further ev aluate the performance on less simi- lar slices, w e consider far slice pairs which include the middle-adjacent pairs that are 300 µ m apart and the non-adjacent pairs (Fig. 2 c, d). RAFT-UP main tains high lab el accuracy as slice similarit y 8 decreases, and ac hieves the highest a verage accuracy among the three OT-based metho ds (RAFT- UP: 0.792, P ASTE2: 0.680, P ASTE: 0.528, Fig. 2 c), demonstrating robust p erformance b ey ond aligning highly similar pairs. RAFT-UP also pro vides the b est geometry preserv ation on these far pairs (Fig. 2 d, Supplementary Fig. 2), highlighting the benefit of the sup ervised Gromo v-W asserstein comp onen t in main taining spatial structure when aligning less similar slices. W e next ev aluate RAFT-UP on a MERFISH dataset. On adjacent section pairs, RAFT- UP ac hieves consisten tly high lab el matching accuracy across all pairs outp erforming all other metho ds that do not use lab el information (Fig. 3 a, Supplemen tary Fig. 3). On more distant non- adjacen t pairs, RAFT-UP maintains high accuracy and substan tially impro v es o v er unconstrained full mapping and partial OT metho ds (Fig. 3 a). Qualitative corresp ondences further supp ort these quan titative ev aluations. RAFT-UP produces coheren t cross-section mappings at the whole-tissue lev el and preserves the spatial lo calization of low-abundan t p opulations such as ep endymal cells across adjacen t and farther-separated pairs (Fig. 3 b, Supplementary Fig. 3). Finally , RAFT-UP consisten tly ac hieves b etter geometric preserv ation than traditional OT-based approach ov er a wide range of neighborho o d radii for b oth adjacent and far pairs (Fig. 3 c), with the largest gains at small radii whic h is most sensitiv e to lo cal distortions. T ogether, these results demonstrate that RAFT-UP preserv es spatial structures across v arious scales while maintaining high lab el alignment accuracy . In terpretable alignmen t of partially o verlapping slices In practice, particularly when aligning ST slices from different biological replicates, the slices ma y only partially ov erlap due to v ariation in tissue cov erage, tearing, or cropping, and their cell-t yp e comp osition ma y also v ary . Many alignmen t formulations assume near-complete ov erlap or require the degree of ov erlap to b e sp ecified, which can lead to spurious corresp ondences when substan- tial non-ov erlapping regions are present. RAFT-UP mitigates this limitation by in tro ducing t wo in terpretable user-controlled parameters, a minimum gene expression similarity threshold ρ f and a tolerance ρ s for spatial distance distortion. These together enable robust alignment in partial-o verlap scenarios. T o demonstrate this utilit y , we construct several con trolled partial-o verlap examples with kno wn corresp ondences at sp ot level or at cell type level. First, we extract pairs of partially ov erlapping windows from the same DLPFC slice and treated them as tw o inputs for alignment (Fig. 4 a, b). In these within-slice exp eriments, the main c hal- lenge is the partial o verlap of spatial regions. RAFT-UP pro duces corresp ondences concentrated 9 Fig. 4 : Ev aluation of RAFT-UP under partial o verlap. a, b , Partially ov erlapping windo ws extracted from the same DLPFC slice. RAFT-UP alignment only contain alignment in the truly o verlapping region while traditional full OT alignmen t forces full matc hing of the tw o partially ov er- lapping windows. c , Partially o verlapping regions with different geometry and cell-type comp osition across an adjacent DLPFC pair. d , A MERFISH example with a similar geometry but different anatomical region comp ositions. RAFT-UP aligns cells from anatomical regions presen t in b oth windo ws, while lea ving cells from regions absent in one slice largely unmapp ed. in the shared region and does not force extensive matches for the non-o v erlapping portions. In con trast, traditional OT with full matc hing enforces global mass matching and therefore matc hes non-o verlapping p ortions, which could also cause large spatial distortion if the geometries are differ- en t. Notably , RAFT-UP ac hieves the correct partial ov erlapping without requiring a pre-sp ecified o verlap prop ortion. W e further illustrate partial ov erlap across differen t slices with different geometry or composi- tion. F or a pair of DLPFC slices, RAFT-UP obtains coheren t mappings in the region where laminar structure is shared while a voiding forced correspondence in regions lacking clear anatomical coun- terparts in the other slice (Fig. 4 c). A similar behavior is observed in the MERFISH example, 10 where RAFT-UP aligns cells belonging to anatomical regions presen t in both windo ws and lea v es the MP A and MPN cells in slice -0.14 largely unmapped as these regions are absent in slice -0.19 (Fig. 4 d). These results suggest that RAFT-UP pro vides in terpretable alignmen ts in the settings with unkno wn and p otentially small ov erlap. Mo deling spatiotemp oral tra jectory from ST slices across time p oints Fig. 5 : Spatiotemp oral analysis of mouse midbrain dev elopment. a , Alignmen t of the Stereo-seq data of mouse midbrain from E12.5 to E14.5 and from E14.5 to E16.5. Cells are colored b y the exp ert annotations from original study: R GC, radial glia cell; GlioB, glioblast; NeuB, neu- roblast. b , The disturbance of the relative lo cation of cells on the Caudal to Rostral axis through alignmen t. The median v alues of the b o x plots are 0.0793, 0.0542, 0.0596, 0.0325, from left to right resp ectiv ely . Cell corresp ondences with a change of the relative co ordinate greater than 0.1 are sho wn. c , The coun ts and visualization of corresp ondences in the alignments that are inconsisten t with the dominant tra jectory patterns whic h are R GC to GlioB, RGC to NeuB, or unchanging. 11 Next, we apply RAFT-UP to Stereo-seq data of mouse midbrain profiled at E12.5, E14.5, and E16.5 to mo del spatiotemporal tra jectories across dev elopmental stages. The dataset includes exp ert annotations of three ma jor cell states including radial glial cells (RGC), glioblasts (GlioB), and neuroblasts (NeuB), with exp ected developmen tal tra jectory from R GC to GlioB and NeuB. W e use RAFT-UP to p erform pairwise alignmen ts b etw een consecutive time p oin ts and visualize the inferred cross-time corresp ondence among the cells (Fig. 5 a). The resulting mapping links cells across time points while preserving their spatial organization within eac h slice. W e quan titatively examine ho w the inferred corresp ondences preserv e the large-scale spatial ordering along the caudal- rostral axis. F or eac h slice, we define a normalized caudal-rostral co ordinate and compute the c hange in this co ordinate for eac h mapp ed cell after aligning to the next time p oin t. Across b oth time pairs, RAFT-UP produces small co ordinate shifts (Fig. 5 b). Additionally , corresp ondences with large coordinate changes ( > 0 . 1) are relatively rare and are mainly concentrated in regions with substan tial shap e evolution. W e also examine consistency b etw een inferred corresp ondences and the ma jor dev elopmental patterns suggested b y the original study . Corresp ondences of the form R GC to GlioB, R GC to NeuB and same-type matches are treated as consistent, and we count edges that do not fall into these categories (Fig. 5 c). The RAFT-UP mappings sho w relatively sparse inconsistent cell correspondences. W e then compare these results with those obtained using SpaT rack [ 22 ]. RAFT-UP exhibits smaller disturbances along the caudal-rostral axis for b oth time pairs, particularly with muc h few er outliers, indicating improv ed preserv ation of within-slice spatial arrangement (Fig. 5 b). At the cell state lev el, RAFT-UP also pro duces few er correspondences that are inconsistent with the ma jor developmen tal transitions (Fig. 5 c). T ogether, these results suggest that RAFT-UP pro duces cross-time corresp ondences that are more spatially preserved and more consisten t with exp ected dev elopmental structure. Spatially preserving analysis of cell-cell communication across slices Cell-cell communication (CCC) is imp ortant for prop er functions of tissues and organs. Because CCC is inherently constrained by spatial proximit y , analyses that compare communication patterns across tissue slices are particularly sensitiv e to geometric distortions introduced during alignment. RAFT-UP is therefore w ell suited for this task, as it enables explicit control on the preserv ation of 12 Fig. 6 : Comparativ e analysis of cell-cell communication. a , COMMOT is applied indep en- den tly to each slice to infer cell-cell comm unication (CCC). CCC inferred on slice A is transferred to slice B using the RAFT-UP sp ot-to-sp ot mapping, enabling direct comparison to CCC inferred on slice B, and vice v ersa. b , Ligand–receptor pairs are rank ed b y their relative W asserstein distance b et ween CCC inferred directly on S2 and CCC inferred on S1 mapp ed onto S2 (CCC@S1 → S2), as w ell as b etw een CCC inferred directly on S1 and CCC inferred on S2 mapp ed onto S1 (CCC@S2 → S1) c , RAFT-UP alignment b etw een the tw o slices where lines indicate matched sp ots. d , Examples of ligand-receptor pairs with concordant spatial patterns across slices (FGF1-F GFR2 and SEMA3E- PLXND1), shown for receiv ed (Rec’d) and sen t (Sen t) CCC inferred on each slice and mapp ed in b oth directions. e , Examples of ligand-receptor pairs with differen t patterns across slices (WNT5A- FZD3 and PDGFB-PDGFRA). spatial proximit y among cells through the alignment (Supplemen tary Fig. 4). Here, we use RAFT-UP to comparativ ely analyze CCC across tissue slices. W e first independently infer CCC on eac h DLPFC slice using COMMOT [ 39 ], a spatially aw are CCC inference to ol for ST data. F or eac h ligand-receptor (LR) pair, COMMOT outputs a sp ot- wise directed net work in which the weigh t on edge ( i, j ) represents the inferred strength of signaling 13 from sp ot i to spot j . Using the transport plan obtained by RAFT-UP from slice A to slice B, w e map these communication fields across slices in b oth directions. Sp ecifically , receiver and sender fields inferred on slice A are transp orted onto slice B through the transp ose of the row-normalized transp ort plan, while fields inferred slice B are transp orted to slice A through the column-normalized transp ort plan. (Fig. 6 a). F or each LR pair, we quantify agreemen t b etw een a mapp ed field and the corresp onding field inferred on the target slice using a relative W asserstein dissimilarity . F or example, to compare the receiver field mapp ed from slice A to Slice B, R A → B , with the one inferred directly on slice B, R B , w e compute d relW ( R A → B , R B ) = W 1 ( R A → B , R B ) median k W 1 ( e R k A → B , R B ) , (1) where e R k A → B are the mapp ed fields using a randomly p erm uted RAFT-UP map, and W 1 is the 1-W asserstein distance. This normalization addresses the tendency of ra w W asserstein distances to increase for sparse fields and b etter rev eals structural agreemen ts b etw een fields. The LR pairs are then rank ed using this dissimilarity score (Fig. 6 b,c). Among the most conserved LR pairs, F GF1-FGFR2 sho ws a clear spatially structured pattern that is highly preserv ed after mapping, with both sender and receiver fields enric hed in the white matter regions (Fig. 6 d, Supplemen tary Fig. 5). This matc hes prior knowledge that F GF receptor sig- naling in oligo dendro cytes regulates m yelin growth and m yelin sheath thickness, where FGFR2 was implicated as a key receptor in this process [ 40 ]. The LR pair PSAP-GPR37 is also consistent across the slices in the same white matter region (Supplementary Fig. 6), whic h is consistent with evidence that prosap osin/prosaptide can signal through GPR37-family receptors and that GPR37 influences oligo dendrocyte differen tiation and cen tral nervous system my elination [ 41 ]. Another highly consis- ten t LR pair is SEMA3E-PLXND1 (Fig. 6 d, Supplementary Fig. 7). This signaling path wa y has a kno wn function of spatial organization with a do cumented role in forebrain axon tract developmen t and circuit wiring sp ecificity [ 42 ]. The LR pair WNT5A-FZD3 has also been implicated in spatial organization during cortical wiring [ 43 ]. In con trast to the highly consisten t SEMA3E-PLXND1 pat- tern, WNT5A-FZD3 is substantially less consistent across the t wo slices (Fig. 6 e, Supplementary Fig. 8). This difference may indicate that these guidance related signaling pathw ays can w ork in dif- feren t spatial scales with SEMA3E-PLXND1 forming a more coheren t pattern while WNT5A-FZD3 app ears more spatially lo calized and thus less consisten t across adjacent slices. PDGFB-PDGFRA 14 also exhibits a high dissimilarity across the t wo slices (Fig. 6 e, Supplemen tary Fig. 9). The CCC fields are broadly distributed, but their strongest regions o ccur in differen t lo cations. PDGFRA is a canonical mark er of oligo dendro cyte precursor cells that p ersist in the adult brain and con- tin ue to generate oligodendro cytes, and PDGF signaling regulates oligodendro cyte dev elopment[ 44 ]. This suggests that PDGF signaling remains broadly active in adult DLPFC tissue, but its spatial comm unication field is less organized across adjacen t slices. Discussion W e in tro duced RAFT-UP , an optimal-transp ort framew ork for aligning spatial transcriptomics (ST) slices when explicit corresp ondences are unav ailable and the exten t of ov erlap is unkno wn. In contrast to approac hes that rely on rigid transformations or produce only laten t-space agreement follo wed b y post hoc matc hing, RAFT-UP directly outputs an explicit probabilistic coupling b etw een spots. Metho dologically , RAFT-UP balances transcriptomic similarity with spatial structure through a fused supervised Gromo v-W asserstein (FsGW) formulation that (1) blocks implausible sp ot pairings using a feature cutoff ( ρ f ) and (2) constrains spatial distance distortion through an interpretable tolerance ( ρ s ). These constraints naturally yield a semi-balanced solution that can leav e unmatched mass when regions do not o verlap, av oiding the need to pre-sp ecify an ov erlap fraction. T o scale to large slices, RAFT-UP employs a downsample-align-upsample w orkflow: an FsGW coupling is computed on geometrically uniform landmarks and then lifted to full resolution via a sup ervised OT step. Across datasets spanning differen t tec hnologies and biological systems, RAFT-UP consistently ac hieves a fa vorable trade-off b etw een matching accuracy and geometric preserv ation. On Visium DLPF C sections, RAFT-UP attains high la yer-wise alignmen t accuracy while maintaining strong neigh b orho o d preserv ation, and the b enefits are most pronounced for non-adjacen t slice pairs where unconstrained transp ort plans are more lik ely to in tro duce long-range crossings or many-to-one assignmen ts. On MERFISH sections, RAFT-UP similarly ac hiev es strong lab el concordance while preserving fine structures, including lo w-abundance populations. Con trolled partial-ov erlap exp er- imen ts on window ed subregions show that RAFT-UP concentrates mass in the truly shared area and a voids forced matc hes in non-o verlapping tissue, unlik e full-mass alignmen t approaches. An imp ortant adv antage of RAFT-UP is its explicit and in terpretable con trol ov er spatial dis- tance preserv ation during alignment. This is particularly v aluable for analyses in whic h spatial 15 pro ximity is mechanistically constrained, such as spatio-temp oral dev elopment of cells and cell-cell comm unication. W e illustrated this adv an tage in tw o studies. In the Stereo-seq mouse midbrain dev elopmental series, RAFT-UP pro duced cross-time corresp ondences while maintaining global spatial ordering of cells. In the DLPFC cell-cell communication analysis, the geometry-preserving alignmen t enabled reliable cross-slice comparison of the inferred signaling fields b y limiting artificial c hanges in spatial range and neighborho o d structure. RAFT-UP do es require sev eral user-sp ecified parameters, but the main controls are directly in terpretable and tend to behav e consistently across datasets once they are calibrated to platform resolution and the exp ected biological differences betw een slices. The feature cutoff ρ f determines ho w strictly p oten tial corresp ondences m ust agree in gene expression, whereas the spatial tolerance ρ s con trols the allow able distortion of within-slice distances. The mixing w eight α pro vides a contin uous w ay to balance transcriptomic similarity against geometric consistency . As in other constrained alignmen t settings, excessively stringent thresholds may exclude true corresp ondences and lead to under-alignmen t, while ov erly p ermissive c hoices can admit spurious links, especially when the slices differ b y anisometric deformations or when the learned feature representation is affected by noise. Sev eral directions may further extend the scop e of RAFT-UP . On the algorithmic side, faster solv ers and multiscale implemen tations could improv e scalability to very large, densely sampled slices, and joint alignment of m ultiple slices could enable direct three-dimensional reconstruction without relying on combining pairwise results. F rom a statistical p ersp ective, incorp orating uncer- tain ty quantification for the transp ort plan would strengthen downstream comparisons and supp ort more reliable h yp othesis testing. Finally , integrating RAFT-UP with dynamical optimal transp ort mo dels offers a natural path tow ard contin uous spatiotemp oral inference while explicit con trol- ling geometric distortion, whic h is crucial in analysis with strong spatial constraints, including comm unication, migration, and niche dynamics. Metho ds F ull deriv ations, optimization algorithms, and implementation details for the optimal transp ort solv ers including fused supervised Gromo v-W asserstein and sup ervised OT, are provided in the Supplemen tary Information. 16 RAFT-UP mo del W e consider t w o spatial transcriptomics (ST) slices, A and B, with n 1 and n 2 sp ots, respectively . RAFT-UP represents an alignment b etw een slices in the form of a nonnegative coupling matrix P ∗ ∈ R n 1 × n 2 , where P ∗ ij represen ts the matc hing weigh t b etw een sp ot i in slice A and sp ot j in slice B. Giv en (1) a cross-slice cost matrix C ∈ R n 1 × n 2 describing spatial gene expression dissimilarit y b et ween sp ots across the slices and (2) intra-slice distance matrices D 1 ∈ R n 1 × n 1 and D 2 ∈ R n 2 × n 2 quan tifying spatial distances b etw een sp ots within the same slice, RAFT-UP obtains the mapping P ∗ b y solving the follo wing fused supervised Gromo v-W asserstein (FsGW) optimal transp ort problem. P ∗ = arg min P ∈ U ( ≤ a , ≤ b ; C , M ) α ⟨ C , P ⟩ F + (1 − α ) ⟨M , P ⊗ P ⟩ F + γ ∥ a − P1 n 2 ∥ 1 + ∥ b − P T 1 n 1 ∥ 1 , (2) U ( ≤ a , ≤ b ; C , M ) = { P ∈ R n 1 × n 2 + P1 n 2 ≤ a , P T 1 n 1 ≤ b , P rt = 0 if C rt > ρ f , P ij P kl = 0 if | D 1 ik − D 2 j l | > ρ s } . (3) Here, M represents the disturbance of spatial distances through the alignmen t with M ij k l = | D 1 ik − D 2 j l | 2 . The w eight parameter α con trols the relativ e contributions of the cross-slice cost ( ⟨ C , P ⟩ F ) and disagreemen t b etw een in tra-slice structure ( ⟨M , P ⊗ P ⟩ ). F or the feasible set (Eq. 3 ), the first t wo conditions P1 n 2 ≤ a , P T 1 n 1 ≤ b preven t the transp ort plan from creating mass, while allowing unmatc hed mass so the alignment can accommo date only partially ov erlapping slices. The third constrain t P rt = 0 if C rt > ρ f prohibits transport b etw een spot pairs whose feature dissimilarit y exceeds the cutoff ρ f . In practice, when C captures biological differences well, choosing ρ f according to cell type separation can largely prev ent cell type mismatches. The last constraint con trols the exten t to whic h the geometry is preserved. Sp ecifically , it prohibits sim ultaneous assigning p ositive masses to P ij and P kl if the difference b etw een the within-space distances D 1 ik and D 2 j l exceeds the threshold ρ s . The third te rm in Eq. 2 , ∥ a − P1 n 2 ∥ 1 + ∥ b − P T 1 n 1 ∥ 1 , encourages a more extensive coupling by p enalizing untransported mass. This is useful b ecause, under the combined constraints, a fully mass-preserving plan ma y not exist, and the total transp orted mass is therefore determined implicitly by the optimization. When dealing with alignmen t of spatiotemp oral data, to accoun t for the spatial gro wth, a relative spatial preserv ation constrain t is used instead: P ij P kl = 0 if | D 1 ik − D 2 j l | > ρ s min { D 1 ik , D 2 j l } . T ogether, RAFT-UP infers an alignment b etw een slices that balances gene-expression agreement with preserv ation of intra-slice spatial structure, while automatically 17 determining the coupled mass. The exten t of feature and geometric consistency is controlled by the in terpretable thresholds ρ f and ρ s . Details of algorithms for solving the FsGW problem and their deriv ation are in Supplementary Note. Data prepro cessing and construction of cost matrices W e ev aluate our method on the DLPFC Visium dataset (12 slices in total), the MERFISH dataset (5 slices), and the mouse embry o Stereo-seq dataset (3 slices), as well as synthetic windo wed slices deriv ed from these datasets. The gene cost matrix for synthetic windo wed slices from the same slice is computed from PCA embeddings with 50 PCs using Euclidean distance. F or mouse embry o Stereo-seq data, we adopt the gene cost matrix from SpaT rack [ 22 ], whic h w as constructed using the squared Euclidean distance of PCA embeddings with 50 PCs. F or DLPFC Visium data, w e first filter out genes with total counts fewer than 50 and then identify spatially v ariable genes b y running SOMDE (with 5 no des per dimension) [ 45 ] indep endently on eac h slice and retaining the top 3000 ranked genes. F or MERFISH, we used all measured genes. After gene selection, each slice w as normalized b y total coun ts with a target of 10000 counts, log1p-transformed, and scaled (zero mean and unit v ariance). When aligning a pair of slices, we further filter the genes by keeping genes that are present in b oth preprocessed slices. A joint graph neural net work is then used to generate comparable lo w-dimensional features for the t w o slices to b e aligned for DLPFC Visium datasets and MERFISH dataset. F or eac h slice, a spatial neigh b orho o d graph is initiated as an alpha complex 1-sk eleton [ 46 ], with the radius set to the a verage distance to the sp ots’ k nearest neigh b ors ( k =15). The graph is then densened to increase lo cal connectivity by connecting no des within three hops in the initial graph yielding the final spatial graph G α . F ull definition of alpha complex and graph construction details are in Supplementary Note. Next, w e use a graph con volutional enco der f θ (shared across the t wo slices) to map sp ot features to h -dimensional embeddings. f θ consists of tw o GCN [ 47 ] lay ers f (1) θ , f (2) θ with PReLU activ ations H ( i ) = f ( i ) θ ( H ( i − 1) , G ) = PReLU( ˜ D − 1 2 ˜ A ˜ D − 1 2 H ( i − 1) W ( i − 1) ) , i = 1 , 2 , (4) where H (0) is the input features, ˜ A is the adjacency matrix of the spatial graph G including self connections, and ˜ D is the diagonal degree matrix with ˜ D ii = P j ˜ A ij . Both hidden lay ers are set to 18 100 dimensions and we take Z : = H (2) as the embedding on whic h we compute the cross-slice costs. The GCN is trained using the Deep Graph Infomax (DGI) framework [ 35 ], jointly across the tw o slices. Let X ∈ R n × d b e the d -dimensional inputs of the n no des, DGI constructs a corrupted view of the original graph by p ermuting no de features ˜ X = π ( X ) where π is a random p ermutation on ro ws of a matrix. Let { h i } n i =1 = f θ ( X, G ) and { ˜ h i } n i =1 = f θ ( ˜ X , G ) b e the node embeddings from with the original inputs and permutated inputs, respectively . DGI then constructs a global summary of the original graph s = σ ( 1 n P i h i ) where σ is sigmoid function, and minimizes/maximizes the concordance b et ween no de embeddings of p erturb ed/unp erturb ed graph with the summary s with the follo wing loss function L DGI ( X, G ; θ, W ) = − 1 n X i h log D W ( h i , s ) + log(1 − D W ( ˜ h i , s )) i , (5) where D W ( h , s ) = σ ( h T W s ) is a discriminator with trainable W . The GCN is then trained by minimizing the total losses of slice A and slice B min θ,W L DGI ( X A , G A ; θ , W ) + L DGI ( X B , G B ; θ , W ) . (6) W e use Adam optimizer with 3500 ep o chs and learning rate of 0 . 0002. After training, we obtain the sp ot embeddings Z A = f θ ( X A , G A ) , Z B = f θ ( X B , G B ), and we use the Euclidean distance b etw een the em b eddings as the cross-slice gene-expression cost C . Do wnsampling W e use t wo downsampling strategies, one for Visium data to exploit the near-hexagonal grid and one for general ST data, with both ensuring even cov erage of the slice. F or Visium data with sp ot lo cations X , a coarse hexagonal grid mask with p oin ts X c is first constructed according to a target grid size, typically chosen as an integer m ultiple of the grid spacing in original data. The coarse grid p oin ts are then reduced to a subset X c ∗ ⊂ X c b y k eeping those that are the nearest neighbor of at least one p oint in X . Finally , we c ho ose do wnsampled spots X ∗ ⊂ X by including the p oints in X that are nearest neigh b ors of p oints in X c ∗ . Here, the nearest neigh b ors are only consider across the t wo p oint sets, X and X c . 19 F or other ST data that resembles irregular grids, w e use a max-min do wnsampling approach follo wing [ 36 ], whic h is designed to promote even spacing among the downsampled p oints while retaining extremal p oints to b etter respect the geometry . Specifically , the do wnsampled set is ini- tiated b y randomly selecting a point x 0 ∈ X , forming X (0) ∗ = { x 0 } . Then p oints are added to the collection iterativ ely b y selecting x i = arg max x j ∈ X \ X ( i − 1) ∗ min x k ∈ X ( i − 1) ∗ D ( x j , x k ) ! , (7) where D is the Euclidean distance of the spatial lo cations of spots. The do wnsampled set is then up dated to X ( i ) ∗ = X ( i − 1) ∗ ∪ { x i } . This pro cess is rep eated un til a predefined num b er of do wnsampled p oin ts is reac hed. Reco v ery of full mapping Once, w e obtain the alignmen t matrix b etw een the downsampled slices, we reconstruct the alignmen t b et ween the full slices by utilizing the relativ e lo cation of all spots to do wnsampled spots (anchors) and the correspondence b et ween the anchors. Let P ∈ R N 1 × N 2 + b e the optimal transp ort b et ween the subsampled slices each with N 1 and N 2 subsampled spots. T o a void fuzzy alignment, we first binarize P b y filtering out all zero ro ws and columns, replacing the largest en try of eac h ro w b y 1 and changing all other en tries to 0, resulting in a ˆ N 1 × ˆ N 2 binary matrix ˆ P . W e then compute tw o distance matrices ˆ D 1 ∈ R n 1 × ˆ N 1 + , ˆ D 2 ∈ R n 2 × ˆ N 2 + , describing the spatial distances b etw een the n 1 and n 2 sp ots in full slices to the ˆ N 1 and ˆ N 2 anc hor sp ots in slice A and slice B, respectively . These tw o distance matrices represents the lo cation of each sp ot relative to the anchor sp ots within eac h slice. W e further use ˆ P to find such relative lo cation across slices. This is ac hieved by computing ˆ D 1 → 2 = ˆ D 1 ˆ P row ∈ R n 1 × ˆ N 2 + , ˆ D 2 → 1 = ˆ D 2 ˆ P T col ∈ R n 2 × ˆ N 1 + , (8) where ˆ P row and ˆ P col are ro w and column normalized ˆ P with each ro w or column summing to 1. When the i th ro w of ˆ D 1 → 2 and the j th row of ˆ D 2 are similar, sp ot i in slice A and sp ot j in slice B ha ve similar lo cations under the co ordinate system represen ted b y the anc hor sp ots in slice B and therefore should hav e a small alignment cost. W e therefore construct the full cross-slice matrices 20 using ( C 1 ) ij = X k ∈K 1 i ˆ D 1 ik − ˆ D 2 → 1 j k 2 1 2 , ( C 2 ) ij = X l ∈K 2 j ˆ D 1 → 2 il − ˆ D 2 j l 2 1 2 , (9) Here K 1 i = T opK ˆ D 1 ( i, :) , k 1 , K 2 j = T opK ˆ D 2 ( j, :) , k 2 fo cuses on only lo cal anch ors to av oid the disruption by long-range spatial distances, where T opK( · , k ) returns the indices of the k smallest v alues in a v ector corresp onding the k -nearest-neighbour of a given sp ot. Finally , w e use C t = ( C 1 + C 2 ) / 2 as a cross-slice spatial cost relying on the optimal alignment b etw een the subsampled slices. Com bined with the cross-slice feature cost C (as described in Data prepro cessing and construction of cost matrices), we solv e the follo wing sup ervised optimal transp ort to obtain the alignment betw een full slices P ∗ = arg min P ∈ U ( ≤ a , ≤ b ; C , C t ) ⟨ P , α C + (1 − α ) C t ⟩ F − ϵH ( P ) + γ ( ∥ a − P1 n 2 ∥ 1 + ∥ b − P T 1 n 1 ∥ 1 ) , (10) U ( ≤ a , ≤ b ; C , C t ) = n P ∈ R n 1 × n 2 + P1 n 2 ≤ a , P T 1 n 1 ≤ b , P rt = 0 if C rt > ρ f , P ij = 0 if ( i, j ) ∈ I ρ t o , (11) where I ρ t : = n ( i, j ) |∃ k ∈ K 1 i , | ˆ D 1 ik − ˆ D 2 → 1 j k | > ρ t or ∃ l ∈ K 2 j , | ˆ D 1 → 2 il − ˆ D 2 j l | > ρ t o , α is default to 0 . 9, ρ f is the cross-slice feature cutoff o v er C and ρ t is the cross-slice spatial cutoff o ver C t in this reco very step. See Supplementary Note for details of sup ervised optimal transp ort algorithms. Ev aluation metrics T o ev aluate alignmen t qualit y , w e use an alignmen t accuracy , whic h quan tifies ho w w ell aligned sp ots preserv e cortical lay er identit y across slices. Let P ∈ R n 1 × n 2 denote the alignmen t matrix b etw een slice A and slice B, where P ij represen ts the matching weigh t b etw een spot i in slice A and sp ot j in slice B. F or each sp ot i in slice A, w e define a hard assignment j ∗ ( i ) = arg max j P ij , 21 the alignmen t accuracy is then defined as 1 n 1 X i 1 ℓ A ( i )= ℓ B ( j ∗ ( i )) , (12) where 1 denotes the indicator function and ℓ is the lay er label of the spot. In addition to alignment accuracy , we introduce a neigh b orho o d preserv ation metric, called global Geometric Preserv ation Rate (GPR), to assess the spatial p erformance of the alignmen t matrix P . The in tuition is that if sp ot i in slice A is aligned with sp ot j in slice B, then the k -nearest neighbors of i should map consistently to the k -nearest neighbors of j . This metric provides a wa y to ev aluate whether lo cal geometric structures are preserv ed under the alignment. F ormally , w e first define the set of v alid indices in slice A V = ( i n 2 X j =1 P ij > ϵ mass ) , whic h ensures that only sp ots with sufficient alignment mass are considered. F or each v alid spot i ∈ V , w e identify its matc hed set of sp ots in slice B, V i = { j | P ij > ϵ entry } . The preserv ation rate of sp ot i is then defined as a weigh ted a verage ov er its matches j ∈ V i : preserv ation-rate[ i ] = P j ∈ V i P ij · pair-preserv ation-rate[ ij ] P j ∈ V i P ij , where the pairwise preserv ation rate compares the neighborho o d of i with that of j : pair-preserv ation-rate[ ij ] = 1 | neigh b ors[ i ] | X k ∈ neighbors[ i ] P l ∈ neighbors[ j ] P kl P n 2 l =1 P kl . The neighorhoo ds are defined as sp ots within Euclidean distance ≤ r including the sp ot itself, and neigh b ors of spots in slice A only contains sp ots in V with non-zero mapping. 22 Finally , the global geometric preserv ation rate (GPR) of the alignmen t matrix P with neigh b orho o ds defined with radius r is obtained b y a veraging the preserv ation rates across all v alid sp ots: GPR( P , r ) = 1 | V | X i ∈ V preserv ation-rate[ i ] . (13) This metric captures ho w w ell the alignment preserves lo cal neigh b orho o d structures, providing a complemen tary view to accuracy b y directly quan tifying the geometric consistency of the alignmen t. Cell-cell comm unication analysis In the cell-cell communication (CCC) analysis of DLPF C data, ligand-receptor pairs with known in teractions through secreted ligands are obtained from CellChatDB [ 48 ]. Ligands and receptor genes expressed in less than 1% of sp ots are filtered out. Then CCC analysis is p erformed using COMMOT [ 39 ] with a spatial distance cutoff of 500 µ m. When comparing the spatial patterns of CCC across aligned slices, we only consider ligand-receptor (LR) pairs with more than 10% of sp ots with non- zero receiv ed signal and non-zero sen t signal. Consider slice A with n 1 sp ots and slice B with n 2 sp ots. Given a COMMOT communication matrix W A ∈ R n 1 × n 1 of a LR pair, the receiver field and sender field in A are obtained by R A ( j ) = P i W A ( i, j ) and S A ( i ) = P j W A ( i, j ), resp ectiv ely . The field, the example R A , is mapped to slice B through the RAFT-UP mapping P ∈ R n 1 × n 2 b y R A → B = ¯ P T R A , where ¯ P is the row-normalized P . Data a v ailabilit y The h uman DLPFC data with annotations [ 49 ] is do wnloaded from gith ub https:// b enc hmarkst- repro ducibility .readthedocs.io/en/latest/Data%20av ailability .h tml and zeno do https: //zeno do.org/records/10698880 , and the original data is av ailable at https://researc h.lib d.org/ spatialLIBD/ . The MERFISH data with annotations [ 50 ] is downloaded from zeno do https:// zeno do.org/records/10698909 and the original data is a v ailable at h ttps://datadry ad.org/dataset/ doi:10.5061/dry ad.8t8s248 . The Stereo-seq mouse midbrain at E12.5,E14.5 and E16.5 time points [ 5 ] is downloaded from https://spatrac k.readthedo cs.io/en/latest/noteb o oks/04.ST data of mouse% 20midbrain with m ultiple times.html and the original data is av ailable at https://db.cngb.org/ stomics/datasets/STDS0000058/data . The curated ligand-receptor pairs are taken from CellChatDB [ 48 ] a v ailable at https://gith ub.com/sqjin/CellChat . 23 Co de a v ailabilit y RAFT-UP is av ailable as an op en-source Python pac k age at https://gith ub.com/L1feiyu/raftup rep o . Ac kno wledgemen t This w ork w as supp orted b y NSF grant DMS2142500 (Y.Z.), NSF grant DMS2151934 (Z.C.) and NIH gran t R01GM152494 (Z.C.). Comp eting In terests The authors declare no comp eting interests. References [1] Tian, L., Chen, F. & Macosk o, E. Z. The expanding vistas of spatial transcriptomics. Natur e Biote chnolo gy 41 , 773–782 (2023). [2] Rao, A., Barkley , D., F ran¸ ca, G. S. & Y anai, I. Exploring tissue architecture using spatial transcriptomics. Natur e 596 , 211–220 (2021). [3] St ˚ ahl, P . L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Scienc e 353 , 78–82 (2016). [4] Ro driques, S. G. et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Scienc e 363 , 1463–1467 (2019). [5] Chen, A. et al. Spatiotemp oral transcriptomic atlas of mouse organogenesis using dna nanoball- patterned arra ys. Cel l 185 , 1777–1792 (2022). [6] Shah, S., Lubeck, E., Zhou, W. & Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hipp o campus. Neur on 92 , 342–357 (2016). [7] Co deluppi, S. et al. Spatial organization of the somatosensory cortex rev ealed by osmfish. Natur e metho ds 15 , 932–935 (2018). 24 [8] Xia, C., F an, J., Emanuel, G., Hao, J. & Zhuang, X. Spatial transcriptome profiling b y mer- fish reveals sub cellular rna compartmentalization and cell cycle-dep endent gene expression. Pr o c e e dings of the National A c ademy of Scienc es 116 , 19490–19499 (2019). [9] Chen, C., Kim, H. J. & Y ang, P . Ev aluating spatially v ariable gene detection metho ds for spatial transcriptomics data. Genome Biolo gy 25 , 18 (2024). [10] Armingol, E., Baghdassarian, H. M. & Lewis, N. E. The diversification of methods for studying cell–cell in teractions and communication. Natur e R eviews Genetics 25 , 381–400 (2024). [11] Y uan, Z. et al. Benchmarking spatial clustering metho ds with spatially resolv ed transcriptomics data. Natur e Metho ds 21 , 712–722 (2024). [12] Jones, A., T ownes, F. W., Li, D. & Engelhardt, B. E. Alignment of spatial genomics data using deep gaussian pro cesses. Natur e metho ds 20 , 1379–1387 (2023). [13] Liu, W. et al. Probabilistic embedding, clustering, and alignment for integrating spatial transcriptomics data with precast. Natur e c ommunic ations 14 , 296 (2023). [14] Clifton, K. et al. Stalign: Alignmen t of spatial transcriptomics data using diffeomorphic metric mapping. Natur e c ommunic ations 14 , 8123 (2023). [15] Xu, H. et al. Spacel: deep learning-based characterization of spatial transcriptome arc hitectures. Natur e Communic ations 14 , 7603 (2023). [16] Xu, C. et al. Deepst: iden tifying spatial domains in spatial transcriptomics by deep learning. Nucleic A cids R ese ar ch 50 , e131–e131 (2022). [17] Guo, T. et al. Spiral: integrating and aligning spatially resolved transcriptomics data across differen t exp eriments, conditions, and technologies. Genome Biolo gy 24 , 241 (2023). [18] Zhou, X., Dong, K. & Zhang, S. In tegrating spatial transcriptomics data across different conditions, technologies and developmen tal stages. Natur e Computational Scienc e 3 , 894–906 (2023). [19] Villani, C. et al. Optimal tr ansp ort: old and new V ol. 338 (Springer, 2008). 25 [20] Peyr ´ e, G., Cuturi, M. et al. Computational optimal transport: With applications to data science. F oundations and T r ends ® in Machine L e arning 11 , 355–607 (2019). [21] Schiebinger, G. et al. Optimal-transport analysis of single-cell gene expression iden tifies dev elopmental tra jectories in reprogramming. Cel l 176 , 928–943 (2019). [22] Shen, X. et al. Inferring cell tra jectories of spatial transcriptomics via optimal transp ort analysis. Cel l Systems 16 (2025). [23] Demetci, P ., San torella, R., Sandstede, B., Noble, W. S. & Singh, R. Scot: single-cell m ulti-omics alignmen t with optimal transp ort. Journal of Computational Biolo gy 29 , 3–18 (2022). [24] M´ emoli, F. Gromo v–wasserstein distances and the metric approach to ob ject matc hing. F oundations of c omputational mathematics 11 , 417–487 (2011). [25] Moriel, N. et al. Nov osparc: flexible spatial reconstruction of single-cell gene expression with optimal transport. Natur e pr oto c ols 16 , 4177–4200 (2021). [26] Cang, Z. & Nie, Q. Inferring spatial and signaling relationships betw een cells from single cell transcriptomic data. Natur e Communic ations 11 , 1–13 (2020). [27] Mages, S. et al. T acco unifies annotation transfer and decomp osition of cell identities for single-cell and spatial omics. Natur e biote chnolo gy 41 , 1465–1473 (2023). [28] V ay er, T., Chap el, L., Flamary , R., T av enard, R. & Courty , N. F used gromo v-w asserstein distance for structured ob jects. A lgorithms 13 , 212 (2020). [29] Zeira, R., Land, M., Strzalko wski, A. & Raphael, B. J. Alignment and in tegration of spatial transcriptomics data. Natur e Metho ds 19 , 567–575 (2022). [30] Liu, X., Zeira, R. & Raphael, B. J. P artial alignment of multislice spatially resolved transcriptomics data. Genome R ese ar ch 33 , 1124–1132 (2023). [31] Klein, D. et al. Mapping cells through time and space with moscot. Natur e 638 , 1065–1075 (2025). 26 [32] Cang, Z., Nie, Q. & Zhao, Y. Supervised optimal transp ort. SIAM Journal on Applie d Mathematics 82 , 1851–1877 (2022). [33] Cang, Z., W u, Y. & Zhao, Y. Sup ervised gromo v–w asserstein optimal transp ort with metric- preserving constrain ts. SIAM Journal on Mathematics of Data Scienc e 7 , 301–328 (2025). [34] Cang, Z., Ning, X., Nie, A., Xu, M. & Zhang, J. SCAN-IT: Domain segmen tation of spatial transcriptomics images by graph neural net work. Pr o c e e dings of the British Machine Vision Confer enc e (BMV C) 406 (2021). [35] V eliˇ cko vi´ c, P . et al. Deep graph infomax. International Confer enc e on L e arning R epr esentations (2019). [36] de Silv a, V. & Carlsson, G. E. T op ological estimation using witness complexes. Eur o gr aphics Symp osium on Point-Base d Gr aphics 157–166 (2004). [37] Hu, Y. et al. Benc hmarking clustering, alignment, and in tegration metho ds for spatial transcriptomics. Genome Biolo gy 25 , 212 (2024). [38] Maynard, K. R. et al. T ranscriptome-scale spatial gene expression in the h uman dorsolateral prefron tal cortex. Natur e neur oscienc e 24 , 425–436 (2021). [39] Cang, Z. et al. Screening cell–cell communication in spatial transcriptomics via collectiv e optimal transport. Natur e Metho ds 20 , 218–228 (2023). [40] F urusho, M., Dupree, J. L., Na ve, K.-A. & Bansal, R. Fibroblast gro wth factor receptor sig- naling in oligo dendro cytes regulates m yelin sheath thic kness. Journal of Neur oscienc e 32 , 6631–6641 (2012). [41] Meyer, R. C., Giddens, M. M., Schaefer, S. A. & Hall, R. A. Gpr37 and gpr37l1 are receptors for the neuroprotective and glioprotectiv e factors prosaptide and prosap osin. Pr o c e e dings of the National A c ademy of Scienc es 110 , 9529–9534 (2013). [42] Chauvet, S. et al. Gating of sema3e/plexind1 signaling b y neuropilin-1 switches axonal repulsion to attraction during brain dev elopment. Neur on 56 , 807–822 (2007). 27 [43] Li, L., Hutc hins, B. I. & Kalil, K. Wn t5a induces sim ultaneous cortical axon outgro wth and repulsiv e axon guidance through distinct signaling mechanisms. Journal of Neur oscienc e 29 , 5873–5883 (2009). [44] Rivers, L. E. et al. Pdgfra/ng2 glia generate my elinating oligo dendro cytes and piriform pro jection neurons in adult mice. Natur e neur oscienc e 11 , 1392–1401 (2008). [45] Hao, M., Hua, K. & Zhang, X. Somde: a scalable metho d for identifying spatially v ariable genes with self-organizing map. Bioinformatics 37 , 4392–4398 (2021). [46] Edelsbrunner, H. & M ¨ uc k e, E. P . Three-dimensional alpha shap es. A CM T r ansactions On Gr aphics (TOG) 13 , 43–72 (1994). [47] Kipf, T. N. & W elling, M. Semi-sup ervised classification with graph con v olutional netw orks. arXiv pr eprint arXiv:1609.02907 (2016). [48] Jin, S. et al. Inference and analysis of cell-cell communication using cellchat. Natur e c ommunic ations 12 , 1088 (2021). [49] Pardo, B. et al. spatiallib d: an r/bio conductor pack age to visualize spatially-resolved transcriptomics data. BMC genomics 23 , 434 (2022). [50] Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Scienc e 362 , eaau5324 (2018). 28
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment