A Kolmogorov-Arnold Surrogate Model for Chemical Equilibria: Application to Solid Solutions
The computational cost of geochemical solvers is a challenging matter. For reactive transport simulations, where chemical calculations are performed up to billions of times, it is crucial to reduce the total computational time. Existing publications …
Authors: Leonardo Boledi, Dirk Bosbach, Jenna Poonoosamy
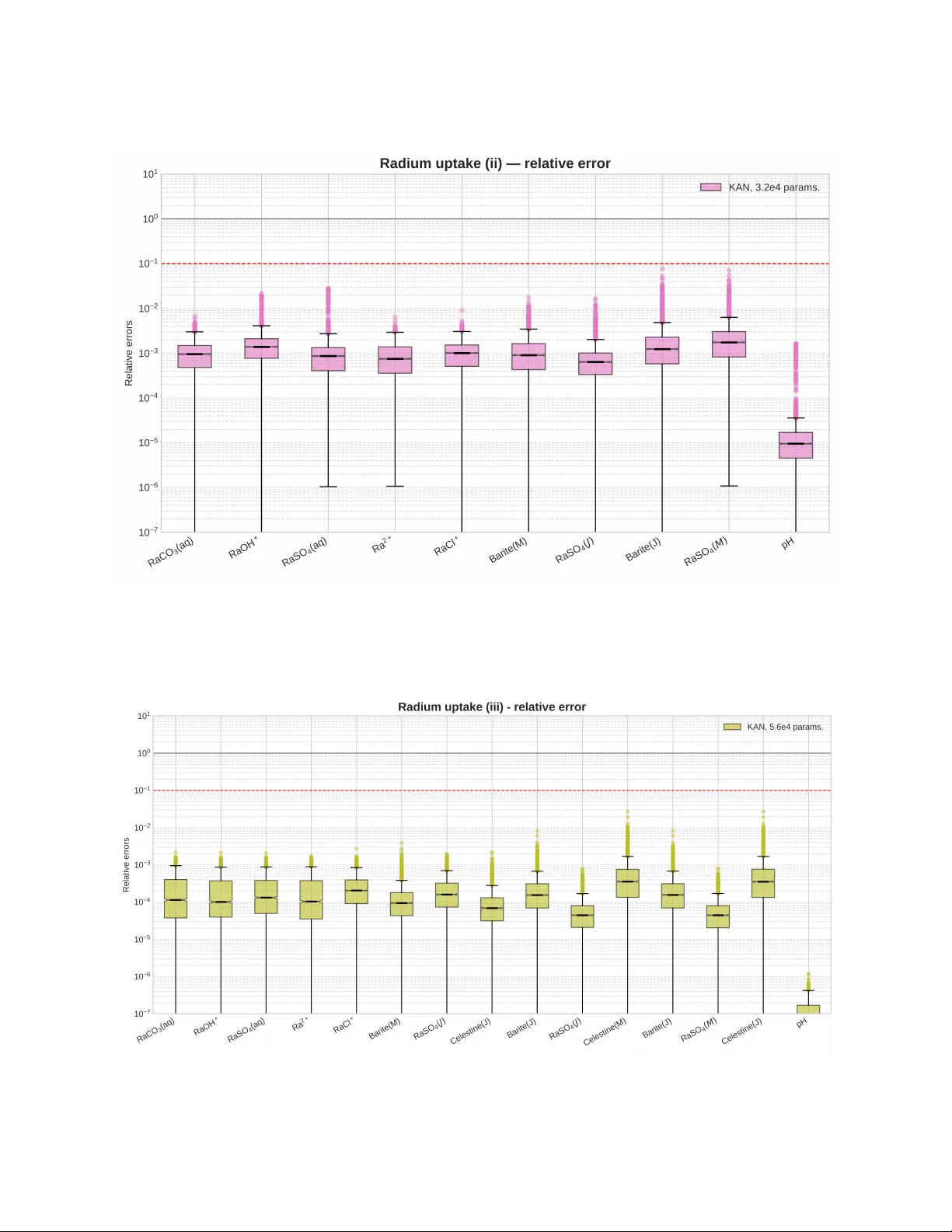
A K O L M O G O R OV - A R N O L D S U R R O G A T E M O D E L F O R C H E M I C A L E Q U I L I B R I A : A P P L I C A T I O N T O S O L I D S O L U T I O N S Leonardo Boledi 1,* , Dirk Bosbach 1 , Jenna P oonoosamy 1 1 Institute of Fusion Energy and Nuclear W aste Management (IFN-2), Forschungszentrum Jülich GmbH * Corresponding author: l.boledi@fz-juelich.de A B S T R AC T The computational cost of geochemical solvers is a challenging matter . For reactiv e transport simula- tions, where chemical calculations are performed up to billions of times, it is crucial to reduce the total computational time. Existing publications hav e explored v arious machine-learning approaches to determine the most effecti ve data-dri ven surrogate model. In particular , multilayer perceptrons are widely employed due to their ability to recognize nonlinear relationships. In this work, we focus on the recent K olmogorov-Arnold networks, where learnable spline-based functions replace classical fixed acti v ation functions. This architecture has achiev ed higher accuracy with fe wer trainable param- eters and has become increasingly popular for solving partial differential equations. First, we train a surrogate model based on an e xisting cement system benchmark. Then, we move to an application case for the geological disposal of nuclear waste, i.e., the determination of radionuclide-bearing solids solubilities. T o the best of our knowledge, this work is the first to in vestigate co-precipitation with radionuclide incorporation using data-driv en surrogate models, considering increasing levels of thermodynamic complexity from simple mechanical mixtures to non-ideal solid solutions of binary (Ba,Ra)SO 4 and ternary (Sr ,Ba,Ra)SO 4 systems. On the cement benchmark, we demonstrate that the K olmogorov-Arnold architecture outperforms multilayer perceptrons in both absolute and relati ve error metrics, reducing them by 62% and 59%, respectiv ely . On the binary and ternary radium solid solution models, K olmogorov-Arnold networks maintain median prediction errors near 1 × 10 − 3 . This is the first step to ward emplo ying surrogate models to speed up reacti ve transport simulations and optimize the safety assessment of deep geological waste repositories. 1 Introduction Computational methods have become a fundamental research direction in the geoscience domain. By simulating the interaction between coupled physical, chemical, and biological processes, reactive transport modelling (R TM) enables scientists to address critical challenges across di verse fields [ 1 , 2 ]. In subsurface ener gy applications, reactiv e transport models are employed to study CO 2 sequestration, geothermal reservoirs, and enhanced oil and gas recov ery [ 3 , 4 , 5 , 6 ]. Additional applications include the analysis of subsurface contaminant migration and electrokinetic desalination [7, 8, 9]. For deep geological repositories of radioacti ve w aste, R TM resolves coupled, nonlinear interactions between transport and geochemical reactions at material interfaces and in host rock, controlling the long-term e volution of pH, porosity , corrosion products, and radionuclide migration under experimentally inaccessible repository conditions [ 10 , 11 ]. These models rely on numerous parameters and competing reaction pathways. Consequently , global sensiti vity analysis is required to identify the processes and parameters that dominate safety-relev ant outcomes, thereby providing a quantitative basis for uncertainty propagation and for focusing safety and performance assessment on the most influential mechanisms [ 12 ]. Comprehensi ve global sensitivity analyses within the safety assessment framework increasingly require the e xecution of lar ge numbers of reacti ve transport simulations. This can only be achie ved through a combination of physics-based models and data-dri ven surrogates [13]. Despite recent advancements, computational cost remains a major challenge in R TM, as the coupling of chemical and flo w solvers requires solving chemical equilibrium calculations millions to billions of times. Namely , chemical L. Boledi et al. Pr eprint submitted to ArXiv equilibrium and/or kinetic calculations in volv e iterati ve methods that are called for each mesh cell and at ev ery time step [ 14 ]. Thus, the chemical solver is the most expensi ve component of the simulation, and it can require up to 10,000x the computational cost of the fluid counterpart [ 15 ]. Consequently , significant research efforts ha ve focused on improving the speed of reacti ve transport simulations in recent years. De Lucia et al. [ 16 ], for instance, de veloped a distributed hash table approach to cache the results of the geochemical solver , and integrated it in the POET simulator [ 17 ]. Another notable example is found in the solv er Reaktoro , which employs on-demand machine learning (ML) to extrapolate new chemical states [ 15 ]. W ithin this framew ork, a speed-up of one to three orders of magnitude has been achiev ed on flows in heterogeneous porous media [18]. A different research direction consists in le veraging data-dri ven methods to fully replace the geochemical solver with a surrogate model. This enables faster prediction of geochemical states without requiring modifications to the underlying solver . Laloy and Jaques de veloped surrogates based on Gaussian processes, polynomial chaos e xpansion, and neural networks (NNs) to predict the kinetic sorption of uranium and the leaching of cement matrices [ 19 , 20 ]. Similarly , Demirer et al. simulated the hydrothermal dolomitization of a fractured carbonate reserv oir via an artificial NN [ 21 ], and multiple ML algorithms hav e been tested on a time-dependent cation exchange problem [ 22 ]. More comprehensiv ely , Prasianakis et al. benchmarked decision trees, Gaussian processes, and NNs against each other for cement hydration and degradation, as well as uranium sorption in claystone systems [ 23 ]. In most of the aforementioned publications, deep NNs exhibit the lowest prediction error , making them a strong candidate for geochemical surrogate models. Given the rapid advancement of deep learning, in vestigating its latest innov ations and their applicability to computational geochemistry is warranted. Among these, K olmogorov-Arnold networks (KANs) offer a promising approach for complex geochemical systems. KANs were first introduced in 2024 [ 24 ], and ha ve since been employed across multiple fields, including fluid dynamics [ 25 ], time series analysis [ 26 ], and molecular property prediction [ 27 ]. By replacing classical fixed activ ation functions with spline-based trainable functions, Liu et al. achie ved a higher prediction accuracy compared to multilayer perceptrons (MLPs) [ 24 ]. Due to their fle xibility , KANs hav e also been adapted to dif ferent NN types, such as con volutional and graph-based architectures [ 28 , 29 ]. Despite reports of higher computational times and occasional training instabilities, [ 30 , 31 ], KANs have demonstrated high effecti veness in solving partial differential equations when combined with physics-informed NNs [ 32 , 33 ]. Their ability to capture complex non-linear relationships with fe w trainable parameters makes them a strong candidate for chemical equilibrium calculations. In this contribution, we in vestigate the applicability of KANs as surrogate models for chemical reactions. First, we consider the hydration and e volution of the cementitious system published in [ 23 ], and compare the performance of KANs against classical MLPs. W e then extend the analysis to sulfate precipitation with radium incorporation, addressing increasing le vels of thermodynamic comple xity from simple mechanical mixtures to non-ideal solid solutions (SSs), including the binary (Ba,Ra)SO 4 and ternary (Sr ,Ba,Ra)SO 4 systems. In particular , we show the accurac y of the KAN surrogate and its computational speedup with respect to the reference solver GEM-Selektor [34, 35]. This paper is structured as follo ws: Section 2 introduces the chemical systems and the data-generation process with GEM-Selektor . The computational methods to create the surrogate model, including the MLP and the KAN, are discussed in Section 3. The performance of the KAN surrogate is analyzed for the cement dissolution benchmark in Section 4. Then, we show three cases of radium sulfate precipitation up to the ternary non-ideal SS model. In Section 5, we summarize our findings and discuss potential future directions. 2 Chemical Equilibrium Model and Solver W e now discuss the application case for our surrogate model and the solver employed to generate the training data. Our study in vestigates the geochemical modelling of radium (Ra) incorporation into sulfate SSs. 226 Ra is a safety-relev ant radionuclide for the disposal of spent nuclear fuel and will dominate the main radioactiv e dose contributor after about 100,000 years [ 36 , 37 ]. Its retention can be achiev ed through co-precipitation into sulfate. The Ra–sulfate system was selected for our in vestigation because its thermodynamic properties and solid-solution mixing beha vior are well constrained by a substantial body of experimental and numerical work spanning a wide range of temperatures and ionic strengths [ 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 ]. In particular , we e valuate the aqueous Ra concentrations predicted from solid-solution thermodynamics by systematically increasing the complexity of the mixing model. This is achiev ed by considering three conceptual representations of Ra incorporation, ranging from (i) idealized mechanical mixtures to (ii) non-ideal binary and (iii) ternary SSs. 2 L. Boledi et al. Pr eprint submitted to ArXiv 2.1 Solid Solution Models In case study (i), Ra incorporation is described using a hypothetical SS treated as a mechanical mixture of pure sulfate end members. In this representation, the Gibbs energy of the solid phase ∆ G M M is defined as the mole-fraction-weighted sum of the standard Gibbs energies of formation of the end members, namely ∆ G M M = n X i =1 X i G ◦ i . (1) Here, X i is the mole fraction of the i -th end member ( i = BaSO 4 or RaSO 4 ). This formulation represents the limiting case of non-interacting solids and is thermodynamically equiv alent to the coexistence of pure phases. In case study (ii), the real mixing behavior of BaSO 4 and RaSO 4 is considered and described as a non-ideal regular SS, following [47]. The total Gibbs energy of the SS is e xpressed as ∆ G real total = ∆ G M M + ∆ G id mix + ∆ G ex , (2) that is the sum of the mechanical mixture term, the ideal Gibbs energy of mixing, and the excess Gibbs energy of mixing. The ideal Gibbs energy of mixing is gi ven by ∆ G id mix = R T n X i =1 X i ln X i , (3) where R is the gas constant and T denotes the temperature. Non-ideal interactions are accounted for through the excess Gibbs energy term, which is e xpressed using the Margules interaction parameters ∆ G ex = n X i = j X i X j w ij . (4) The term w ij denotes the binary interaction parameter between the end members i and j . The acti vity coefficients of the end members are calculated using the Thomson–W aldbaum model assuming a regular solution behavior , with a Margules interaction parameter w RaBa = 2470 J mol − 1 [47]. In case study (iii), the SS model is extended to the ternary RaSO 4 BaSO 4 SrSO 4 system. Follo wing [ 47 ], ternary mixing is described using a regular solution model with pairwise binary interaction parameters w RaBa = 2470 J mol − 1 , w SrRa = 1750 J mol − 1 , and w SrBa = 750 J mol − 1 . Equations (1) and (3) are reformulated to include the third end member , while the excess Gibbs ener gy term accounts for all binary interactions among the three sulfate components. Solid-solution-acqueous-solution equilibria are modeled using the GEM-Selektor software [ 35 ], which incorporates the TSolMod library for the mixing in SSs [34] and the PSI–Nagra 12/07 chemical thermodynamic database [48, 49]. 2.2 Solver Setup In GEM-Selektor , phase equilibria are determined by direct minimization of the total Gibbs energy of the system, which is defined by its bulk elemental composition, temperature, pressure, standard molar Gibbs ener gies of all chemical species, and mixing parameters for solution phases. Thermodynamic data for solid sulfates are taken from T able 2 in [ 47 ], while data for aqueous species are obtained from the PSI–Nagra database [ 49 ]. This database incorporates temperature and pressure dependencies derived primarily from the Helgeson–Kirkham–Flowers equation of state for aqueous ions and complexes, as well as for the water solvent, as implemented in the SUPCR T98 database [ 50 ]. Mixing in the ternary SS is described using the built-in ternary regular solution model of [ 34 ]. For aqueous species, an electrolyte ion-association model consistent with the SUPCR T92 database is applied together with the built-in e xtended Debye–Hückel model [51]. Model BaSO 4 (µ mol ) NaCl ( mmol ) RaBr 2 (µ mol ) SrSO 4 ( mmol ) T ( ◦ C) (i) 50 – 500 50 – 500 50 – 500 - 25.0 (ii) 50 – 500 50 50 – 500 - 20.0 – 90.0 (iii) 50 – 500 50 – 500 50 – 500 5 – 50 25.0 T able 1: Input v ariables for the GEM-Selektor simulations. Note that we consider different temperature v alues only in case (ii). The input configuration for the geochemical solver consists of 1 kg of water and 1 mol of atmospheric air , referred to as "AtmAirNit" in GEM-Selektor . Calculations are performed at P = 1 bar . T able 1 shows the range of the remaining 3 L. Boledi et al. Pr eprint submitted to ArXiv Figure 1: Sketch of an MLP (left) and a KAN (right) with two hidden layers. In KANs, instead of training for weights and biases, learnable activ ation functions are placed on each edge. variables for each SS model. The selected values are inspired by the radium incorporation simulation in [ 52 ]. Over this range, we select points via Sobol sampling to generate the datasets for the upcoming surrogate models [ 53 ]. The thermodynamic dataset for the (Ba,Sr,Ra)SO 4 H 2 O system, including the temperature dependence of RaSO 4 solubility , is taken from [46]. 3 Surrogate Models In this section, we introduce the ML models used to accelerate the chemical equilibrium solv er . Specifically , we employ NNs to predict the outcome of the chemical reaction, i.e., output concentrations and pH, based on the system’ s initial conditions. W e emphasise that this approach generates a mapping between input and output data without requiring knowledge of the underlying physics, which we refer to as a surrogate model [ 54 ]. This strategy is dif ferent from, e.g., the aforementioned approach from Leal et al. [ 15 ], which le verages pre vious reaction outcomes to speed up the upcoming computations. Once the surrogate model is built, it is meant to fully replace the original solver . W e now describe the two main algorithms of this study , MLPs and KANs. 3.1 Multilayer P erceptr ons Let us consider a vector of observations x ∈ X ⊆ R p + , with p ∈ N , which represents the initial state of a chemical system. Let y ∈ Y ⊆ R q + , with q ∈ N , be the vector of outputs, such as the equilibrium concentration and speciation. An NN is a mathematical function F θ : X → Y depending on a set of parameters θ ∈ Θ that associates to any observation point x the output point y . It comprises computational units, the so-called neurons, which take the form h ( x ) = f ( w ⊤ x + b ) , (5) where f is the real-valued acti vation function, w is the vector of weights, and b represents the bias associated with the neuron. During the training process, the weights and biases associated with each neuron are selected to minimize the prediction error . An MLP is a feedforward fully-connected NN consisting of at least one hidden layer [ 23 ]. That is, an NN without directed loops or cycles across neurons and where every neuron of a given layer is connected to all the neurons of the pre vious layer , see Figure 1. Mathematically , the extension of Equation (5) to an MLP of n hidden layers can be expressed as a nested composition of af fine transformations and acti vation functions, which tak es the form y = F θ ( x ) = f n +1 W ( n +1) f n W ( n ) f n − 1 W ( n − 1) . . . f 2 W (2) f 1 W (1) x + b (1) + b (2) + . . . b ( n − 1) + b ( n ) + b ( n +1) . (6) Here, f i represent the vector of real v alued functions from R c h to R c h for i = 1 , . . . , n + 1 , W ( i ) is the weight matrix, and c h denotes the number of neurons in the h -th hidden layer . A more detailed analysis of the MLP architecture is beyond the scope of this work and can be found, e.g., in [55, 56]. 4 L. Boledi et al. Pr eprint submitted to ArXiv The network is implemented and trained in the Pytorch Lightning framew ork, which allows straightforward extension to GPU acceleration [ 57 ]. For the selection of hyperparameters such as the number of layers, neurons, and the batch size, we employ the Bayesian optimiser Optuna [58]. 3.2 Kolmogor ov-Arnold Networks The main idea behind the KAN lies in the Kolmogoro v-Arnold representation theorem, which states that every multiv ariate continuous function can be written as a finite composition of continuous functions of a single variable and the binary operation of addition [24]. Based on this, Liu et al. proposed the alternativ e neuron formulation ϕ ( x ) = w f f ( x ) + w s X j c j B j,d ( x ) , (7) where x represents a scalar input to simplify the notation, and w f and w s are learnable scaling parameters. The first term denotes a classical fix ed acti vation function (commonly the SiLU acti vation), while the second term is a learnable spline component. Specifically , B j,d ( x ) denotes the j -th B-spline basis function of degree d [ 59 ], and c j are the learnable spline coefficients optimised during training to minimise prediction error . The extension of Equation (7) to a KAN of n hidden layers can be expressed as the composition f ( x ) = ( Φ n +1 ◦ Φ n ◦ · · · ◦ Φ 2 ◦ Φ 1 )( x ) , (8) where Φ i denotes the i -th KAN layer . Here, Φ i = { ϕ i,p,q } represents the matrix of 1D activ ation functions connecting input neuron p to output neuron q in layer i , as defined in [ 24 ]. For further details, the reader is referred to the original publication. A ke y distinction from MLPs is that KANs place learnable acti vation functions on the edges between neurons rather than using fixed activ ations at the nodes, see Figure 1 for a visual comparison. The degree d and number of control points for these spline-based acti vations can be fle xibly chosen by the user . This flexibility enables KANs to achie ve lower prediction errors compared to MLPs with fe wer trainable parameters [24]. For the implementation and training, we lev erage the efficient-kan package [ 60 ], which offers a KAN model compatible with Pytorch Lightning . Hyperparameters, including the number of layers, neurons per layer , and spline degree, are selected using the Bayesian optimisation library Optuna [58]. 4 Results After having introduced the surrogate model, we demonstrate the capability of the KAN across four test cases. W e start by examining the cement hydration benchmark, then we mov e to radium uptake into SSs containing barium and strontium. 4.1 Cement System CaO SiO 2 H 2 O T o assess the performance of the KAN against classical approaches, we first consider the benchmark published in [ 23 ]. Here, the authors e xamined the hydration of the CaO SiO 2 H 2 O cementitious system under isothermal conditions at 25 °C. The input is 3-dimensional (CaO, SiO 2 , and H 2 O), and the output is 18-dimensional. The output covers the aqueous species after equilibration, as well as the amounts and composition of the solid phases and SSs. As the training data is published [ 61 ], we leverage the 50,000 sample points in 10_PC_02_LHS_50000_54854_01_ s1_G.csv for our dataset. W e refer to [ 23 ] for details on the employed geochemical solver and the data generation process. T o train the NNs, we randomly select 40,000 points from the dataset, while the remaining part is split in half for the v alidation and test phases, respectiv ely . As preprocessing, log transformation and min-max scaling are applied to both the input and output data. In their study , Prasianakis et al. trained a 5-layer MLP with 192 neurons per layer and Mish acti vation functions, which we k eep as the reference model. For the KANs, we consider two models of dif ferent sizes. The first network consists of 4 hidden layers with 28 neurons per layer , 7th-order acti v ation functions, and 10 grid points, resulting in approximately one-third of the trainable parameters compared to the MLP model. The second KAN consists of 5 layers with 40 neurons per layer , 8-th order acti v ation functions, and 12 grid points. All networks are trained ov er 200 epochs, with a batch size of 192 and the mean squared error loss function. Finally , a learning rate scheduler is employed to a void local minima, starting with a v alue of 0.01. If the validation error does not impro ve in 10 iterations, the learning rate is reduced by 90% . Figure 2 sho ws the root mean squared error (RMSE) on each output variable. The blue bars denote the MLP model according to [ 23 ], while the green and orange bars represent the KAN. Note that we cannot retrie ve the same error v alue 5 L. Boledi et al. Pr eprint submitted to ArXiv Figure 2: RMSE on the test set for the cement hydration case. The reference MLP model (blue) is sho wn against two KANs of different sizes (green and orange). V ariable RMSE MLP RMSE KAN Impro vement (%) pH 3 . 3071 × 10 − 3 1 . 8907 × 10 − 3 42.83 MassW ater 4 . 1398 × 10 − 4 1 . 5640 × 10 − 4 62.22 Ca(aq) 5 . 3386 × 10 − 6 1 . 8701 × 10 − 6 64.97 Si(aq) 2 . 5183 × 10 − 6 1 . 0015 × 10 − 6 60.23 O(aq) 1 . 0262 × 10 − 5 3 . 9045 × 10 − 6 61.95 H(aq) 1 . 0571 × 10 − 5 3 . 9398 × 10 − 6 62.73 Ca(s) 4 . 7758 × 10 − 3 8 . 2431 × 10 − 4 82.74 Si(s) 9 . 1093 × 10 − 4 3 . 2470 × 10 − 4 64.35 O(s) 5 . 7943 × 10 − 3 1 . 8594 × 10 − 3 67.91 H(s) 5 . 8698 × 10 − 3 1 . 8339 × 10 − 3 68.76 Portlandite 1 . 7666 × 10 − 2 6 . 4487 × 10 − 3 63.50 AmorfSi 1 . 7160 × 10 − 3 1 . 4347 × 10 − 3 16.39 mCSHQ 1 . 1664 × 10 − 4 3 . 8269 × 10 − 5 67.19 Ca(ss) 1 . 0327 × 10 − 3 3 . 8317 × 10 − 4 62.90 Si(ss) 9 . 0186 × 10 − 4 2 . 4104 × 10 − 4 73.27 H 2 O(ss) 1 . 6592 × 10 − 3 5 . 9939 × 10 − 4 63.88 V(s) 9 . 3427 × 10 − 2 2 . 8549 × 10 − 2 69.44 Gel_water 8 . 3947 × 10 − 4 2 . 7253 × 10 − 4 67.54 T able 2: Prediction errors on the test set for the cement hydration test case. The MLP and KAN models with 1 . 5 × 10 5 parameters are shown, together with the percentage impro vement of the RMSE. as in [ 23 ] since the training process is not fully reproducible, but the o verall beha vior is comparable, as the formation of minerals and the total solid volume are once again the most challenging quantities to predict. In terms of absolute error, the KANs outperform the MLP model across all output v ariables, except for amorphous silica in the smaller network. In particular , if we pick a KAN with the same number of trainable parameters, an av erage error improvement of 62% is achiev ed, see T able 2. When ev aluating chemical reactions, we often encounter output variables with different orders of magnitude. Thus, it is paramount to examine the relati ve error of the predictions. Following [ 23 ], the relativ e root mean squared error (RRMSE) is defined as RRMSE = s 1 N X i ( e i ) 2 , with e i = ( y i − ˆ y i ) /y i if y i , ˆ y i = 0 , 1 if y i = 0 and ˆ y i = 0 , 0 if y i = 0 and ˆ y i = 0 , (9) 6 L. Boledi et al. Pr eprint submitted to ArXiv Figure 3: RRMSE on the test set for the cement hydration case. The reference MLP model (blue) is shown against two KANs of different sizes (green and orange). V ariable RRMSE MLP RRMSE KAN Impro vement (%) pH 2 . 9427 × 10 − 4 1 . 7009 × 10 − 4 42.20 MassW ater 6 . 3279 × 10 − 3 1 . 9846 × 10 − 3 68.64 Ca(aq) 5 . 4218 × 10 − 3 2 . 2426 × 10 − 3 58.64 Si(aq) 1 . 1215 × 10 − 2 5 . 1705 × 10 − 3 53.90 O(aq) 6 . 7081 × 10 − 3 2 . 3244 × 10 − 3 65.35 H(aq) 5 . 3405 × 10 − 3 2 . 3007 × 10 − 3 56.92 Ca(s) 3 . 4789 × 10 − 3 1 . 0662 × 10 − 3 69.35 Si(s) 1 . 7738 × 10 − 3 6 . 6102 × 10 − 4 62.74 O(s) 1 . 6295 × 10 − 3 6 . 6046 × 10 − 4 59.47 H(s) 1 . 8466 × 10 − 3 7 . 6938 × 10 − 4 58.34 Portlandite 9 . 4112 × 10 − 2 4 . 8741 × 10 − 2 48.21 AmorfSi 6 . 4440 × 10 − 2 2 . 1477 × 10 − 2 66.67 mCSHQ 1 . 3699 × 10 − 3 5 . 7155 × 10 − 4 58.28 Ca(ss) 1 . 5273 × 10 − 3 7 . 5066 × 10 − 4 50.85 Si(ss) 1 . 7567 × 10 − 3 6 . 0620 × 10 − 4 65.49 H 2 O(ss) 1 . 3894 × 10 − 3 6 . 4193 × 10 − 4 53.80 V(s) 1 . 7991 × 10 − 3 7 . 2056 × 10 − 4 59.95 Gel_water 1 . 6498 × 10 − 3 6 . 991 × 10 − 4 57.63 T able 3: Prediction errors on the test set for the cement hydration test case. The MLP and KAN models with 1 . 5 × 10 5 parameters are shown, together with the percentage impro vement of the RRMSE. where y i and ˆ y i denote the true v alues and the NN predictions, respecti vely . Since the formation of minerals, e.g., portlandite, occurs only under certain system conditions, Equation (9) av oids di vision by zero in the error computation. Figure 3 sho ws the RRMSE on each output variable. The blue bars denote the MLP model according to [ 23 ], while the green and orange bars represent the KANs. In terms of relative error , the KANs consistently outperform the MLP model across all output v ariables, ev en when selecting the netw ork with fe wer trainable parameters. The bigger network achiev es an av erage improv ement of 59% , as shown in T able 3. As a final test, we examine the training time of both architectures on an NVIDIA Quadro GV100 GPU. T able 4 shows the training times of the networks discussed above, plus an additional MLP with 4 hidden layers and 132 neurons per layer for comparison. Shukla et al. reported significantly slower training times for KANs compared to MLPs [ 30 ], attributing this to the backpropagation step becoming less ef ficient as the comple xity of the activ ation functions increases. Our results confirm their findings: the KAN takes almost four times longer to train for the smallest network configuration, with this gap widening for larger netw orks due to the increased complexity of learnable spline-based 7 L. Boledi et al. Pr eprint submitted to ArXiv Model T rainable parameters T raining time ( s ) MLP 5 . 6 × 10 4 153 MLP 1 . 5 × 10 5 162 KAN 5 . 6 × 10 4 600 KAN 1 . 5 × 10 5 968 T able 4: Comparison of training times between KANs and MLPs for the cement hydration test case across dif ferent network sizes. Figure 4: Relative error on the test set for the radium uptake case with mechanical mixing. The errors are plotted for three KANs trained on datasets of different sizes, where m denotes the exponent of the Sobol sampler . In red, we show the number of predictions with an error abov e 10% . activ ations. W e underline, howe ver , that training is a one-time cost in the modeling workflo w (solver setup, data generation, network setup and training, etc.), whereas the higher accurac y achiev ed by KANs translates to impro ved predictions across millions or billions of subsequent ev aluations in reactive transport simulations. It is evident then that the KAN architecture is preferable as a surrogate model for the e xamined cement system. W e underline that the objectiv e of this benchmark is to inv estigate how accurate KANs are against a pre-published NN model. While the average RRMSE remains high when predicting portlandite and amorphous silica, the applicability of this specific surrogate in a real-world scenario is be yond the scope of this work. 4.2 Radium Uptake (i) - Mechanical Mixing For the upcoming test cases, we move to an application of interest, namely the formation of Ra SSs. W e start by considering the simplest system without any “real” SS model, i.e., the so-called mechanical mixing. The input is 3-dimensional (BaSO 4 , NaCl, and RaBr 2 ), and the output is 8-dimensional. Here, we only track the radium species and the pH as quantities of interest. First, we compare the ef fect size on the surrogate model accurac y . W ith the solv er GEM-Selektor , three datasets of size 2 m are generated, where m = 15 , 16 , 17 . The input data are chosen according to a Sobol sampler , then split in half to obtain the training and v alidation sets. From the latter , 5000 points are subtracted for testing. This time, the KAN architectures are selected with the hyperparameter-optimization tool Optuna . The optimizer finds the best network configuration up to a maximum of 4 hidden layers, 24 neurons per layer , 8th-order activ ation functions, and 15 grid points. All networks are trained ov er 100 epochs, with a batch size of 192 and the mean squared error loss function. A learning rate scheduler is employed as in Section 4.1, b ut with an 80% reduction factor . 8 L. Boledi et al. Pr eprint submitted to ArXiv Figure 5: Relati ve error on the test set for the radium uptak e case with mechanical mixing. The classical MLP model (blue) is sho wn against two KANs of dif ferent sizes (green and orange). In red, we show the number of predictions with an error abov e 10% . Figure 4 sho ws the relati ve error for all the output v ariables and dataset sizes. All the error values are combined in a box plot for a more detailed examination. While the median errors are all below 1 × 10 − 3 , it is also important to check how man y predictions are accurate enough for the application of the surrogate model. Note that ev en with 32,768 ( 2 15 ) data points, only 37 e valuations out of 5000 exceed the 10% error threshold. Additionally , both the median error and the number of high-error points decrease as the size of the employed dataset increases. As training points can be easily generated with GEM-Selektor , the model can be refined ev en further up to the desired accuracy . W e now keep the KAN model trained on the 131,072 ( 2 17 ) points dataset as a reference and compare it once again with an MLP . The hyperparameters are selected with Optuna up to a maximum of 10 layers and 64 neurons per layer . Additionally , a KAN with less than half of the trainable parameters is considered. Figure 5 sho ws the relativ e error for all the output v ariables on the dif ferent NNs. Once again, both KANs exhibit a lo wer median error on all the output variables (green and orange), ev en when considering the small-size network. W e underline that the KANs sho w fewer high-error predictions, as lo w as one tenth, and only for one single output. As in the cement hydration test case, the KAN architecture performs best. 4.3 Radium Uptake (ii) - (Ba,Ra)SO 4 Solid Solution After having pro ven the ef fecti veness of KANs on the reference problems, we mov e to wards more complex chemical systems by considering SS models. In this example, the two-component (Ba,Ra)SO 4 SS is examined. The input is 3-dimensional (BaSO 4 , RaBr 2 , and T ) and the output is 10-dimensional, where T is the system’ s temperature. Again, we only track the radium species and the pH as quantities of interest. A dataset of 262,144 ( 2 18 ) points is generated using the GEM-Selektor solver . The dataset splitting process follows as in Section 4.2. A KAN with three hidden layers, 24 neurons per layer, 8th-order acti vation functions, and 12 grid points is selected, resulting in ca. 32,200 trainable parameters. The network is trained o ver 100 epochs, with a batch size of 192 and the mean squared error loss function. Figure 6 shows the relativ e errors for all the output variables. Note that the median errors are all below the v alue 2 × 10 − 3 and no predictions are above the 10% error threshold. Thus, the KAN prov es to be an ef fectiv e surrogate model for the two-component SS as well. W e underline that, for the first time, temperature dependency is considered as a variable in the surrog ate model, in contrast to recent publications such as [62]. 9 L. Boledi et al. Pr eprint submitted to ArXiv Figure 6: Relativ e error on the test set for the radium uptake case with the two-component SS model. Figure 7: Relativ e error on the test set for the radium uptake case with the three-component SS model. 10 L. Boledi et al. Pr eprint submitted to ArXiv 4.4 Radium Uptake (iii) - (Sr ,Ba,Ra)SO 4 Solid Solution For the most comple x test case, we examine the ternary (Sr ,Ba,Ra)SO 4 SS model with the addition of strontium. The input is four-dimensional (BaSO 4 , NaCl, RaBr 2 , SrSO 4 ) and the output is 15-dimensional. Only the radium-containing species and the pH are tracked. W e prepare a dataset of 524,000 ( 2 19 ) points using GEM-Selektor , which is then split according to Section 4.2. A KAN with four hidden layers, 25 neurons per layer , 10th-order activ ation functions, and 12 grid points is selected, resulting in approximately 56,000 trainable parameters. The network is trained o ver 100 epochs, with a batch size of 192 and the mean squared error loss function. Figure 7 shows the relative errors for all the output v ariables. The median errors are all below 1 × 10 − 3 , and no predictions exceed the 10% error threshold. Even with the added complexity of strontium in the system, the KAN surrogate model provides accurate predictions of the chemical equilibrium. Model Run time GEMS (s) Run time KAN (s) Impr ovement (%) (i) Mech. mix 2.45 0.176 92.82 (ii) Binary SS 1.29 0.162 87.44 (iii) T ernary SS 4.13 0.260 93.71 T able 5: Comparison of run times between GEM-Selektor and the KAN surrogates for the radium uptake case studies. The time required to perform 5,000 equilibria calculations is displayed. Finally , we in vestigate the computational ef ficiency of the surrogate models against the original solver GEM-Selektor . T able 5 sho ws the run times for 5,000 equilibrium calculations on an AMD Ryzen Threadripper PR O 5965WX 24-Cores processor . As expected, the computational cost increases with the model complexity , with the ternary SS requiring the most time. Note, howe ver , that the KAN surrogate consistently achiev es time reductions exceeding 87% across all cases. Despite the increased complexity requiring ca. 50,000 trainable parameters in case (iii), the surrogate model achiev es a 93 . 7% reduction in computational time—a speedup factor of approximately 16 × . 4.5 Discussion Having assessed the performance of KAN surrogate models, we highlight the follo wing findings: Surrogate model accuracy : KANs achiev e a higher prediction accuracy than MLPs across all tested scenarios. KANs exhibit lo wer median errors on the test set and significantly fe wer high-error predictions, see Figure 5. W ith enough training data, the KAN surrogate can successfully replace the chemical equilibrium solver . For the most complex ternary SS model, no prediction error exceeds the 10% threshold (see Figure 7), with median errors consistently below 1 × 10 − 3 . Surrogate model speedup: Despite longer training times, KANs demonstrate e xcellent computational ef ficiency . KAN surrogates achie ve 87-93% reductions in computational time compared to GEM-Selektor across the radium uptake test cases (see T able 5). Parameter efficiency: A notable advantage of KANs is their ability to achie ve superior accuracy with fe wer trainable parameters compared to MLPs, reducing the memory requirements for training and storage. Both in the cement hydration benchmark and in the radium uptake (i) case, KAN architectures with fe wer parameters outperformed larger MLP models. Importantly , all networks were trained on identical datasets, and we did not observe the data inefficienc y issues reported in [30]. T raining stability: Shukla et al. reported loss div ergence when training KANs to solve partial differential equations [ 30 ]. Howe ver , in the examined geochemical equilibrium applications, we observed no such instabilities. The transition from MLPs required no modifications or stabilization of the training process. T raining performance: The main limitation of KANs is increased training time. KANs not only train slower than MLPs, but the gap widens significantly with the number of trainable parameters, i.e., the complexity of the spline-based activ ation functions (see T able 4). In summary , the application of KANs as surrogate models for chemical equilibrium calculations demonstrates clear advantages o ver classical MLP architectures. The primary limitation—increased training time—represents a one-time in vestment of at most ten minutes in our test cases. This modest cost is far outweighed by the superior prediction accuracy (up to 62% error reduction). 11 L. Boledi et al. Pr eprint submitted to ArXiv This study focused exclusiv ely on one-time predictions of chemical equilibria. Integrating surrogates into reactive transport simulations introduces additional challenges. When chemical reactions are solved at each computational cell and time step, violations of mass conserv ation can lead to error accumulation ov er time. T o address this challenge, Silv a et al. enforced char ge balance as a penalty term in the loss function [ 22 ]. An alternati ve approach w ould incorporate the Gibbs energy minimization formulation directly into the loss function, analogous to physics-informed NNs for partial dif ferential equations. Inv estigating these strate gies for R TM represents a crucial ne xt step to ward deplo ying KAN-based surrogates. 5 Conclusions In this work, we in vestigated KANs as surrogate models for computing chemical equilibria. W e first tested KANs against classical MLPs on a cement hydration benchmark. Then, we examined three models of radium incorporation into sulfate SSs up to the ternary (Sr,Ba,Ra)SO 4 system. T o our knowledge, this represents the first surrogate model for radionuclide co-precipitation, including temperature-dependent systems. On the cement hydration benchmark, KANs reduce absolute and relati ve errors by 62% and 59%, respecti vely , compared to MLP architectures. Importantly , KANs achiev e this accuracy with fe wer trainable parameters, outperforming larger MLP models on identical datasets. This performance extends to more complex systems: for the binary and ternary SS models, KANs maintained median prediction errors near 1 × 10 − 3 with no predictions exceeding the 10% error threshold. Beyond accuracy , KANs achiev e 87-93% reductions in ev aluation time compared to GEM-Selektor across all test cases. While KANs require longer training times than MLPs—at most ten minutes in our test cases—this represents a one-time in vestment that is ne gligible within the modelling workflo w . The combination of superior accurac y and parameter ef ficiency make KANs a promising tool to accelerate R TM. Future work should address the extension to time-dependent simulations, including coupling with flow solv ers to tackle more complex geochemical scenarios. Additionally , strategies to enforce conservation of mass and pre vent error accumulation could further improv e the performance of KANs. This work represents an important step tow ard ef ficient real-time uncertainty quantification and high-resolution simulations for complex geochemical systems rele v ant to nuclear waste management and en vironmental applications. Open Research Section The employed datasets, as well as the GEM-Selektor input files and Python generation scripts, are available at https://doi.org/10.5281/zenodo.18682412 . Declaration of Generative AI and AI-assisted T ools The authors declare that they have used Claude (version Sonnet 4.5) and ChatGPT (version 5.2) to spell-check the manuscript and refine sentences. W e emphasize that AI tools have generated no content. Conflict of Interest Declaration The authors declare there are no conflicts of interest for this manuscript. Acknowledgements The authors gratefully ackno wledge The Helmholtz Association of German Research Centers (HGF) and the Federal Ministry of Research, T echnology and Space (BMFTR), Germany for supporting this work within the frame of the Innov ation Pool project “DTN - Digital twins in nuclear waste disposal” and the Helmholtz Research Program “Nuclear W aste Management, Safety and Radiation Research” (NUSAFE). The authors would like to thank Ryan Santoso for his constructiv e criticism on the manuscript. 12 L. Boledi et al. Pr eprint submitted to ArXiv References [1] C. Steefel, D. Depaolo, P . Lichtner, Reacti ve transport modeling: An essential tool and a new research approach for the Earth sciences, Earth and Planetary Science Letters 240 (3-4) (2005) 539–558. doi:10.1016/j.epsl. 2005.09.017 . [2] C. I. Steefel, Reactiv e T ransport at the Crossroads, Revie ws in Mineralogy and Geochemistry 85 (1) (2019) 1–26. doi:10.2138/rmg.2019.85.1 . [3] Q. Kang, P . C. Lichtner , H. S. V iswanathan, A. I. Abdel-Fattah, Pore Scale Modeling of Reactiv e T ransport In volv ed in Geologic CO2 Sequestration, T ransport in Porous Media 82 (1) (2010) 197–213. doi:10.1007/ s11242- 009- 9443- 9 . [4] D. Liu, R. Agarwal, Y . Li, S. Y ang, Reactiv e transport modeling of mineral carbonation in unaltered and altered basalts during CO2 sequestration, International Journal of Greenhouse Gas Control 85 (2019) 109–120. doi:10.1016/j.ijggc.2019.04.006 . [5] A. Y apparov a, G. D. Miron, D. A. Kulik, G. K osakowski, T . Driesner , An adv anced reactiv e transport simulation scheme for hydrothermal systems modelling, Geothermics 78 (2019) 138–153. doi:10.1016/j.geothermics. 2018.12.003 . [6] S. Erol, T . Akın, A. Ba¸ ser , Ö. Saraço ˘ glu, S. Akın, Fluid-CO2 injection impact in a geothermal reservoir: Evaluation with 3-D reacti ve transport modeling, Geothermics 98 (2022) 102271. doi:10.1016/j.geothermics.2021. 102271 . [7] A. V isser , H. P . Broers, R. Heerdink, M. F . P . Bierkens, T rends in pollutant concentrations in relation to time of recharge and reactiv e transport at the groundwater body scale, Journal of Hydrology 369 (3) (2009) 427–439. doi:10.1016/j.jhydrol.2009.02.008 . [8] B. Leterme, P . Blanc, D. Jacques, A reactive transport model for mercury fate in soil—application to different anthropogenic pollution sources, En vironmental Science and Pollution Research 21 (21) (2014) 12279–12293. doi:10.1007/s11356- 014- 3135- x . [9] J. M. Paz-García, B. Johannesson, L. M. Ottosen, A. N. Alshaw abkeh, A. B. Ribeiro, J. M. Rodríguez-Maroto, Modeling of electrokinetic desalination of bricks, Electrochimica Acta 86 (2012) 213–222. doi:10.1016/j. electacta.2012.05.132 . [10] L. Montenegro, J. Samper , A. Mon, L. De W indt, A.-C. Samper , E. García, A non-isothermal reacti ve transport model of the long-term geochemical e volution at the disposal cell scale in a hlw repository in granite, Applied Clay Science 242 (2023) 107018. doi:https://doi.org/10.1016/j.clay.2023.107018 . [11] F . Claret, N. I. Prasianakis, A. Baksay , D. Lukin, G. Pepin, E. Ahusborde, B. Amaziane, G. Bátor , D. Becker , A. Bednár, M. Béreš, S. Bérešová, Z. Böthi, V . Brendler, K. Brenner , J. B ˇ rezina, F . Chave, S. V . Churakov , M. Hokr , D. Horák, D. Jacques, F . Jankovský, C. Kazymyrenko, T . K oudelka, T . K ovács, T . Krej ˇ cí, J. Kruis, E. Laloy , J. Landa, T . Ligurský, T . Lipping, C. López-Vázquez, R. Masson, J. C. L. Meeussen, M. Mollaali, A. Mon, L. Montenegro, B. Pisani, J. Poonoosamy , S. I. Pospiech, Z. Saâdi, J. Samper, A.-C. Samper-Pilar , G. Scaringi, S. Sysala, K. Y oshioka, Y . Y ang, M. Zuna, O. K olditz, EURAD state-of-the-art report: development and improv ement of numerical methods and tools for modeling coupled processes in the field of nuclear waste disposal, Frontiers in Nuclear Engineering 3 (2024). doi:10.3389/fnuen.2024.1437714 . [12] J. Samper , C. López-Vázquez, B. Pisani, A. Mon, A. C. Samper-Pilar , F . J. Samper-Pilar , Global sensitivity analysis of reactiv e transport modelling for the geochemical evolution of a high-le vel radioacti ve waste repository , Applied Geochemistry 180 (2025) 106286. doi:10.1016/j.apgeochem.2025.106286 . [13] O. Kolditz, D. Jacques, F . Claret, J. Bertrand, S. V . Churakov , C. Debayle, D. Diaconu, K. Fuzik, D. Garcia, N. Graebling, B. Grambo w , E. Holt, A. Idiart, P . Leira, V . Montoya, E. Niederleithinger , M. Olin, W . Pfingsten, N. I. Prasianakis, K. Rink, J. Samper , I. Szöke, R. Szöke, L. Theodon, J. W endling, Digitalisation for nuclear waste management: predisposal and disposal, En vironmental Earth Sciences 82 (1) (2023) 42. doi:10.1007/ s12665- 022- 10675- 4 . [14] A. M. M. Leal, D. A. Kulik, W . R. Smith, M. O. Saar, An overvie w of computational methods for chemical equilibrium and kinetic calculations for geochemical and reacti ve transport modeling, Pure and Applied Chemistry 89 (5) (2017) 597–643. doi:10.1515/pac- 2016- 1107 . [15] A. M. M. Leal, S. Kyas, D. A. Kulik, M. O. Saar , Accelerating Reactiv e Transport Modeling: On-Demand Machine Learning Algorithm for Chemical Equilibrium Calculations, T ransport in Porous Media 133 (2) (2020) 161–204. doi:10.1007/s11242- 020- 01412- 1 . 13 L. Boledi et al. Pr eprint submitted to ArXiv [16] M. De Lucia, M. Kühn, A. Lindemann, M. Lübke, B. Schnor , POET (v0.1): speedup of many-core parallel reactiv e transport simulations with f ast DHT lookups, Geoscientific Model De velopment 14 (12) (2021) 7391– 7409, publisher: Copernicus GmbH. doi:10.5194/gmd- 14- 7391- 2021 . [17] M. Lübke, M. De Lucia, S. Petri, B. Schnor , A Fast MPI-Based Distributed Hash-T able as Surrogate Model for HPC Applications, in: M. H. Lees, W . Cai, S. A. Cheong, Y . Su, D. Abramson, J. J. Dongarra, P . M. A. Sloot (Eds.), Computational Science – ICCS 2025, Springer Nature Switzerland, Cham, 2025, pp. 233–240. doi:10.1007/978- 3- 031- 97635- 3_28 . [18] S. Kyas, D. V olpatto, M. O. Saar, A. M. M. Leal, Accelerated reactive transport simulations in heterogeneous porous media using Reaktoro and Firedrake, Computational Geosciences 26 (2) (2022) 295–327. doi:10.1007/ s10596- 021- 10126- 2 . [19] E. Laloy , D. Jacques, Emulation of CPU-demanding reactive transport models: a comparison of Gaussian processes, polynomial chaos expansion, and deep neural networks, Computational Geosciences 23 (5) (2019) 1193–1215. doi:10.1007/s10596- 019- 09875- y . [20] E. Laloy , D. Jacques, Speeding Up Reactive T ransport Simulations in Cement Systems by Surrogate Geochemical Modeling: Deep Neural Networks and k-Nearest Neighbors, T ransport in Porous Media 143 (2) (2022) 433–462. doi:10.1007/s11242- 022- 01779- 3 . [21] E. Demirer , E. Coene, A. Iraola, A. Nardi, E. Abarca, A. Idiart, G. de Paola, N. Rodríguez-Morillas, Improving the Performance of Reactiv e T ransport Simulations Using Artificial Neural Networks, Transport in Porous Media 149 (1) (2023) 271–297. doi:10.1007/s11242- 022- 01856- 7 . [22] V . L. S. Silva, G. Regnier , P . Salinas, C. E. Heaney , M. D. Jackson, C. C. Pain, Rapid modelling of reactive transport in porous media using machine learning: limitations and solutions, arXiv:2405.14548 [cs] (2025). doi:10.48550/arXiv.2405.14548 . [23] N. I. Prasianakis, E. Laloy , D. Jacques, J. C. L. Meeussen, G. D. Miron, D. A. Kulik, A. Idiart, E. Demirer , E. Coene, B. Cochepin, M. Leconte, M. E. Savino, J. Samper-Pilar , M. De Lucia, S. V . Churakov , O. K olditz, C. Y ang, J. Samper , F . Claret, Geochemistry and machine learning: methods and benchmarking, En vironmental Earth Sciences 84 (5) (2025) 121. doi:10.1007/s12665- 024- 12066- 3 . [24] Z. Liu, Y . W ang, S. V aidya, F . Ruehle, J. Halverson, M. Solja ˇ ci ´ c, T . Y . Hou, M. T egmark, KAN: K olmogorov- Arnold Networks, arXi v:2404.19756 [cs] (2025). doi:10.48550/arXiv.2404.19756 . [25] C. Guo, L. Sun, S. Li, Z. Y uan, C. W ang, Physics-informed Kolmogorov–Arnold network with Chebyshev polynomials for fluid mechanics, Physics of Fluids 37 (9) (2025) 095120. doi:10.1063/5.0284999 . [26] K. Xu, L. Chen, S. W ang, K olmogorov-Arnold Networks for T ime Series: Bridging Predictive Power and Interpretability, arXiv:2406.02496 [cs] (2024). doi:10.48550/arXiv.2406.02496 . [27] L. Li, Y . Zhang, G. W ang, K. Xia, K olmogorov–Arnold graph neural networks for molecular property prediction, Nature Machine Intelligence 7 (8) (2025) 1346–1354, publisher: Nature Publishing Group. doi:10.1038/ s42256- 025- 01087- 7 . [28] A. D. Bodner , A. S. T epsich, J. N. Spolski, S. Pourteau, Con volutional K olmogorov-Arnold Networks, arXiv:2406.13155 [cs] (2025). doi:10.48550/arXiv.2406.13155 . [29] M. Kiamari, M. Kiamari, B. Krishnamachari, GKAN: Graph K olmogorov-Arnold Networks, arXi v:2406.06470 [cs] (2024). doi:10.48550/arXiv.2406.06470 . [30] K. Shukla, J. D. T oscano, Z. W ang, Z. Zou, G. E. Karniadakis, A comprehensiv e and F AIR comparison between MLP and KAN representations for dif ferential equations and operator networks, Computer Methods in Applied Mechanics and Engineering 431 (2024) 117290. doi:10.1016/j.cma.2024.117290 . [31] Y . Hou, T . Ji, D. Zhang, A. Stefanidis, Kolmogoro v-Arnold Netw orks: A Critical Assessment of Claims, Performance, and Practical V iability, arXiv:2407.11075 [cs] (2025). doi:10.48550/arXiv.2407.11075 . [32] J. D. T oscano, V . Oommen, A. J. V ar ghese, Z. Zou, N. Ahmadi Daryakenari, C. W u, G. E. Karniadakis, From PINNs to PIKANs: recent advances in physics-informed machine learning, Machine Learning for Computational Science and Engineering 1 (1) (2025) 15. doi:10.1007/s44379- 025- 00015- 1 . [33] B. Jacob, A. A. Ho ward, P . Stinis, SPIKANs: separable physics-informed K olmogorov–Arnold networks, Machine Learning: Science and T echnology 6 (3) (2025) 035060, publisher: IOP Publishing. doi:10.1088/2632- 2153/ ae05af . [34] T . W agner , D. A. Kulik, F . F . Hingerl, S. V . Dmytriev a, GEM-SELEKTOR GEOCHEMICAL MODELING P A CKA GE: TSolMod LIBRAR Y AND D A T A INTERF ACE FOR MUL TICOMPONENT PHASE MODELS, The Canadian Mineralogist 50 (5) (2012) 1173–1195. doi:10.3749/canmin.50.5.1173 . 14 L. Boledi et al. Pr eprint submitted to ArXiv [35] D. A. Kulik, T . W agner , S. V . Dmytrie va, G. K osako wski, F . F . Hingerl, K. V . Chudnenko, U. R. Berner , GEM- Selektor geochemical modeling package: revised algorithm and GEMS3K numerical kernel for coupled simulation codes, Computational Geosciences 17 (1) (2013) 1–24. doi:10.1007/s10596- 012- 9310- 6 . [36] T . Zhang, K. Gregory , R. W . Hammack, R. D. V idic, Co-precipitation of Radium with Barium and Strontium Sulfate and Its Impact on the Fate of Radium during T reatment of Produced W ater from Unconv entional Gas Extraction, En vironmental Science & T echnology 48 (8) (2014) 4596–4603. doi:10.1021/es405168b . [37] SKB, RD&D Programme 2022. Programme for research, de velopment and demonstration of methods for the management and disposal of nuclear waste, T ech. Rep. TR-22-11, Svensk Kärnbränslehantering AB (2022). [38] F . Brandt, E. Curti, M. Klinkenberg, K. Rozo v , D. Bosbach, Replacement of barite by a (Ba,Ra)SO 4 solid solution at close-to-equilibrium conditions: A combined experimental and theoretical study , Geochimica et Cosmochimica Acta 155 (2015) 1 – 15. doi:10.1016/j.gca.2015.01.016 . [39] F . Brandt, M. Klinkenberg, J. Poonoosamy , J. W eber , D. Bosbach, The Effect of Ionic Strength and Sr aq upon the Uptake of Ra during the Recrystallization of Barite, Minerals 8 (11) (2018). doi:10.3390/min8110502 . [40] F . Brandt, M. Klinkenberg, J. Poonoosamy , D. Bosbach, Recrystallization and Uptake of 226 Ra into Ba-Rich (Ba,Sr)SO 4 Solid Solutions, Minerals 10 (9) (2020) 1 – 28. doi:10.3390/min10090812 . [41] M. Klinkenber g, F . Brandt, U. Breuer , D. Bosbach, Uptake of Ra during the Recrystallization of Barite: A Microscopic and T ime of Flight-Secondary Ion Mass Spectrometry Study, En vironmental Science and T echnology 48 (12) (2014) 6620 – 6627. doi:10.1021/es405502e . [42] M. Klinkenber g, J. W eber , J. Barthel, V . V inograd, J. Poonoosamy , M. Kruth, D. Bosbach, F . Brandt, The solid solution–aqueous solution system (Sr ,Ba,Ra)SO 4 + H 2 O: A combined experimental and theoretical study of phase equilibria at Sr-rich compositions, Chemical Geology 497 (2018) 1 – 17. doi:10.1016/j.chemgeo.2018.08. 009 . [43] J. Poonoosamy , A. Kaspor , C. Schreinemachers, D. Bosbach, O. Cheong, P . M. K owalski, A. Obaied, A radio- chemical lab-on-a-chip paired with computer vision to unlock the crystallization kinetics of (Ba,Ra)SO 4 , Scientific Reports 14 (2024) 9502. doi:10.1038/s41598- 024- 59888- 6 . [44] J. W eber, J. Barthel, M. Klink enberg, D. Bosbach, M. Kruth, F . Brandt, Retention of 226 Ra by barite: The role of internal porosity , Chemical Geology 466 (2017) 722 – 732. doi:10.1016/j.chemgeo.2017.07.021 . [45] V . V inograd, F . Brandt, K. Rozo v , M. Klinkenber g, K. Refson, B. W inkler, D. Bosbach, Solid-aqueous equilibrium in the BaSO 4 RaSO 4 H 2 O system: First-principles calculations and a thermodynamic assessment, Geochimica et Cosmochimica Acta 122 (2013) 398 – 417. doi:10.1016/j.gca.2013.08.028 . [46] V . V inograd, D. K ulik, F . Brandt, M. Klinkenber g, J. W eber , B. W inkler , D. Bosbach, Thermodynamics of the solid solution - Aqueous solution system (Ba,Sr,Ra)SO 4 + H 2 O: II. Radium retention in barite-type minerals at elev ated temperatures, Applied Geochemistry 93 (2018) 190 – 208. doi:10.1016/j.apgeochem.2017.10.019 . [47] V . V inograd, D. Kulik, F . Brandt, M. Klinkenberg, J. W eber , B. Winkler , D. Bosbach, Thermodynamics of the solid solution - Aqueous solution system (Ba,Sr ,Ra)SO 4 + H 2 O: I. The effect of strontium content on radium uptake by barite, Applied Geochemistry 89 (2018) 59 – 74. doi:10.1016/j.apgeochem.2017.11.009 . [48] W . Hummel, U. Berner, E. Curti, F . J. Pearson, T . Thoenen, Nagra/PSI Chemical Thermodynamic Data Base 01/01, Univ ersal-Publishers, 2002. [49] T . Thoenen, W . Hummel, U. Berner, E. Curti, The PSI/Nagra Chemical Thermodynamic Database 12/07 (2014). [50] H. Helgeson, D. Kirkham, G. Flo wers, Theoretical prediction of the thermodynamic behavior of aqueous electrolytes by high pressures and temperatures; IV, Calculation of activity coef ficients, osmotic coefficients, and apparent molal and standard and relativ e partial molal properties to 600 degrees C and 5kb, American Journal of Science 281 (10) (1981) 1249 – 1516. doi:10.2475/ajs.281.10.1249 . [51] J. W . Johnson, E. H. Oelkers, H. C. Helgeson, SUPCR T92: A software package for calculating the standard molal thermodynamic properties of minerals, gases, aqueous species, and reactions from 1 to 5000 bar and 0 to 1000°C, Computers and Geosciences 18 (7) (1992) 899 – 947. doi:10.1016/0098- 3004(92)90029- Q . [52] Y . W ang, P . Alt-Epping, G. Deissmann, Y . Y ang, J. Hu, D. Bosbach, J. Poonoosamy , Contrasting coprecipitation and recrystallization mechanisms for Ra immobilization via (Ba,Ra)SO 4 solid solution formation in fractured crystalline rocks: Insights from 3D reactiv e transport modeling, Geochimica et Cosmochimica Acta (2026). doi:10.1016/j.gca.2026.01.045 . [53] I. M. Sobol’, On the distribution of points in a cube and the approximate ev aluation of integrals, USSR Computa- tional Mathematics and Mathematical Physics 7 (4) (1967) 86–112. doi:10.1016/0041- 5553(67)90144- 9 . 15 L. Boledi et al. Pr eprint submitted to ArXiv [54] B. Williams, S. Cremaschi, No vel T ool for Selecting Surrogate Modeling T echniques for Surface Approxi- mation, in: Computer Aided Chemical Engineering, V ol. 50, Elsevier , 2021, pp. 451–456. doi:10.1016/ B978- 0- 323- 88506- 5.50071- 1 . [55] C. G. Hounmenou, K. E. Gne you, R. L. Glele Kakaï, A Formalism of the General Mathematical Expression of Multilayer Perceptron Neural Networks (2021). doi:10.20944/preprints202105.0412.v1 . [56] K. Y . Chan, B. Abu-Salih, R. Qaddoura, A. M. Al-Zoubi, V . Palade, D.-S. Pham, J. D. Ser , K. Muhammad, Deep neural networks in the cloud: Re view, applications, challenges and research directions, Neurocomputing 545 (2023) 126327. doi:10.1016/j.neucom.2023.126327 . [57] W . Falcon, The PyT orch Lightning team, PyT orch Lightning (2019). doi:10.5281/zenodo.3828935 . URL https://github.com/Lightning- AI/lightning [58] T . Akiba, S. Sano, T . Y anase, T . Ohta, M. K oyama, Optuna: A Next-generation Hyperparameter Optimization Framew ork, in: Proceedings of the 25th A CM SIGKDD International Conference on Kno wledge Discov ery & Data Mining, KDD ’19, Association for Computing Machinery , New Y ork, NY , USA, 2019, pp. 2623–2631. doi:10.1145/3292500.3330701 . [59] H. Prautzsch, W . Boehm, M. Paluszn y , Bézier and B-Spline T echniques, Mathematics and V isualization, Springer, Berlin, Heidelberg, 2002. doi:10.1007/978- 3- 662- 04919- 8 . [60] C. Blealtan, A. Dash, efficient-kan (2024). URL https://github.com/Blealtan/efficient- kan [61] N. Prasianakis, E. Laloy , D. Jacques, J. Meeussen, C. T ournassat, G. Miron, D. Kulik, A. Idiart, E. Demirer , E. Coene, B. Cochepin, M. Leconte, M. Savino, J. Samper II, M. De Lucia, S. Churakov , O. Kolditz, C. Y ang, J. Samper , F . Claret, Geochemistry and Machine Learning: Methods and Benchmarking (2025). doi:10.5281/ zenodo.14904784 . [62] H. Peng, A. Rajyaguru, E. Curti, D. Grolimund, S. V . Churakov , N. I. Prasianakis, Machine Learning-Enhanced Modeling of Calcium Carbonate Nucleation in Porous Media Under Counter-Diffusion Conditions, W ater Re- sources Research 61 (11) (2025) e2025WR040484. doi:10.1029/2025WR040484 . 16
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment