Sample-Efficient Adaptation of Drug-Response Models to Patient Tumors under Strong Biological Domain Shift
Predicting drug response in patients from preclinical data remains a major challenge in precision oncology due to the substantial biological gap between in vitro cell lines and patient tumors. Rather than aiming to improve absolute in vitro predictio…
Authors: Camille Jimenez Cortes, Philippe Lal, a
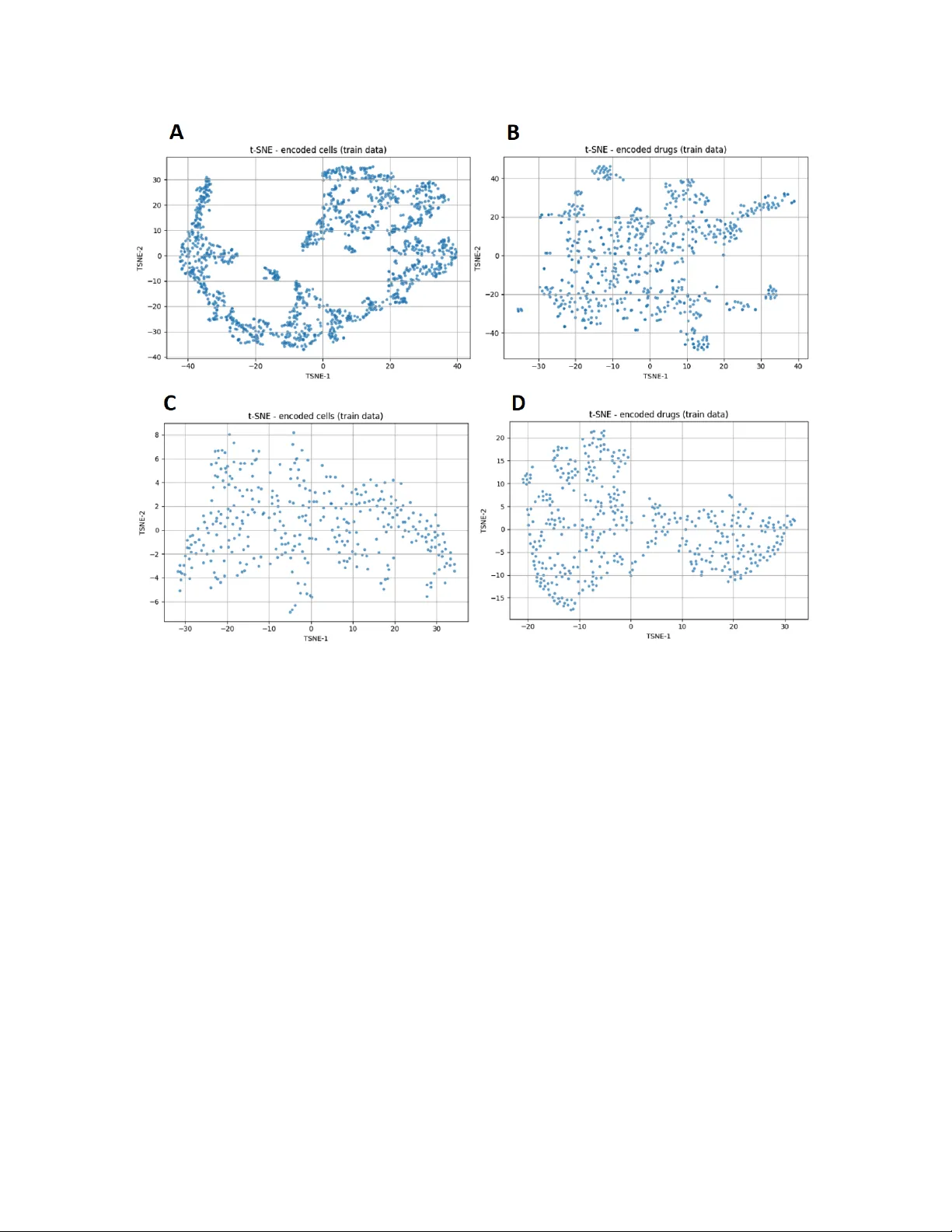
Sample-Ecien t A daptation of Drug-Resp onse Mo dels to P atien t T umors under Strong Biological Domain Shift Camille Jimenez Cortes, Philipp e Lalanda, German V ega Univ ersité Grenoble Alp es (UGA), F rance Abstract Predicting drug resp onse in patien ts from preclinical data remains a ma jor c hallenge in precision oncology due to the substan tial biological gap b et ween in vitro cell lines and patient tumors. Rather than aiming to impro ve absolute in vitro prediction accuracy , this w ork examines whether explicitly separating represen tation learning from task sup ervision enables more sample-ecien t adaptation of drug-resp onse mo dels to patien t data under strong biological domain shift. W e prop ose a staged transfer-learning framew ork in whic h cellular and drug represen tations are rst learned indep enden tly from large collections of unlabeled pharmacogenomic data using auto encoder-based representation learning. These representations are then aligned with drug-resp onse lab els on cell-line data and subsequen tly adapted to patient tumors using few-shot sup ervision. Through a systematic ev aluation spanning in-domain, cross-dataset, and patien t-level settings, w e show that unsup ervised pretraining provides limited b enet when source and target domains ov erlap substan tially , but yields clear gains when adapting to patient tumors with very limited lab eled data. In particular, the prop osed framework achiev es faster p erformance improv emen ts during few-shot patien t-level adaptation while main taining comparable accuracy to single-phase baselines on standard cell-line b enc hmarks. Ov erall, these results demonstrate that learning structured and transferable representations from unlabeled molecular proles can substantially reduce the amount of clinical sup ervision required for eective drug-resp onse prediction, oering a practical pathw ay tow ard data-ecien t preclinical-to-clinical translation. In tro duction Predicting drug resp onse from molecular proles is a cen tral goal of precision oncology , with the promise of guiding therapeutic decisions based on tumor-specic c haracteristics. Large-scale pharmacogenomic screening eorts on cancer cell lines ha v e enabled the developmen t of increasingly accurate machine-learning mo dels by pairing molecular features with measured drug sensitivities 1 . These resources hav e b ecome a cornerstone for computational drug-resp onse prediction, pro viding standardized, data-ric h en vironments for mo del training and benchmarking. Despite this progress, translating mo dels trained on in vitro cell lines to patien t tumors remains a ma jor c hallenge. Cell lines represent simplied biological systems that dier substantially from primary tumors in terms of cellular heterogeneity , micro en vironmental con text, and clinical confounders 2 . As a result, predictiv e mo dels that p erform well on cell-line b enc hmarks often exhibit limited generalization when applied directly to patient data. This preclinical-to-clinical gap contin ues to constrain the practical impact of drug-resp onse prediction (DRP) mo dels in real-world oncology settings. F rom a machine-learning p ersp ectiv e, this limitation can b e framed as a problem of domain shift. Mo dels are t ypically trained in a source domain dened by cell-line molecular proles and screening conditions, while deplo yment occurs in a target domain corresponding to patient tumors. Dierences in data distributions b et w een these domains—arising from biological v ariabilit y , exp erimen tal proto cols, and measurement platforms—violate the assumptions under which standard sup ervised learning guaran tees generalization 3,4 . A ddressing such shifts is therefore essential for reliable patient-lev el prediction. A growing b ody of w ork has explored transfer learning and domain adaptation (DA) strategies for DRP , including adv ersarial alignment, discrepancy minimization, and representation learning approac hes 5,6 . Among these, auto encoder-based metho ds ha ve attracted particular interest due to their abilit y to learn compact latent representations from high-dimensional molecular data 7 . Sev eral models jointly enco de cellular omics and drug features using auto encoders coupled with sup ervised predictors, achieving strong p erformance on cell-line b enc hmarks. F or example, DeepDRA integrates m ulti-omics cell proles with drug descriptors and molecular ngerprints through mo dalit y-sp ecic auto encoders trained end-to-end with a prediction net work 8 . How ev er, such single-phase training strategies tightly couple represen tation learning and task sup ervision and rely almost exclusively on lab eled cell–drug pairs, thereby limiting the exploitation of large collections of unlab eled molecular data. In clinical settings, labeled patien t data are inheren tly scarce, making it unrealistic to expect large sup ervised training sets in the target domain. Consequen tly , the key practical question is not whether a mo del ac hieves state-of-the-art performance when trained and ev aluated on cell lines, but whether it can b e adapted ecien tly to a new biological domain using only a small num b er of lab eled patient samples. F ew-shot learning pro vides a natural framework to address this challenge by enabling rapid adaptation of pretrained mo dels with minimal sup ervision 9 . While few-shot approaches ha ve shown promise for DRP in patient-deriv ed tumor cells and xenograft mo dels, it remains unclear under which conditions representation learning genuinely impro ves adaptation eciency rather than merely enhancing in-domain accuracy . In this work, we inv estigate whether explicitly separating representation learning from task sup ervision can enable sample-ecient adaptation of DRP mo dels to new domains, even when direct in vitro p erformance remains comparable to existing approac hes. Our fo cus is therefore not on maximizing in-domain accuracy on cell-line b enc hmarks, but on minimizing the amount of target-domain sup ervision required to ac hieve reliable adaptation under strong biological domain shift. W e h yp othesize that unsup ervised pretraining on large collections of unlab eled molecular proles can yield structured and transferable representations that accelerate learning in the target domain, while oering limited b enet when source and target distributions o verlap substantially 10 . T o test this hypothesis, we prop ose a staged transfer-learning framew ork based on auto encoder-driven represen tation learning and ev aluate it across a sp ectrum of settings characterized by increasing levels of distribution shift, ranging from in-domain leav e-out proto cols to cross-dataset transfer and patient-lev el few-shot adaptation. Through this analysis, we aim to clarify when representation learning impro ves DRP in practice and to quantify its impact on data eciency under clinically realistic constraints. Con tributions and pap er organization. This work mak es three main contributions. First, w e prop ose a staged transfer-learning framework that explicitly separates unsup ervised representation learning, task-sp ecic alignment, and few-shot clinical adaptation for DRP . Second, we demonstrate through a systematic exp erimen tal analysis that, although unsup ervised pretraining yields limited gains for direct in vitro prediction, it substan tially improv es few-shot adaptation to patient tumors, reducing the n umber of lab eled target samples required for eective transfer. Third, we link these p erformance patterns to laten t-space geometry , providing mec hanistic insight into when and why represen tation learning is b enecial under strong biological domain shift. The remainder of the pap er is organized as follo ws. Section 2 reviews related w ork on DRP and domain shift. Section 3 presents the proposed framew ork. Section 4 describ es the exp erimen tal setup. Section 5 rep orts the results, and Section 6 discusses their implications, limitations, and future directions. Related W ork A central challenge in applying mac hine-learning models to biomedical data is their sensitivity to changes in data distribution. Mo dels are typically trained in controlled, data-rich environmen ts (source domains) but are often deploy ed in settings where data distributions dier substan tially (target domains), such as indep enden t cohorts or patien t p opulations. These discrepancies, commonly referred to as domain shift, arise from biological heterogeneity , exp erimen tal proto cols, and p opulation-sp ecic eects, and can sev erely degrade predictiv e p erformance. F ormally , a domain is dened as a joint distribution D = P ( X, Y ) o ver inputs X and lab els Y 3,4 . Domain shift o ccurs when source and target domains dier, i.e., P ( X S , Y S ) = P ( X T , Y T ) , due to changes in input distributions (cov ariate shift), lab el distributions (lab el shift), or their conditional relationship (concept shift) 11,12 . In biomedical applications, such shifts are particularly pronounced and often unav oidable. DRP exemplies this problem. Models trained on large-scale in vitro pharmacogenomic screens operate in a domain that diers fundamentally from that of patient tumors. While cell-line datasets provide abundant and standardized molecular proles, patient data in tro duce additional sources of v ariabilit y , including tumor micro en vironmen t eects, intra-tumor heterogeneity , and clinical confounders. As a result, models optimized on cell-line data frequently fail to generalize to patient cohorts, motiv ating approaches explicitly designed to cop e with domain shift. T w o metho dological frameworks hav e b een widely studied to address this issue: D A and Domain Generalization (DG). DA fo cuses on transferring knowledge from source domains to a sp ecic target domain that is accessible during training, often under limited or absent target lab els 5,13 . In contrast, DG aims to learn mo dels that generalize to unseen target domains by exploiting v ariabilit y across m ultiple source domains 3,14 . Both frameworks seek representations that capture task-relev ant information while discarding domain-sp ecic v ariabilit y , a goal that is particularly challenging in high-dimensional biomedical settings and naturally motiv ates representation learning approaches. In this w ork, we fo cus on a DA setting, in which the target domain (patien t tumors) is kno wn at training time but lab eled data are extremely limited. As such, our ob jectiv e is not to learn domain-in v ariant representations for unseen targets, as in DG, but to enable sample-ecient adaptation under strong biological domain shift. Imp ortan tly , this setting does not assume explicit distribution alignmen t b et w een source and target domains, but rather emphasizes rapid sp ecialization using limited target-domain sup ervision. Within D A and DG, existing approaches can b e broadly grouped into three families. Statistical alignment metho ds reduce discrepancies b etw een domains by minimizing div ergence measures such as Maximum Mean Discrepancy or co v ariance dierences 15–17 . A dversarial approac hes enforce domain inv ariance through domain discriminators trained in opp osition to feature extractors, as in Domain-A dversarial Neural Netw orks 5 . Reconstruction-based approaches rely on auto enco der arc hitectures to learn latent represen tations that preserve essen tial input structure while attenuating domain-sp ecic noise 18–20 . Among these, auto encoder-based metho ds are particularly attractive for biomedical data due to their ability to handle high dimensionality and heterogeneous mo dalities. A uto encoders hav e b een widely adopted for multi-omics integration and DRP . Early studies show ed that auto encoders can eectively compress molecular proles into informative laten t spaces 21,22 . Subsequen t w ork extended this paradigm by jointly enco ding cellular omics and drug features to mo del cell–drug in teractions, including metho ds such as MOLI, DeepCDR, and DeepDRA 8,23,24 . In particular, DeepDRA emplo ys modality-specic autoenco ders trained join tly with a prediction netw ork in a single-phase supervised setting, learning representations directly optimized for DRP . While these approaches demonstrate strong p erformance on cell-line b enc hmarks, their tightly coupled training strategy limits the exploitation of unlab eled molecular data and may reduce robustness under strong domain shift. Representation learning and task sup ervision are learned simultaneously from lab eled cell–drug pairs, which constrains the diversit y of biological states captured in the latent space and ma y hinder transfer to domains far from the training distribution, such as patient tumors. More recen t studies hav e explored explicit DA strategies for DRP , including adversarial alignment and discrepancy minimization b et ween pharmacogenomic datasets or b et ween preclinical and clinical data 6,25 . Although these metho ds can improv e cross-domain p erformance, they often require access to target-domain data during training and rely on assumptions ab out lab el distributions that may not hold in clinical settings. Despite these adv ances, few works explicitly inv estigate when representation learning is b enecial under domain shift or disen tangle the resp ectiv e roles of representation learning and task supervision. This gap motiv ates training paradigms that separate unsup ervised representation learning from downstream sup ervision, lev erage large collections of unlab eled molecular proles, and enable sample-ecient adaptation to patien t data under strong biological shift. Figure 1. ST aR-DR framework. Cell and drug features are indep enden tly enco ded in to latent represen tations and com bined for drug-resp onse prediction (DRP). T raining includes unsup ervised pretraining on CTRP–GDSC, sup ervised alignment on cell-line resp onse data, and few-shot adaptation to TCGA. Prop osal As discussed in the previous sections, DRP mo dels trained on in vitro cell-line data are exp osed to a strong domain shift when applied to patien t tumors. While large pharmacogenomic screens provide abundan t molecular proles and drug-resp onse measurements, patient-lev el data remain scarce and heterogeneous. This discrepancy limits the direct applicability of fully sup ervised mo dels and motiv ates training strategies that explicitly account for distribution shifts while minimizing clinical sup ervision requirements. T o address this challenge, we prop ose ST aR-DR (Staged T ransfer of Representations for Drug Resp onse) ,a staged transfer learning framework that explicitly separates representation learning, task alignmen t, and clinical adaptation. The core idea is to exploit large collections of unlab eled molecular data to learn structured and transferable representations of cells and drugs, to align these representations with pharmacological signal using cell-line supervision, and nally to adapt the resulting mo del to the patien t domain using limited lab eled clinical data. The prop osed framework is comp osed of three main comp onen ts: (i) a cell enco der that maps high-dimensional molecular proles of cancer cells to a compact laten t representation, (ii) a drug enco der that em b eds molecular descriptors and ngerprints into a separate laten t space, and (iii) a ligh tw eight prediction head that combines these representations to estimate drug sensitivity . Rather than training these comp onen ts jointly in a single sup ervised phase, the mo del follows a three-stage training strategy designed to improv e robustness under strong domain shift. In the rst phase (P1), the cell and drug enco ders are pretrained indep enden tly using auto enco ders in a self-sup ervised manner. This phase relies exclusively on unlab eled molecular data and aims to learn laten t spaces that capture fundamen tal biological and chemical v ariabilit y while remaining agnostic to the do wnstream prediction task. By using reconstruction as the learning signal, the enco ders are encouraged to mo del intrinsic properties of cells and drugs instead of dataset-sp ecic correlations that may not transfer across domains. In the second phase (P2), the pretrained enco ders are jointly ne-tuned with a light w eight classier using lab eled cell–drug resp onse pairs from large-scale pharmacogenomic screens. This step aligns the latent represen tations with pharmacological signal while preserving the structure learned during unsup ervised pretraining. Imp ortan tly , the prediction head is inten tionally kept simple, so that p erformance gains primarily reect the quality and transferability of the learned representations rather than increased model complexit y . In the third phase (P3), the mo del is adapted to the clinical setting using a few-shot learning strategy on patien t data. A small num b er of labeled patien t–drug response pairs are used to specialize the pretrained and task-aligned mo del to the patient domain. Given the strong biological shift b et ween cell lines and tumors, adaptation primarily fo cuses on the cellular representation, while drug representations are largely preserv ed to av oid ov ertting in the presence of limited clinical data. This design enables sample-ecient patient-lev el adaptation without requiring explicit domain-alignment constraints or extensive retraining. An ov erview of the prop osed framework and its three training stages is illustrated in Figure 1. Overall, this staged design enables ST aR-DR to lev erage unlabeled molecular data that w ould otherwise remain un used, mitigate the impact of strong distribution shifts b et w een preclinical and clinical settings, and supp ort robust and sample-ecien t adaptation to patient data. Exp erimen tal setup This section describes the datasets, mo deling choices, and ev aluation proto cols used to assess the prop osed staged transfer-learning framework under increasing levels of distribution shift. Datasets and mo dalities W e consider three datasets reecting progressiv ely stronger domain shifts. T wo large-scale preclinical cell-line resources are used for training and v alidation: CTRP–GDSC 26,27 , which serves as the primary training dataset, and CCLE 28 , used as an indep enden t b enc hmark for cross-dataset ev aluation. Patien t-lev el transfer is assessed on TCGA 29 . CTRP and GDSC pair molecular proles of cancer cell lines with drug sensitivit y measuremen ts, enabling sup ervised learning on cell–drug resp onse pairs. CCLE provides complementary m ulti-omics characterizations of cancer cell lines collected under distinct exp erimental proto cols, making it suitable for ev aluating cross-dataset generalization within the in vitro domain. In contrast, TCGA comprises multi-omics and clinical data from patient tumors and is used to assess adaptation under strong preclinical-to-clinical domain shift. Pro cessed molecular features and binarized drug-resp onse lab els were obtained from the public DeepDRA rep ository . Cellular proles comprise gene expression and somatic m utation data, while drug representations include molecular descriptors and structural ngerprints. Gene expression features are represented as con tinuous v alues p er gene, capturing the transcriptional state of each sample, whereas somatic mutations are enco ded as binary indicators at the gene level (1 for mutated, 0 for wild-type). On the drug side, molecular structure is represen ted using Morgan ngerprints, enco ded as high-dimensional binary v ectors capturing circular substructures, together with con tinuous physicochemical and top ological molecular descriptors. Mo dalit y-sp ecic feature matrices are aligned by in tersecting common entities across datasets and concatenated to form the nal cell and drug input spaces. After prepro cessing and alignment, CTRP–GDSC contains 373 cell lines and 690 drugs (28,833 cell–drug pairs), CCLE contains 470 cell lines and 23 drugs (633 pairs), and TCGA includes 714 patients and 32 drugs (2,485 pairs), with gene expression as the only av ailable molecular modality . T o ensure cross-domain comparabilit y , the feature schema learned on CTRP–GDSC is p ersisted and reused to reindex CCLE and TCGA. Drug-resp onse labels are binarized, with resistant pairs enco ded as class 0 and sensitiv e pairs as class 1. Prepro cessing All features are indep enden tly normalized using p er-feature min–max scaling, and missing v alues are imputed with zeros. During training, an additional normalization step is applied using statistics computed on the training split only and reused for v alidation and testing to preven t data leakage. Class im balance is addressed exclusiv ely at training time using random undersampling. In the CTRP–GDSC dataset, drug–response lab els are moderately im balanced, with 10,057 sensitiv e and 18,776 resistan t cell–drug pairs. After a stratied train–v alidation split, undersampling is applied to the training subset only to balance class prop ortions, while v alidation and test sets retain the original lab el distribution to ensure un biased p erformance ev aluation. Mo del arc hitecture The model consists of tw o mo dalit y-sp ecic autoenco ders—one for cellular features and one for drug features—follo wed by a light weigh t multila y er p erceptron (MLP) classier op erating on the concatenated laten t representations. Latent dimensionalities are set to 700 for cells and 50 for drugs, reecting the higher complexit y of m ulti-omics cellular proles relative to drug features. Autoenco ders are trained using mean squared error reconstruction loss and optimized with Adam (learning rate 10 − 3 , weigh t deca y 10 − 8 ). The prediction head is a one-hidden-lay er MLP with 128 ReLU units, follo wed by a sigmoid output lay er optimized using binary cross-en tropy (BCE) loss. All mo dels are implemented in PyT orch, trained with a batc h size of 64 for 25 ep o c hs, and random seeds are xed to 42 to ensure repro ducibility . T raining proto col T raining follows the staged framework describ ed earlier. In Phase 1, cell and drug auto encoders are pretrained indep enden tly on CTRP–GDSC using unlab eled data to learn transferable representations. In Phase 2, pretrained enco ders are join tly ne-tuned with the MLP classier on lab eled CTRP–GDSC cell–drug pairs to align representations with pharmacological signal. In Phase 3, the resulting mo del is adapted to TCGA using a few-shot learning proto col. Adaptation primarily targets the cellular enco der, while the drug enco der is k ept xed to av oid ov ertting given the limited num b er of comp ounds in TCGA. Baseline mo del F or comparison, we implement a single-phase sup ervised auto encoder–MLP baseline adapted from DeepDRA. This baseline matc hes the proposed framew ork in arc hitecture and training settings but is trained end-to-end on lab eled CTRP–GDSC data without unsup ervised pretraining or patien t-level adaptation. T o ensure a fair comparison, data normalization and class balancing are p erformed strictly within the training split. W e refer to this baseline as AE–MLP . W e delib erately restrict comparisons to a single-phase auto enco der–MLP baseline (AE–MLP) that matches the prop osed framework in architecture, optimization, and training proto col. This design choice isolates the eect of the training strategy—namely the explicit separation b et ween representation learning and task sup ervision—from confounding factors related to mo del capacity or architectural dierences. Comparisons with heterogeneous state-of-the-art mo dels would primarily reect dierences in architecture, mo dalit y co verage, or optimization, rather than the specic contribution of staged represen tation learning to adaptation eciency , which is the fo cus of this study . Ev aluation proto col and metrics Mo del p erformance is ev aluated using b oth pair-lev el and group-aw are proto cols. In addition to standard pair-lev el splits, w e employ Leav e-Cell-Out (LCO) and Leav e-Drug-Out (LDO) settings to assess generalization to unseen cell lines and nov el comp ounds, resp ectively . P erformance is reported using R OC–AUC and PR–A UC, whic h provide threshold-free assessments robust to class im balance. Balanced accuracy is additionally rep orted for in-domain and cross-dataset ev aluations. Results W e ev aluate the prop osed staged framew ork across a range of experimental settings designed to progressively increase the magnitude of distribution shift b et ween training and ev aluation data. Rather than fo cusing solely on absolute in-domain performance, our analysis emphasizes adaptation eciency , i.e., how rapidly p erformance improv es as limited lab eled data b ecome av ailable in the target domain. In-domain robustness under leav e-out proto cols W e rst ev aluate in-domain generalization on CTRP–GDSC using three complementary cross-v alidation proto cols designed to prob e dierent generalization regimes: (i) a standard pair-level split, (ii) LCO, in whic h cell lines are disjoint betw een training and v alidation sets, and (iii) LDO, in which compounds are disjoin t b et ween training and v alidation sets. As shown in Figure 2, ST aR-DR and AE-MLP achiev e similarly high p erformance under the standard pair-lev el split, indicating comparable tting capacity when training and ev aluation distributions are closely aligned. This result conrms that separating representation learning from task sup ervision do es not inheren tly improv e in-domain p erformance when sucient lab eled cell–drug pairs are av ailable. Under the LCO protocol, p erformance remains close to the pair-level baseline for b oth metho ds, indicating that generalization to unseen cell lines within the same pharmacogenomic dataset is relatively straightforw ard. This b eha vior is largely structural: in CTRP–GDSC, drug-resp onse patterns are predominantly driven b y drug-sp ecic eects, which are largely conserved across cell lines. As a result, holding out cell lines preserves the dominan t drug-related signal, leading to limited eective distribution shift despite the absence of sp ecic cellular proles during training. In con trast, p erformance degrades substan tially under LDO for b oth approaches, highligh ting the intrinsic dicult y of extrap olating drug-resp onse predictions to previously unseen compounds. By removing entire drugs from the training set, LDO suppresses the dominan t source of predictive structure, forcing mo dels to extrap olate beyond the observ ed c hemical space. Imp ortantly , no systematic performance gap is observ ed b et w een the staged framework and the single-phase baseline across an y of the three in-domain proto cols, indicating that unsupervised pretraining do es not provide a consisten t adv antage for direct in vitro prediction or for generalization within closely related cell-line domains. T ogether, these results establish a critical reference p oin t for the remainder of the study: the primary con tribution of the prop osed framework do es not lie in improving absolute p erformance on cell-line b enc hmarks, but rather in its ability to supp ort more ecien t adaptation under stronger domain shift, as examined in subsequent cross-dataset and patient-lev el ev aluations. Figure 2. In-domain cross-v alidation p erformance under lea ve-out proto cols. Five-fold cross-v alidation on the CTRP–GDSC dataset using cell-line gene expression and mutation proles combined with drug descriptors and Morgan ngerprints. Bars rep ort mean ± s.d. balanced accuracy (left) and area under the precision–recall curv e (A UPRC, righ t) across three ev aluation settings: standard pair-lev el split (Baseline), Leav e-Drug-Out (LDO), and Leav e-Cell-Out (LCO). Figure 3. Cross-dataset p erformance on CCLE. Mo dels trained on CTRP–GDSC are ev aluated on CCLE. (A–B) ROC (top) and Precision–Recall (b ottom) curves av eraged ov er 10 runs. (A) ST AR-DR. (B) Single-phase baseline (AE-MLP). Shaded regions indicate ± 1 s.d. (C) Balanced accuracy (mean ± s.d.). Figure 4. F ew-shot adaptation to TCGA under strong domai n shift. ROC–A UC (blue) and area under the precision–recall curve (AUPR C, orange) as a function of the num b er of lab eled TCGA patien t samples used for adaptation. (A) ST aR-DR. (B) Single-phase baseline (AE-MLP). Curves rep ort the mean o ver 5 indep enden t runs; shaded bands indicate ± 1 standard deviation. Cross-dataset generalization to CCLE W e next assess cross-dataset generalization by training mo dels on CTRP–GDSC and ev aluating them on CCLE. This setting introduces a mo derate level of distribution shift arising from dierences in cell-line panels, exp erimen tal proto cols, and data prepro cessing pip elines, while remaining conned to the in vitro cell-line domain. As shown in Figure 3, both approac hes achiev e comparable p erformance on ROC-A UC, PR-AUC, and balanced accuracy on CCLE. These quantitativ e results are supp orted by a PCA-based analysis of cellular molecular proles (Figure 5), which sho ws that CTRP–GDSC and CCLE o ccup y closely ov erlapping regions in feature space, indicating a relatively limited shift b et ween the tw o datasets. T ak en together, these ndings suggest that when the source and target domains share substantial biological and statistical ov erlap, unsup ervised representation pretraining provides little adv antage ov er standard sup ervised training. In such settings, tightly coupled representation learning and task sup ervision app ear sucien t to capture the relev an t predictive structure. This observ ation further supp orts the central premise of this study: the b enets of the prop osed staged framework are unlikely to b e revealed by cross-dataset cell-line benchmarks alone, but instead emerge under stronger distribution shifts, as examined in the patien t-level adaptation exp erimen ts that follow. P atien t-level adaptation under strong domain shift W e nally turn to patien t-level transfer on TCGA, which constitutes a substan tially more challenging and clinically relev ant ev aluation setting. As illustrated in Figure 5, TCGA lies far outside the cell-line manifold, reecting pronounced biological dierences b et w een primary tumors and in vitro cancer mo dels in terms of cellular heterogeneity , micro en vironmen tal context, and clinical confounders. Consistent with this strong domain shift, zero-shot transfer from CTRP–GDSC to TCGA yields weak p erformance for b oth metho ds. Figure 5. PCA of cell-line molecular proles across datasets. Centroids are connected b y Mahalanobis distances computed in PCA space (2D, p ooled cov ariance). Distances are 0.597 b et ween CTRP+GDSC and CCLE, 18.821 b et ween CTRP+GDSC and TCGA, and 18.609 b et w een CCLE and TCGA, based on gene expression and mutation proles. W e therefore fo cus on few-shot adaptation, in whic h a limited num b er of labeled TCGA patient–drug pairs are used to ne-tune pretrained models. As shown in Figure 4, predictive p erformance impro v es progressiv ely with increasing target-domain supervision for b oth approaches. How ever, across all few-shot regimes, the staged framew ork consistently achiev es higher ROC–A UC and PR–AUC than the single-phase baseline. Strikingly , with as few as 20 labeled TCGA samples, the staged framew ork already attains substan tially higher p erformance, indicating a faster rate of improv ement as a function of target-domain sup ervision. Imp ortan tly , this adv antage cannot b e attributed to increased model capacity or arc hitectural complexit y , as b oth metho ds share iden tical downstream predictors. Instead, the observ ed gains reect the quality and transferabilit y of the representations learned during unsup ervised pretraining, which enable more ecient sp ecialization to patient-lev el data under strong biological domain shift. Laten t-space analysis and represen tation q ualit y T o gain insight into the observed p erformance dierences, we examine the structure of the learned laten t spaces. As shown in Figure 6, cellular embeddings pro duced by ST aR-DR exhibit a more compact and organized structure than those learned by the AE-MLP . This increased compactness and regularit y reect the broader cov erage of biological v ariability acquired during unsup ervised pretraining on large collections of unlab eled molecular proles. In contrast, dierences b et ween drug embeddings remain comparatively modest. This observ ation is consisten t with the more limited div ersity of av ailable comp ound data and with the similar performance of b oth approaches under the LDO ev aluation proto col. T aken together, these results characterize the b eha vior of the prop osed framew ork across increasing levels of distribution shift. Figure 6. Laten t-space structure of cellular and drug represen tations. t-SNE visualizations of cell and drug embeddings learned by ST AR-DR pretraining phase (A)(B) and the single-phase baseline (AE-MLP) (C)(D). Quan titative analysis is p erformed using the mean k -nearest-neigh b or radius ( k = 10 ) and the co ecien t of v ariation, rev ealing more compact and b etter organized cellular representations after unsup ervised pretraining, while dierences in drug embeddings remain limited. Discussion This study examined the role of unsup ervised representation learning in drug-resp onse prediction across progressiv ely stronger domain shifts, with a sp ecic emphasis on sample-ecient adaptation to patient tumors. Rather than seeking improv emen ts in absolute in vitro accuracy , we adopted adaptation eciency as the primary ev aluation criterion, reecting the constraints of clinical settings where lab eled patient data are inheren tly limited. Limits of in-domain and cross-dataset b enchmarks. Across b oth in-domain ev aluations on CTRP–GDSC and cross-dataset generalization to CCLE, ST aR-DR did not exhibit systematic adv an tages o ver AE-MLP . These ndings indicate that when source and target domains share substan tial biological and statistical ov erlap, tightly coupled representation learning and task sup ervision are sucient to capture the relev an t predictive structure. In such regimes, unsup ervised pretraining oers limited additional b enet, suggesting that improv ements observed in cell-line b enc hmarks alone should not b e interpreted as evidence of impro ved clinical transferability . A daptation under strong biological domain shift. P atient-lev el transfer to TCGA represents a fundamen tally dierent setting characterized b y pronounced biological domain shift. Primary tumors dier from in vitro mo dels along m ultiple dimensions, including cellular heterogeneit y , micro en vironmen tal inuences, and clinical confounders, whic h join tly limit zero-shot generalization from preclinical data. In this regime, ST aR-DR exhibits more ecient adaptation when limited lab eled patien t data become av ailable, highligh ting the importance of explicitly separating representation learning from task-sp ecic sup ervision under clinically realistic conditions. Mec hanistic in terpretation. Analysis of the learned latent spaces provides insight into why such dierences emerge under strong domain shift. As illustrated in Figure 6, unsupervised pretraining yields more compact and structured cellular representations, indicating broader and more regular cov erage of biological v ariability in molecular feature space. These prop erties facilitate rapid sp ecialization during few-shot adaptation, oering a mechanistic explanation for the improv ed adaptation eciency observed at the patient level. In contrast, drug representations exhibit comparatively minor dierences b et ween training strategies, consistent with the limited div ersity of a v ailable comp ound data and the similar b eha vior observ ed under LDO ev aluation. Implications for mo del ev aluation. T ogether, these ndings suggest that the practical v alue of represen tation learning in DRP cannot b e reliably assessed through in-domain or cross-dataset cell-line b enc hmarks alone. Ev aluation strategies fo cused exclusively on absolute in vitro p erformance risk underestimating the utility of mo dels designed for clinical translation. By contrast, assessing p erformance through adaptation eciency under strong biological domain shift pro vides a more informativ e and clinically meaningful p erspective, directly aligned with real-world deploymen t constrain ts. Accordingly , our comparison against a carefully matched single-phase baseline is sucient to assess the impact of staged represen tation learning on adaptation eciency , indep enden t of architectural complexity or task-sp ecic design choices, and directly aligned with the clinically motiv ated ob jectiv e of data-ecient patient-lev el adaptation. Limitations and outlo ok. Sev eral limitations remain. The biological gap b etw een cell lines and patient tumors contin ues to constrain zero-shot transfer, indicating that representation learning alone is insucient to fully bridge the preclinical-to-clinical divide. Moreov er, gains on the drug side remain mo dest, underscoring the need for richer chemical representations and more diverse comp ound datasets. F uture work may b enet from integrating additional molecular mo dalities, incorp orating ligh tw eight domain-alignment mechanisms, or combining representation learning with causal or mechanistic mo deling approac hes to further improv e robustness under clinical domain shift. T ak en together, these ndings indicate that the primary v alue of unsup ervised representation learning for DRP lies not in improving in vitro b enchmark p erformance, but in enabling data-ecient adaptation to patien t tumors under strong biological domain shift, thereby substantially reducing the amoun t of clinical sup ervision required for eective preclinical-to-clinical translation. Conclusion This w ork shows that the primary v alue of unsup ervised representation learning in drug-resp onse prediction lies not in improving direct in vitro accuracy , but in enabling sample-ecient adaptation to new biological domains. When training and ev aluation domains ov erlap substantially , as in cell-line b enc hmarks and closely related pharmacogenomic screens, single-phase sup ervised mo dels remain sucient and unsup ervised pretraining provides limited additional b enet. In contrast, under strong biological domain shift b et ween in vitro mo dels and patient tumors, explicitly separating represen tation learning from task sup ervision yields more transferable representations and supp orts eective adaptation with very few lab eled patien t samples. By leveraging large collections of unlab eled cellular proles, ST aR-DR learns ric her and more structured cellular represen tations that b etter capture the biological v ariabilit y encountered in patient tumors. This impro ved organization facilitates faster adaptation in few-shot regimes, even when zero-shot transfer and in vitro p erformance remain comparable to existing approaches. Imp ortantly , these gains are ac hieved without increasing model complexit y , underscoring the cen tral role of data eciency rather than architectural sophistication. A t the same time, imp ortan t challenges remain. Impro vemen ts on the drug side are mo dest, reecting the limited div ersity of av ailable comp ound data, and the biological gap b et ween cell lines and patient tumors con tinues to constrain zero-shot generalization. These observ ations indicate that representation learning alone cannot fully bridge the preclinical-to-clinical divide and must b e complemented by ric her chemical information and additional biological context. Ov erall, this study argues that ev aluating drug-resp onse mo dels through the lens of adaptation eciency pro vides a more clinically meaningful p erspective than fo cusing solely on in-domain accuracy . By shifting emphasis from b enc hmark p erformance to data-ecien t clinical adaptation, staged representation learning oers a practical step tow ard more realistic and clinically actionable preclinical-to-clinical translation. References 1. Cai Z, Poulos R C, Liu J, Zhong Q. Machine learning for m ulti-omics data in tegration in cancer. iScience. 2022;25(2):103798. 2. El Naqa I, Krishnaswam y S, Haib e-Kains Bea. A systematic assessment of deep learning metho ds for drug response prediction: from in vitro to clinical applications. Briengs in Bioinformatics. 2023;24(1):bbac517. 3. Blanchard G, Lee G, Scott C. Generalizing from several related classication tasks to a new unlab eled sample. In: Adv ances in Neural Information Processing Systems. vol. 24; 2011. p. 2178-86. 4. Blanchard G, Deshmukh A, Dogan U, Lee G, Scott C. Domain generalization by marginal transfer learning. Journal of Machine Learning Research. 2021;22(2):1-55. 5. Ganin Y, Lempitsky V. Unsup ervised domain adaptation by backpropagation. In: Pro ceedings of the 32nd International Conference on Machine Learning; 2015. p. 1180-9. 6. Jiang P , Lu Y, Zhou Qea. T ransCDR: a deep learning mo del for enhancing the generalizability of cancer drug resp onse prediction through transfer learning and m ultimo dal data fusion for drug represen tation. Briengs in Bioinformatics. 2023;24(4):bbad156. 7. Partin A, Patel S, T ennakoon CHea. Deep learning metho ds for drug resp onse prediction in cancer: Predominan t and emerging trends. F rontiers in Medicine. 2023;10:1086097. 8. Mohammadzadeh-V ardin T, Sey A, Razzaghi Aea. DeepDRA: Drug repurp osing using multi-omics data integration with auto encoders. PLoS One. 2024;19(5):e0307649. 9. Ma W, T udor R, Luo Y ea. F ew-shot learning creates predictive mo dels of drug resp onse that translate from high-throughput screens to individual patients. Nature Biomedical Engineering. 2021;5(6):555-67. 10. Theo doris CV, Stein MB, W ang Y ea. T ransfer learning enables predictions in netw ork biology . Nature. 2023;614(7948):375-82. 11. Ben-David S, Blitzer J, Crammer K, Kulesza A, Pereira F, V aughan J. A theory of learning from dieren t domains. Machine Learning. 2010;79(1–2):151-75. 12. Zhao H, des Combes R T, Zhang K, Gordon GJ. On learning in v ariant representations for domain adaptation. In: Proceedings of the 36th In ternational Conference on Mac hine Learning; 2019. p. 7523-32. 13. Ben-David S, Blitzer J, Crammer K, Pereira F. Analysis of representations for domain adaptation. In: A dv ances in Neural Information Pro cessing Systems. vol. 19; 2006. p. 137-44. 14. Muandet K, Balduzzi D, Schölk opf B. Domain generalization via inv ariant feature representation. In: Pro ceedings of the 30th International Conference on Machine Learning. vol. 28; 2013. p. 10-8. 15. T zeng E, Homan J, Zhang N, Saenko K, Darrell T. Deep domain confusion: Maximizing for domain in v ariance. arXiv preprint arXiv:14123474. 2014. 16. Long M, W ang J, Ding G, Sun J, Y u PS. T ransfer joint matching for unsup ervised domain adaptation. In: Pro ceedings of the IEEE Conference on Computer Vision and Pattern Recognition; 2014. p. 1410-7. 17. Sun B, F eng J, Saenko K. Return of frustratingly easy domain adaptation. In: Pro ceedings of the AAAI Conference on Articial Intelligence. vol. 30; 2016. p. 2058-65. 18. Bousmalis K, T rigeorgis G, Silb erman N, Krishnan D, Erhan D. Domain separation netw orks. In: A dv ances in Neural Information Pro cessing Systems. vol. 29; 2016. p. 343-51. 19. Ghifary M, Kleijn WB, Zhang M, Balduzzi D, Li W. Deep reconstruction-classication net works for unsup ervised domain adaptation. In: Europ ean Conference on Computer Vision; 2016. p. 597-613. 20. Zhuang F, Cheng X, Luo P , P an SJ, He Q. Sup ervised represen tation learning with double enco ding-la y er auto enco der for transfer learning. A CM T ransactions on Intelligen t Systems and T echnology . 2017;9(2):1-17. 21. T an J, Ung M, Cheng C, Greene C. Unsupervised feature construction and knowledge extraction from genome-wide assays of breast cancer with denoising auto encoders. In: Pacic Symp osium on Bio computing; 2015. p. 132-43. 22. Chaudhary K, Poirion O, Lu L, Garmire LX. Deep learning–based multi-omics integration robustly predicts surviv al in liver cancer. Clinical Cancer Research. 2018;24(6):1248-59. 23. Shari-Noghabi H, Zolotarev a O, Collins CC, Ester M. MOLI: m ulti-omics late in tegration with deep neural netw orks for drug resp onse prediction. Bioinformatics. 2019;35(14):i501-9. 24. Zhang F, et al. DeepCDR: a h ybrid graph conv olutional net work for predicting cancer drug response. Bioinformatics. 2021. 25. He D, et al. A con text-aw are deconfounding auto encoder for robust prediction of p ersonalized clinical drug resp onse. Nature Mac hine Intelligence. 2022;4(10):879-92. 26. Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivit y and basal gene expression reveals mechanism of action. Nature Chemical Biology . 2016;12(2):109-16. 27. Y ang W, Soares J, Greninger P , Edelman EJ, Lightfoot H, F orb es S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therap eutic biomark er disco v ery in cancer cells. Nucleic A cids Research. 2013;41(Database issue):D955-61. 28. Barretina J, Cap onigro G, Stransky N, V enkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclop edia enables predictive mo deling of anticancer drug sensitivity . Nature. 2012;483(7391):603-7. 29. Liu J, Lich ten b erg T, Hoadley KA ea. An In tegrated TCGA Pan-Cancer Clinical Data Resource to Driv e High-Qualit y Surviv al Outcome Analytics. Cell. 2018;173(2):400-16.e11.
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment