MBD-ML: Many-body dispersion from machine learning for molecules and materials
Van der Waals (vdW) interactions are essential for describing molecules and materials, from drug design and catalysis to battery applications. These omnipresent interactions must also be accurately included in machine-learned force fields. The many-b…
Authors: Evgeny Moerman, Adil Kabylda, Almaz Khabibrakhmanov
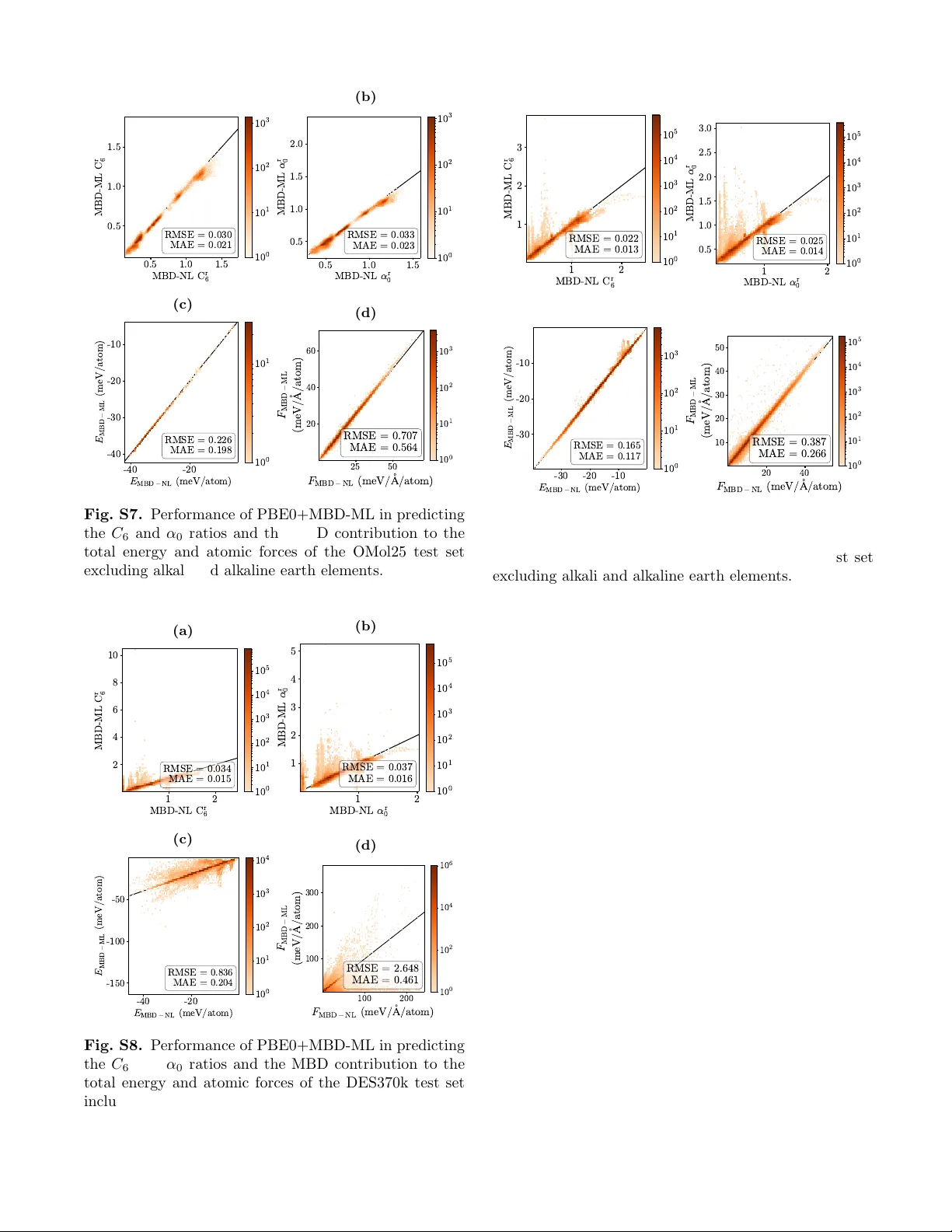
MBD-ML: Man y-b o dy disp ersion from mac hine learning for molecules and materials Evgen y Mo erman, 1 Adil Kab ylda, 1 Almaz Khabibrakhmano v, 1 and Alexandre Tk atc henko 1 , ∗ 1 Dep a rtment of Physics and Materials Scienc e, University of Luxemb our g, L-1511 Luxemb o ur g Cit y, Luxemb our g V an der W aals (vdW) interactions are essen tial for describing molecules and materials, from drug design and catalysis to battery applications. These omnipresent interactions must also b e accu- rately included in machine-learned force fields. The many-bo dy dispersio n (MBD) method stand s out as one of the most accurate and transferable approaches to capture vdW interactions, requir- ing only atomic C 6 co efficien ts and p olarizabilities as i nput. W e present MB D-ML, a pretrained message passing n eural netw ork tha t predicts these atomic prop erties directly from atomic struc- tures. Through seamless integration with libMBD, our metho d enables the immediate calculation of MBD-inclusive total energies, forces, and stress tensors. By elimi nating the need for in termediate electronic stru cture calculations, MBD-ML offers a practical and streamlined to ol that simplifies the incorp oration of sta te-of-the-art vdW interactions in to a n y electronic structure co de, as well as empirical and machine-learned force fields. INTR ODUCTION V an der W aals (vdW) disp ersion interactions pla y a decisive role in the prop erties of molecular crystals, con- densed matter, and biological systems [ 1 , 2 ]. They go v- ern p olymorphism in organ i c crystals [ 3 , 4 ], protein fold- ing [ 5 ], and the cohesion of la yered, p olymeric, and p orous materials [ 6 , 7 ]. In many suc h systems, disp ersion in teractions are not a small p erturbation but a leading con tribution that critically determines relat i ve energies, equilibrium ge omet r i es , and atomistic dynamics. Density functional th eor y (DFT) has b ecome a cor- nerstone of atomistic sim ulations of molecules and solids, and increasingly serves as a reference [ 8 – 10 ] for th e construction of mac hine-learning force fields (MLFFs) [ 11 ]. Ho wev er, most widely used semi-lo cal and h ybrid exchange-correlation functionals, as well as l o cal ML architectures, do n ot account for long-range vdW disp ersion. As a consequence, explicit disp ersion inter- actions are indisp ensable for achieving quantitativ e accu- racy , unless one deals w it h systems where strong cov alent b onds dominate the d es ir e d b ehavior [ 11 , 12 ]. Ov er the past tw o decades, a wide range of vdW meth- o ds hav e b een prop osed. Early and still widely adopted approac hes include the pairwise-additive DFT-D meth- o ds [ 13 ], most notably the D3 correction by Grimme [ 14 ], based on tabulated C 6 co efficien ts and p olarizabilities α 0 . The Tk atchenk o-Sc heffler (TS) [ 15 ] metho d intro du ce d an imp ortan t conceptual ad v ance by maki ng these quan- tities en vironment-dependent through Hirshfeld parti- tioning of the electron density . F urthermore, in con trast to empirical metho ds, the TS metho d, as w ell as its fur- ther extensions, is a self-consistent interatomic vdW den- sit y functional [ 16 ]. Similarly , the exchange-hole dip ole momen t (XDM) metho d [ 17 , 18 ] derives atomic p olar- izabilities from the electronic densit y and incorp orates higher-order multipole contributions to the dispe rs i on en- ergy through C 8 and C 10 co efficien ts obtained from the exc hange-hole multipole momen ts. A fundamental limitation of all pairwi se schemes is their inabilit y to capture collectiv e man y-b ody ef- fects in di sp ersion interactions [ 1 , 2 ]. This shortcom- ing is addressed b y the man y-b o dy disp ersion (MBD) metho d [ 19 ], which mo dels disp ersion interactions usi ng a system of dip ole-coupled quantum harmonic oscillators parametrized to repro duce atomi c α 0 and C 6 co efficien ts. The MBD framew ork naturally accounts for non-additive man y-b o dy effects as well as p olarization anisotropy [ 20 ], and has b een shown to substantially impro v e accuracy for molecular crystals, lay ered materials, and large molecu- lar systems [ 21 – 25 ]. Several v arian ts of MBD, differing in their parametrization and treatment of short-range screening, hav e b een developed [ 21 , 24 – 27 ], including the state-of-the-art MBD-NL formulation [ 25 ]. Despite this ric h v ariet y of vdW metho ds and the demonstrated accuracy of man y-b ody approac hes, the D3 correcti on remains the most widely used in large- scale and high-thr ou ghp ut applications. This includes generation of DFT-based datasets for molecule s [ 9 ] and materials [ 8 ], as w el l as routine applications in com- putational c hemistry and materi als science. While the limitations of D3 are well do cumented [ 24 , 28 – 34 ], its p opularit y is largely driven by its ease of use. D3 is implemented as a standal one library and requires only atomic p ositions and element t yp es, without the need for electronic-structure information or tigh t coupling to D FT co des. F or the MBD metho d, how ev er, the rel i anc e on electronic-structure calculati on s to parametrize the MBD Hamiltonian remains a ma jor b ottlenec k that limits its applicability in large-scale simulations and ML architec- tures. In this work, we remov e this limitation for the MBD metho d by introdu ci n g a m achine learning-based frame- w ork, termed MBD-ML, that enables accurate many- b ody disp ersion calculations usin g only atomic geometry as input. W e achiev e this by training a state-of-the-art message-passing neural netw ork to predict the effective atomic p olarizabilities α 0 and C 6 co efficien ts, using the DFT+MBD-NL metho d [ 25 ] as a reference. By integrat- ing this mo del into the libMBD library [ 35 ] (see Fig. 1 ), 2 w e make it p ossible to compute disp ersion energies, atomic forces, and stress tensors at the MB D -NL level of accuracy without p erforming any electronic-structure calculations. Prev i ou s machine-learned MBD mo dels [ 36 ] emplo yed simple deep -l e arn i ng architectures to p re di ct Hirshfeld volume ratios for the anteceden t MBD@rsSCS metho d and were limi t ed to small molecules containing C, H, N, and O. In contrast, the presen t MBD-ML mo del is applicable to b oth molecules and crystals across ov er 70 chemical elements and is directly integrated in to the libMBD infrastructure. Moreov er, α 0 and C 6 ratios from MBD-NL, which we used for training, are mor e r ob us t and t r an sf er abl e across chemical space, facilitat i n g treat- men t of ionic and metallic comp ounds, which are prob- lematic for MBD@rsSCS relying on Hirshfeld volume ra- tios [ 26 , 37 ]. W e demonstrate the transferability and general per- formance of the MBD-ML framework on subsets of five state-of-the-art data sets (see Fig. 1 ) cov ering small molecules of widely v arying comp ositions (QCML [ 38 ]) and sizes (OMol25 [ 9 ]), biomolecular and drug-like dimers (DES370k [ 39 ]), molecular cry st al s (OMC25 [ 40 ]) and inorganic materials (OMat 24 [ 8 ]). Beyond that we assess the robustness and practical accuracy of M BD - ML b y applyi n g the metho d to the prediction of structures and relative energetics of molecular- cr y st al p olymorphs, and comp ari n g the results directly against refere nc e ab initio M BD - NL calculations. Our results demonstrate that MBD-ML retains the accuracy of MBD-NL, while dramatically extend i ng its u sab i l ity to large-scale ML- based data generation or atomistic mo deling. METHODS The MBD framew ork The MBD metho d [ 19 ] emplo ys coupled quan tum Drude oscillators (QDOs) [ 41 , 42 ] to mo del collective electron density fluctuations, u n de rp i nni ng vdW disp er- sion. In this framework, the electronic resp onse of each atom is represented by a single QDO, whic h captures th e collective fluctuations of all v alence electrons. Each QDO consists of a negatively charged quasiparticle harmoni- cally b ound to a p ositively c harged pseudonucleus. The mapping b etw een atoms and oscillators is established via the static p olarizability α 0 ,i and effective excitation fre- quency ω i = 4 C 6 ,ii / 3 α 2 0 ,i . The interaction b etw een oscillators is describ ed via the long-range dip ole-dip ole coupling tensor T lr . The result- ing Hamiltonian for a system of N coupled QDOs is given b y H MBD ( { R i , α 0 ,i , ω i } ) = X i − 1 2 ∇ 2 ξ i + 1 2 ω 2 i ξ 2 i + 1 2 X ij ω i ω j √ α 0 ,i α 0 ,j ξ i T lr ij ξ j , (1) with ξ i = √ m i x i denoting the mass-weigh ted displace- men ts of the oscillating c harge relati ve to the n uclei p o- sition R i . Since the Hamiltonian ( 1 ) is quadratic in displace- men ts, the many-bo dy disp ersion energy can b e obtained b y direct diagonal i zat i on: E MBD = 3 N X k =1 ˜ ω k 2 − N X i =1 3 ω i 2 , (2) where ˜ ω k are the frequencies of the 3 N collectiv e MBD eigenmo des. The resulting MBD energy is for- mally equiv alen t to the adiabatic-connection fluctua- tion–dissipation (A CFD-RP A) correlation energy [ 20 ], while the Hami l ton i an diagonalization provides a numer- ically efficient route to its ev aluation. The computational complexity of the MBD meth o d scales as O ( N 3 ), match- ing that of Kohn-Sham DFT. How ever, we emphasize that the prefactor is substantially smaller, such that the cost of an MBD calculation constitutes only a small frac- tion of a single DFT self-consistency cycle, even for large systems [ 35 ]. Within this general formalism, several v ariants of the MBD metho d hav e b een developed, which primarily dif- fer in the determination of the atomic p olarizabilities α 0 ,i , disp ersion co efficients C 6 ,ii , and the long-range in- teraction t en sor T lr . Amon g these, the non-lo cal MBD approac h (MBD-NL) [ 25 ] represents the most adv anced and robust formulation, exhibiting uniformly high accu- racy across co v ale nt, ionic, and metallic systems. This is ac hieved by using the Vydrov-V an V o orhis (VV) p olariz- ability functional [ 43 ] to obtain α VV 0 ,i and C VV 6 ,ii co efficien ts that parametrize the MBD Hamiltonian. A key feature of MBD-NL is the explicit remov al of con tributions from jellium-like density regions [ 25 ]. This cutoff i s essential to av oid dou bl e counting of correlation energy , as most exchange-correlation functionals are ex- act for the homogeneous electron gas by construction. F urthermore, to comp ensate for the approximate nature of the VV p olarizabilit y functional, the resulting α VV 0 ,i and C VV 6 ,ii are renormalized using high-quality free-atom reference data: α 0 ,i = α VV 0 ,i α VV , free 0 ,i α ref , free 0 ,i , C 6 ,ii = C VV 6 ,ii C VV , free 6 ,ii C ref , free 6 ,ii , (3) where α ref , free 0 ,i and C ref , free 6 ,ii denote the corresp onding f re e- atom reference v alues. F urther tec hnical de tai l s of the 3 Fig. 1. Overview of the training and the v alidation of the MBD-ML mo del: (top left) MBD-ML was trained on the QCML molecular data set spanning almost the en tire p erio dic system (top right; Re pr o duced from Ref. 38 und er CC BY 4.0). (middle) The mo del has b een integrated in to the li b MB D p ython interface, pym b d, to allo w direct calculation of MBD prop erties fr om the at omi c structure. (b ottom) The mo del w as v alidated on b oth molecules and molecular crystals in predicting fundamental prop erties i n cl u di n g total energy and force contributions and – in the case of organic crystals – to p erform geometry opt im iz at i ons and recov er t h e energy ranking of different p olymorphs. MBD-NL formalism and its implementation can b e found in the original MBD-NL publication [ 25 ]. The combination of the VV p olarizabilit y functional, the jellium cutoff, and normalization to fre e-at om refer- ence data yields a robust and transferable parametriza- tion of the MBD Hamiltonian across diverse c hemical en vironments. In particu l ar, the MBD-NL formulation a voids the “p olarization catastrophe” obser ved for ear- lier MBD v ariants in transition-metal comp ounds [ 26 , 37 ] and significantly reduces ov erbinding error in metals an d ionic solids [ 25 ], achieving balanced accuracy across the p eriodic table. The MBD-ML framework Building on the MBD-NL formalism desc ri b ed ab o ve, w e now in tro duce the MBD-M L mo del, which learns to repro duce its k ey atomic inputs, α r 0 and C r 6 , directly from th e atomic configurati on. MB D -M L utilize s the SO3krates architecture [ 44 ], an equiv ariant message pass- ing neural netw ork that represen ts a general and flex- ible framework for training state-of-the-art MLFFs. A closely related mo del is the recent SO3LR [ 12 ], wh i ch w as trained to map atomic p ositions R , atomic num b ers Z and the total charge Q and spin S to atomic forces F i , partial charges q i and Hirshfe l d ratios h i . Here, we 4 mo dified and re-trained the mo del to predict the α r 0 and C r 6 ratios: α r 0 , C r 6 = SO3krates ( R , Z, Q, S ) . (4) defined as the ratios b etw een the C VV 6 ,ii and α VV 0 ,i atomic co efficien ts computed b y the VV p olarizability functional in a multi-atomic system and their free-atom equiv alents C VV , free 6 ,ii and α VV , free 0 ,i , α r 0 ,i = α VV 0 ,i α VV , free 0 ,i , C r 6 ,ii = C VV 6 ,ii C VV , free 6 ,ii . (5) whic h in the following will b e simply referred to as C r 6 and α r 0 for brevity . In contrast to polarizabilities and dis p ersion co effi- cien ts, broadly v arying in magnitude across the p eriodic table, these unit-less ratios are t ypically confined in the range ≈ 0 − 2 for most elements and chemical environ- men ts, which makes α r 0 and C r 6 naturally suitable tar- gets for machine learning. W e note that the MBD-ML mo del predicts α r 0 and C r 6 separately , while within the SO3LR mo del the analogous quantities are directly con- nected ( α r 0 ,i = h i and C r 6 ,i = h 2 i ). This imp ortan t dis- tinction originates from the underl y i ng MBD-NL metho d and mak es the resulting MBD predictions more robu st for general systems , as it w as explained ab ov e. The v alues of α 0 ,i and C 6 ,ii , required to parametri z e the MBD Hamiltonian ( 1 ), are obtained by multiplying the p re dic ted ratios by the free-atom referenc e quantities according to Eq. ( 5 ). This is done i nternally within the libMBD in terface and hence do es not require any user in terven tion. Th us , the MBD-ML-predicted ratios are sufficient to directly compute all MBD-related quantities, including energies, atomic forces, and stress tensors, via the efficient libMBD routines [ 35 ]. T raining on the atomic ratios rather t h an directly on the MBD prop erties also offers another key adv antage – α r 0 and C r 6 ratios are relatively short-ranged quan- tities, whereas energies and forces can b e very sensi- tiv e to long-range in teractions. Conseque ntly , a semi- lo cal message-passing architecture like SO3krates i s well- suited for this application. In addition, the α r 0 and C r 6 ra- tios are not v ery sensitiv e to the underlying densit y func- tional approximation (DF A), so that application of the mo del in com bination with other exc hange-correlation functionals only requires the mo dification of the damp- ing parameter β , gov erning the short-range b ehavior of the dip olar tensor T lr . In the present work, β was not re-optimized for the MBD-ML mo del and the tab- ulated default v alues were used for the resp ectiv e DF As ( β PBE = 0 . 81 , β PBE0 = 0 . 83) following Ref. [ 25 ]. The current MBD-ML implementation uses SO3krates with tw o message -p assi n g lay ers and a c ut off radius of 4 ˚ A, giving an effective receptiv e fiel d of 8 ˚ A, and w as trained on the QCML dataset [ 38 ], comprising ov er 30 million molecules with up to 8 non-h ydrogen atoms across 79 chemical elements. QCML provides structures, ener- gies, and forces at the PBE0+MBD-NL level, along with MBD-NL p olarizabilities α VV 0 ,i , and C VV 6 ,ii co efficien ts, en- abling direct t r ai ni n g of the MBD-ML mo del. W e refer the reader to Section S1 of the Supplementary Informa- tion for the technical details of the training pro cedure. MODEL V ALID A TION T o quantify the accuracy and identify p otential limita- tions of th e MBD-ML mo del i n predicting MBD correc- tions to total energies, atomic forces, and stress ten sor s (for p erio dic systems), we first ev al uat e the trained mo del on three test sets spanning different chemical com p ound spaces. Beyond the hold-out v alidation on the QCML data, w e assess p erformance on molecular non-cov alent dimers from DES370k [ 39 ], and on molecular crystals us- ing a subset of OMC25 [ 40 ]. Details on the selection and recalculation of the OMol25 and DES370k subsets can b e found in Section S2 and S3, resp ectively . Molecular data sets QCML. The MBD-ML mo del trained on the QCML dataset achiev es strong p erformance for neutral and p os- itively c harged molecules (T ab l e I , Fig. 2 ), with RM- SEs of 0.023 and 0.020 for α r 0 and C r 6 ratios, resp ec- tiv ely . These lo w error s demonstrate that M BD -M L ac- curately captures the quantum mechanical effects that mo dulate atomic p olarizabilities and disp ersion c o effi- cien ts in div erse chemical environmen ts . The accurately predicted ratios yield sub-meV errors in the MBD energy (0 . 229 meV/atom) and force (0 . 440 meV/atom) contribu- tions (Fig. 2 ), indicat i n g its suitability as an accurate drop-in replacement of the ab initio MBD-NL metho d for routine electronic-structu re or MLFF calculations. Additionally , v alidation on the hold-out test set re- v ealed that some negatively c harged molecules are inad- equately de sc ri b ed by the employ ed ab initio treatme nt. The α r 0 and C r 6 ratios v ary substantially with increasing confinement p oten tial cutoff radius in FHI-a ims (Sec- tion S4.1), indicati ng that the underlying VV p olariz- ability functional is hi gh l y sensitive to electronic density tails. This sensitivity arises b ecause additional electrons in anions are often weakly b ound and extremely delo- calized, resulting in a far-reaching, slowly decaying elec- tron densit y . Moreov er, our random insp ection of several pathological cases revealed that all of those anions had negative electron affinities, meaning that they are not stable thermo dynamically (please consult Section S4 for the results). F or such systems, a b ound solution only exists b ecause of the fini t e basis set span. F or these rea- sons, we decided to exclude negatively charged molecules 5 T ABLE I. Performance of MBD-ML mo dels on ratios and MBD prop erties for different test sets i n terms of RMSE and MAE v alues. Errors for α r 0 and C r 6 are unitless. N denotes the num be r of systems in each t es t set. a Excluding negatively charged molecules (due to insufficient accuracy of ab initio treatment; see QCML discussion in the Mo del validation section). b Excluding alk ali and alk aline earth metals (due t o under-representation in the trai ni n g set; see DES370k discussion in the Mo del validation section). QCML a ( N =83226) DES370k a,b ( N =309130) OMOL25 b ( N =825) OMC25 ( N =200) OMA T24 ( N =203) MAE RMSE MAE RMSE MAE RMSE MAE RMSE MAE RMSE R a tio Pr e dictions α r 0 0.013 0.020 0.014 0.025 0.023 0.033 0.025 0.031 0.247 0.429 C r 6 0.014 0.023 0.013 0.022 0.021 0.030 0.017 0.022 0.269 0.895 MBD Pr op erties E MBD (meV/atom) 0.158 0.228 0.117 0.165 0.198 0.226 0.713 0.841 – – F MBD (meV/ ˚ A) 0.302 0.440 0.266 0.387 0.564 0.707 0.786 0.930 – – σ MBD (meV/ ˚ A 3 ) – – – – – – 0.112 0.143 – – a 2.5 5.0 MBD -NL C r 6 2 4 MBD -ML C r 6 RMSE = 0.023 MAE = 0.014 10 0 10 2 10 4 b 2 4 MBD -NL α r 0 1 2 3 4 MBD -ML α r 0 RMSE = 0.020 MAE = 0.013 10 0 10 1 10 2 10 3 10 4 c -30 -20 -10 E MBD − NL ( me V/at . ) -30 -20 -10 E MBD − ML ( me V/at . ) RMSE = 0.228 MAE = 0.158 10 0 10 1 10 2 10 3 d 25 50 F MBD − NL ( me V/ Å ) 20 40 60 80 F MBD − ML ( me V/ Å ) RMSE = 0.440 MAE = 0.302 10 0 10 1 10 2 10 3 10 4 Fig. 2. Performance of PBE0+MBD-ML in predicting the C 6 and α 0 ratios and the MBD contribution to the total energy and atomic forces of the QCML test set. from the v alidation, and w e do not recommend using the QCML data for their vdW energies and r at i os until rig- orous ab initio b enc hmarks and stability-based filtering are p er f orm ed . In terestingly , including anionic molecules deteriorates prediction accuracy for MBD ratios (Sec- tion S4.2) , with er ror s exceeding one order of magnitude, y et MBD ene rgi e s and forces remain nearly unc hanged (Fig. S5). Bey ond these well-kno wn issues of conv erging the elec- tronic s t ru ct u re of molecular anions, charged systems are generally challenging for the state-of-the-art vdW metho ds (MBD-NL, XDM, DFT-D4) [ 45 ], i n particu- lar, c harged-neutral interactions. F or the MBD case, the observ ed interaction energy d i sagr ee ments actually arise from the underl y i ng DF A and can b e mitigated e.g. by com bining MBD wit h density-corrected DF As [ 46 ]. DES370k. T o v alidate the MBD-ML mo del b e- y ond single molecules, we tested it on molec ul ar dimers from the DES 370k dataset [ 39 ], recomputed at the PBE0+MBD-NL le vel as a part of the QCell biomolec- ular d atas et [ 47 ]. F or 309,130 dimers excluding neg- ativ ely charged systems and structures con taining al- k ali or alk aline earth elements (see b elow), MBD- ML achiev es prediction errors for MBD ratios com- parable t o the QCML hold-out set, with sligh tly im- pro ved accuracy for MBD energies and forces (RMSEs of 0 . 165 meV/atom and 0 . 387 meV/ ˚ A, resp ectively; s e e T a- ble I ). Given the su bs t antial differences in chemical com- p ound space b etw een QCML (diverse smal l molecule s) and DES370k (bi omol ec ul ar and drug-like dimers), the MBD-ML mo del prov es transferable and reliable b eyond its training distribution. A more in-depth analysis of the v alidation results sho ws that accuracy substantially deteriorates for struc- tures containing alk ali (Li, Na, K) and alk aline earth elemen ts (Be, Mg, Ca), which are severely under- represented in the QCML training set [ 38 ]. Eac h el e- men t app ears in only 10 − 5 − 10 − 4 % of the 33.5 million QCML structures (approximately 3 − 30 structures p er elemen t), making them the rarest elements alongsi de the lan thanides. Conseq ue ntly , w e exclude these elements from our assessment. A complete o verview of MBD-ML 6 a 0.5 1.0 MBD -NL C r 6 0.25 0.50 0.75 1.00 1.25 MBD -ML C r 6 RMSE = 0.022 MAE = 0.017 10 0 10 1 10 2 10 3 b 0.5 1.0 MBD -NL α r 0 0.50 0.75 1.00 1.25 MBD -ML α r 0 RMSE = 0.031 MAE = 0.025 10 0 10 1 10 2 10 3 c -80 -60 E MBD − NL ( me V/at om) -80 -70 -60 -50 E MBD − ML ( me V/at om) RMSE = 0.841 MAE = 0.713 10 0 10 1 d 25 50 F MBD − NL ( me V/ Å ) 20 40 60 80 F MBD − ML ( me V/ Å ) RMSE = 0.930 MAE = 0.786 10 0 10 1 10 2 10 3 e 0 5 10 σ MBD − NL ( me V/ Å 3 ) 0 5 10 σ MBD − NL ( me V/ Å 3 ) RMSE = 0.143 MAE = 0.112 σ x x , σ y y , σ z z σ x y , σ x z , σ y z Fig. 3. Performance of PBE0+MBD-ML in predicting the C 6 and α 0 ratios and the MBD contribution to the total energy , atomic forces an d stress tensor comp onen ts i n the OMC25 test set . p erformance including these problematic cases is pro- vided in Section S5. Hence, we recommend to use the default version of MBD-ML with care when dealing with suc h under-represented eleme nts. How ev er, this limita- tion can b e ov e rc ome by fine-tuning the m o del on a sp e- cific dataset of interest. Molecular crystals T o further test t he extrap olating ability of the MBD- ML mo del, a random set of 200 molecular crystal struc- tures from the recently published OMC25 data set was c hosen and re comp u te d at the PBE0+MBD-NL lev el of theory . T o maximize the diversit y of the subset, it was ensured that all 200 cr ys t al structures featured unique molecules. Despite b eing trained exclusively on small molecules, the MBD-ML predictions faithfully repro duce the PBE0+MBD-NL reference. The ratios are predicted with a RMSE of 0 . 02 − 0 . 03, and the resulting MBD energies and forces are accurate to within 1 meV/atom and 1 meV/ ˚ A/atom, resp ectiv ely , as illustrated in Fig. 3 . While the magnitude of MBD con tributions to the stress tensor σ MBD range only b et ween 0 and 15 meV/ ˚ A 3 , the MBD-ML mo del predicts b oth diagonal and off-diagonal stress comp onents with a very high accuracy , yielding a RMSE of 0 . 143 meV/ ˚ A 3 (see Fig. 3 ). It is imp ortan t to note that molecular-crystal unit cells often contain more than 100 atoms; consequently , even a mo dest p er-atom error of 0 . 8 meV can translate into a total-energy deviation exceeding 80 meV. How ev er, this consideration only applies to total energies, as such errors cancel out almost exactly for energy differences, resulting in correct p olymorph rankings, as shown in the relev ant section b elo w. T o elucidate the origin of this deviation, w e p erformed a sensitivity analysis of the MBD energy with resp ect to the C r 6 and α r 0 ratios. W e found that b oth systematic bias and random noise in these ratios influence the resulting MBD energy , with the effect of bias b eing appro ximately twice as large as that of noise. T ak en to- gether, the se contributions account for the o verall devi- ation of the MBD-ML energy . Details of this analysis are provided in Section S6 of the Supplementary Infor - mation. Ov erall, the MBD-ML mo del demonstrates robust ac- curacy and transferability across chemical en vironments. Although trained exclusively on small single-molecule structures from the QCML data set, the predictive accu- racy of the mo del extends to p erio dic molecular crystals con taining multiple molecules of v arying complexity . 7 a 0 ° 45 ° 90 ° 135 ° 180 ° 10 − 3 10 − 1 10 1 10 3 MBD-ML | | F MBD − ML | − | F MBD − NL | | | F MBD − NL | b 0 ° 45 ° 90 ° 135 ° 180 ° 10 − 3 10 − 1 10 1 10 3 D3 | | F D 3 | − | F MBD − NL | | | F MBD − NL | c 0 ° 45 ° 90 ° 135 ° 180 ° 10 − 3 10 − 1 10 1 10 3 D4 | | F D 4 | − | F MBD − NL | | | F MBD − NL | 20 40 60 | F MBD − NL | ( me V / Å ) Fig. 4. Comparison of MBD-ML and DFT-D p er f orm anc e on predicting vdW force con tributions compared to MBD- NL. Polar representation of atomic force deviati on s computed from the MBD-ML ( a ), D3 ( b ) and D4 metho d ( c ). The radial co ordinate sp ecifies the relative magn i t ud e deviation of the atomic force vector to the MBD-NL reference in p ercent. Th e angular co ordinate corresp onds to the angular deviation from the reference in degrees. The p oin t colour enco des the absolute magnitu de of the MBD-NL atomic force As a guide to the ey e the radial 100% mark is highlighted in red. MBD-ML FOR CES FOR V AR YING MOLECULE SIZES Ha ving v alidated MBD-ML on fixed chemical environ- men ts, we n ow systematically examine whet he r its force accuracy degrades with inc re asi n g molecular size, and b enc hmark it against the widely used pairwise D3 and D4 corrections. W e selected 825 molecule s from th e OMol25 dataset [ 9 ] with sizes uniformly distributed from 3 to 350 atoms. T o av oid art i f act s, only uncharged, spin- non-p olarized molecules w ere included, excluding sys- tems containing alk ali or alk aline earth elements (p erfor- mance including these rare-in-data elements is rep orted in Section S5). Atomic forces were computed at the PBE0+MBD-NL le vel and compared to predic t ion s from MBD-ML, D 3 [ 14 , 48 ], and D4 [ 49 ]. Fig. 4 shows magni- tude and angu l ar deviati ons relati ve to MBD-NL forces, color-co ded by the magnitude of MBD-NL force vectors. An in-depth statistical analysis app ears in Fig. S12 and T able S2. MBD-ML forces show excellent agreemen t with MBD- NL r ef er en ce s, consistent with QCML and DES370k re- sults. The med i an and mean magnitude devi at i on s are 2–3% wi th a 95 th p ercen tile of 7 . 7%. Angular deviations a verage < 2 ◦ with a 95 th p ercen tile of 5 . 1 ◦ . While Fig. 4 sho ws a few MBD-ML outliers with angular deviations exceeding 150 ◦ , these o ccur exclusiv ely when reference forces are ne arl y zero (deep purple) , maki n g such devia- tions practicall y irrelev an t. In contrast, D3 and D4 are strikingly differen t, with deviations in magnitude and angle b eing approximately ten times larger t h an for MBD-ML – 28 . 2% and 17 . 4 ◦ for D3 and 39 . 5% and 19 . 8 ◦ for D4, resp ectively . This is consistent with previously rep orted disagreements b e- t ween MBD and pairwis e metho ds for atomi c chains [ 50 ], non-co v alen t dimers [ 51 ], and p olymers [ 52 ]. This quali- tative difference also manifests itself in ext r em e outliers (force magnitude deviations ex ce ed in g 100%). D3 pro- duces 3066 such outli e rs (2 . 4% of all atoms) and D4 pro- duces 4025 (3 . 2%) , compared to only 8 atoms (0 . 006%) for MBD-ML. In terestingly , D4 shows larger deviations from MBD- NL for b oth force magnit ud es and angles than D3. The p olar representation in Fig. 4 reveals an imp ortan t quali- tative d iff er en ce : while b oth metho ds show substantially broader deviation distribut i ons than MBD-ML, in the case of D4 large angular dev iat ion s exceeding 65 ◦ pre- dominan tly o ccur for very small force magnitudes (deep purple markers). In con trast, D3 forces ex hi b it sizeable angular deviations for a h igh er fract i on of large vdW forces (green markers) as well. This distinction is criti- cal, as deviations in larger forces are more consequential for practical applications. These res ul t s demonstrate that MBD-ML closely re- pro duces the MBD-NL reference across diverse molecular sizes, while emphas i zi ng the imp ortance of many-bo dy effects b ey ond pairwise approximations. The substantial deviations of DFT-D metho ds from MBD-NL, particu- larly for lar ger forces where accuracy is most critical, un- derscore the imp ortance of many-b ody di sp ersion effec t s that pairwise approaches fundamentally cannot captur e. 8 10 − 4 10 − 3 10 − 2 10 − 1 10 0 10 1 R MS D / Max . d i sp . ( Å ) PBE PBE+MBD-ML Max. disp. Not converged I II I II I II III I II III I II I II I II − 10 0 0 10 0 10 1 10 2 ∆ V ( %) aspirin maleic acid ANP maleic hydrazide pentacene coronene o-diiodobenzene Fig. 5. Structural comparison of p olymorphs re- laxed with PBE and PBE+MBD-ML compared to th e PBE+MBD-NL relaxed structures: (top) RMSD and maxim um atomic disp l ace m ent and (b ottom) ratio of the unit cell volume of the relaxed struct ur es with resp ect to the PBE+MBD-NL-relaxed str uc t ur e. Blue crosses rep- resen t cases where the PBE geometry optimi z at ion did not conv erge. The RMSD and volume ratio is shown f or ev ery p olymorph of each molecular crystal. ANP refers to 2-amino-5-nitropyrimidine. Numerical v alues are tab- ulated in T able S4. MOLECULAR CR YST AL STR UCTURE AND ST ABILITY Predicting the relative stability of molecu l ar crystal p olymorphs is among the most demanding practical tests for a disp ersion method, as energy difference s b etw een forms often fall b elow 1 k J /mol p er molecule. T o assess MBD-ML in this context, we optimized geometries and computed energy ran k in gs at the DFT+MBD-ML level of theory for a set of crystals spanning h ydrogen-b onded, v an der W aals, and mixed interaction t yp es (see T able S3 and Section S8). T ABLE I I. P olymorph energy differ en ce s (kJ/mol p er molecule) from PBE 0+ MB D -NL (∆ E MBD − NL ) and PBE0+MBD-ML (∆ E MBD − ML ). Roman numerals indi- cate the polymorphi c forms b etw een which the energy difference is computed (e.g., I → I I denotes E II − E I ). Systems are group ed by d omi n ant i ntermolecular inter- action. ANP refers to 2-amino-5-nitropyrimidine. In- correct rankings likely due to in comp l et e cancellation of total energy errors are in b old. a Structures were taken from Ref. 54 . Molecule ∆ E MBD − NL (kJ / mol) ∆ E MBD − ML (kJ / mol) Mixe d Inter actions Aspirin I → I I -0.262 -0.291 Hydr o gen Bonde d Maleic acid I → I I 0.670 0.467 ANP I → I I 0.183 -0.292 I → I I I 0.237 0.261 Maleic hydrazide I → I I 0.974 0.870 I → I I I -0.384 0.220 van der Waals P entacene I → I I -0.476 -0.622 Coronene a I → I I 0.272 0.057 o -Diio dob enzene I → I I 0.029 0.015 Geometry optimization W e first assess the quality of the optimized structures b efore ev aluating their energy rankings. PBE+MBD- ML closely repro duces PBE+MBD-NL [ 53 ] crystal struc- tures, with RMSDs of atomic p ositions ranging from 0 . 001 ˚ A to 0 . 05 ˚ A for most structures (Fig. 5 ). Un it cell volumes differ by l ess than 1% from the ab ini- tio reference, demonstrating that MBD-ML forces and stresses are sufficiently accurate to yield vi rt u al ly iden- tical equilibrium geometries. These small deviations are within typical uncertainties of e xp erimental crystal structures and DFT geometry optimizations, confirming MBD-ML’s reliability for structural pr ed i ct i ons . In con- trast, uncorrected PBE substantially ov erestimates unit cell volumes by ov er 20% due to missing attractive dis- p ersion interactions, with RMSDs one to tw o orders of magnitude higher than MBD-ML (Fig. 5 ). This com par - ison underscores the nece ssi ty of correctly accounting for disp ersion interactions in molecular cry s t als . Energy ranking W e ev aluate MBD-ML’s reliability in predicting the energetic ordering of p olymorphs follo wing the estab- lished proto col by Ho ja et al. [ 4 ]. Energie s of st r u c- tures optimized at the PBE+MBD-NL and PBE+MBD- 9 ML levels w ere recompute d using P BE 0+M BD - NL and PBE0+MBD-ML, resp ectiv ely , to determine lattice en- ergy differences. Since MBD-ML is designed as a cost- effective replacement for ab initio MBD-NL, we use PBE0+MBD-NL as our reference rather than exp erimen- tal data, whic h would require free energy calculations with vibrational entropic contributions b eyond the scop e of this work. F or seven of nine p olymorph transitions studied, MBD- ML correctly predicts the energy ranking with errors b e- lo w 0 . 3 kJ/mol p er molecule (T able I I ). These s ys tems span diverse interaction types – v an der W aals, h ydro- gen b onding and mixed, demonstrating MBD-ML’s ro- bustness across c hemical en vironments. Given that typ- ical p olymorph energy differences are on the order of 0 − 7 kJ / mol [ 55 ], the MBD-ML e rr or s are w ell within the range required for reliable p olymorph screening. Tw o cases exhibit larger errors resulting i n in- correct rankings: the I → I I transition of 2-amino-5- nitropyrimidine (0 . 5 kJ/mol p er molecule) and the I → I I I transition of maleic h ydrazide (0 . 6 kJ/mol p er mol ec ul e) . These errors are, how ever, still very subtle and likely a consequence of the cancellation of total energy errors as previously discussed for molecular crystals of the OM C25 data set. LIMIT A TIONS OF THE MODEL So far, the MBD-ML mo del has b een sho wn to b e an accurate and reliable disp ersion metho d for diverse sy s- tems, including organic and inorganic molecules of widely v arying sizes, molecular dimers, and organic cr y st al s. Tw o limitations w arrant attention, how ev er. Molecular anions with un b ound electrons can yield highl y unphys- ical predicted a r 0 and C r 6 ratios, though this re fle c t s the pathological electronic st ru ct u r e of such systems rather than a deficiency of the mo del itself. More substantiv ely , the mo del requires further developmen t for systems con- taining alk ali and alk aline eart h elemen ts and for inor- ganic mat er i als . Whil e the former was discussed in the v alidation results ab ov e, we examine the p erformance of MBD-ML for inorganic materials in more detail here. In particular, signific antly larger a r 0 and C r 6 errors are observ ed on a random subset of 203 materials from the OMat24 dataset [ 8 ], recomputed at the HSE06+M BD - NL level of theory . As shown in T able I , the mean abso- lute errors of 0.247 and 0.269 for these ratios are 10–20 times higher t han for all other chemical classes examined in this study . These errors are sufficiently large t o intro- duce false negative MBD e i genv alues, preven ting calcu- lation of MBD energies and f orc es for most materials in the OMat24 subset. This inadequate p erformance is likely due to the mis- matc h b etw een the complex and diverse atomic environ- men ts in inorganic materials and those represented in 10 1 10 2 10 3 10 4 Number of atoms 10 − 2 10 0 10 2 10 4 Execution time(s) N 3 N 1 . 6 Ratio calculation (MBD-ML) E/F calculation (libMBD) Fig. 6. Computational scaling of the calculation of the a r 0 and C r 6 ratio calculation by the MBD -M L mo del and of the MBD prop ert y calculation via libMBD. the QCML train i ng dataset. By construction, QCML molecules, derived exc l us ively from organic molecule databases, contain at most 8 non-hydrogen atoms [ 38 ], whereas inor gan i c solids consist almost exclusiv ely of non-h ydrogen atoms in extended p erio d i c environmen ts, resulting i n minimal o verlap b et ween the atomic envi- ronmen ts present in the tw o datasets. Crystalline silicon offers a simple illustration. Each silicon atom has tetra- hedral c oordination with four neigh b ours, yet any QCML molecule is highly unlikely to contain five silicon atoms in a comparable co ordination environmen t. Impro ving MBD-ML p erformance in inorganic solids and syst ems containing alk ali and alk aline earth me t al s requires incl ud i ng representativ e subs e t s of such systems in the training data, wh i ch will be pursu ed i n fut ure work. COMPUT A TIO NAL SCALING Alongside accuracy , computational efficiency is a key requirement for an y metho d intended for large- sc ale ap- plications. Although the formal scaling of MBD with system size is cub i c, its cost in practice remains a small fraction of a single DFT self-consistency cycle. Until no w, how ev er, obtaining the a r 0 and C r 6 ratios required a full DFT cal cu l at ion , mak i ng this the true computa- tional b ottleneck. MB D -M L remov es this b ottlenec k, re- ducing the cost of a complete MBD calculation to that of a neur al netw ork inference follow ed by a matrix diag- onalization. T o quantify the resulting p erformance, we c haracterize the scaling of MBD-ML and libMBD as a function of system size. W e computed MBD corrections to t otal energies and 10 forces for water clusters (H 2 O) n with n = 3 − 4321 (up to nearly 13,000 atoms) serially on a single 128-core no de (AMD EPYC Rome); the results are summari ze d in Fig. 6 . F or systems up to 1000 atoms, t he MBD-ML cal- culation time for a r 0 and C r 6 remains approximately con- stan t at only a few seconds. The subsequent MBD energy and force calculations exhibit the theoretically predi ct ed cubic scaling b eyond 60 atoms, and are faster than the ra- tio calculations up to a crossov er p oin t at 250 atoms. F or clusters exceeding 1000 atoms, t h e MBD-ML execution time grows with an approximate p o wer of 1.6, reaching a maximum of 196 seconds for the (H 2 O) 4321 cluster. This analysis demonstrates that integrating MBD-ML in to the libMBD infrastructure enables MBD calculations of unprecedented scale that w ould otherwise b e imprac- tical with state-of-the-art DF As. SUMMAR Y AND CONCLUSIONS W e ha ve develop ed MBD-ML, a pretrained machine learning mo del that serv es as an accurate and efficient replacement for ab initio MBD-NL calculations. By di- rectly predicting p olarizabilit y α r 0 and C r 6 ratios from atomic co ordinates, MBD-ML enables computation of MBD energies, forces, and stresses without resorti n g to an y electronic structure calculations. In tegration with libMBD reduces the entire workflo w to a single func t ion call in the pymbd interface, facilitating incorp oration of MBD-ML into a wide range of at omi st i c simulation work- flo ws, including those built on the Atomic Simulation En vironment ( ASE ) [ 56 ]. W e hav e demonstrated the reliabilit y , accuracy , and transferability of MBD-ML across div erse systems: sin- gle molecules of widely v arying sizes and chemical com- p ositions, molecular dimers, and organic crystals. Com- pared t o ab initio MBD-NL, MBD-ML repro duces ener- gies, forces, and stresses with errors b elow 1 meV/atom, 1 meV/ ˚ A, and 0 . 2 meV/ ˚ A 3 , resp ectively . Molecular crys- tal geometry optimizations further confirm this p erfor- mance, with structural RMSDs of 10 − 3 − 10 − 2 ˚ A and p olymorph energy ranking errors of at most 0 . 6 kJ / mol. This developmen t addresses a practical barrier to the wider adoption of man y-b o dy d is p ersion metho ds, namely that the accuracy of MBD-NL has until no w come at the cost of requiring a full electronic structure calcula- tion. E ar l y implementations in co des such as V ASP and Quan tum Espresso suffered from computational inef- ficiencies [ 35 ]; the libMBD library [ 35 ] largely resolved these, and MBD-ML no w remov es the remaining barrier b y decoupling many-bo dy disp ersion calculations from an y underlying electronic structure co de. Beyond facil- itating MBD-NL adoption in quantum chemistry co des, MBD-ML also enables more accurate disp ersion treat- men ts in machine learning force fields [ 12 ]. Sev eral limitations hav e b een identified for future de - v elopment. The mo del’s applicability to in organ i c solids and systems containing alk ali or alk aline earth metals is currently l i mi t ed by under-representation of these sys- tems in the training data, and can in principle b e im- pro ved by dataset expansion and fine-tuning. The treat- men t of molecular anions p oses a more fundamental c hal- lenge, as accurately describing the electronic structure of negatively charged molecules is a well-kno wn difficulty that, as demonstrated in this w ork, is further exacerbated within t h e MBD formalism. Generating reliable train- ing data for such systems will require additional w ork to iden tify suitable basis set sett i ngs and density function al appro ximations. CODE A V AILABILITY The MB D -M L mo del has been integrated into the Python interface of the libMBD library , pymbd ( https: //github.com/em819/libmbd.git ). All ab initio elec- tronic structure calcul at i on s ha ve b een p erformed with FHI-aims . F or the geometry optimizations, the Atomic Sim ulation Environmen t (ASE) interface [ 56 ] to FHI- aims w as employ ed. RMSDs were computed u si n g the pymatgen [ 57 ] pack age. A CKNOWLEDGEMENTS All calculations were p erformed on t h e Luxembourg national sup ercomputer MeluXina. FUNDING E.M., A.K., A.Kh., and A.T. ackno wledge th e Euro- p ean Research Council under ERC-AdG grant FITMOL (101054629). A.T. ackno wledge the Luxem b ourg Na- tional Research F und under gran t FNR-CORE MBD-in- BMD (18093472). A.K. ackno wledges financial supp ort from the Luxembourg National Research F und (FNR AFR Ph.D. Grant 15720828). CONTRIBUTIONS A.T. conceiv ed and sup ervised the pro ject. A.K. trained the ML mo del. E.M and A.Kh. integrated the MBD-ML mo del into the libMBD library . E.M. p er- formed all calculations and data analysis of the main results and drafted the manuscript with input from all authors. A.Kh. analysed the challenges inv olving m ol ec- ular anions. E.M. and A.K. created the figures. All au- thors discussed the results and contributed to editing the 11 man uscript. ∗ alexandre.tk atc henko@uni.lu [1] J. Herm ann, R. A. DiSt asio Jr, and A. Tk atchenk o, First-principles mo dels for v an der waals interactions in molecules and materials: Concepts, theory , and applica- tions, Chem. Rev. 117 , 4714 (2017) . [2] M. St¨ ohr, T. V an V o orhis, and A. Tk atchenk o, Theory and practice of mo deling v an der waals interactions in electronic-structure calc ulations, Chem. So c. Rev. 48 , 4118 (2019 ) . [3] A. M. Reilly and A. Tk atc henko, Role of dispersion in ter- actions in the polymorphism and en tropic stabilization of the aspirin crystal, Phys. Rev. Lett. 113 , 055701 (2014) . [4] J. Ho ja, H.-Y. Ko, M. A. N eumann, R. Car, R. A . DiS- tasio Jr, and A. Tk atchenk o, Reliable and practical com- putational description of mo lecular crystal p olymorphs, Sci. Adv. 5 , eaau3338 (2019) . [5] M. St¨ ohr and A. Tk atchenk o, Quantum mechanics of pro- teins in explic it water: The role of plasmon-like solute - solven t interactions, Sci. Adv. 5 , eaax0024 (2019) . [6] T. Bj¨ orkman, A. Gulan s, A. Krasheninniko v, and R. Nieminen, Are w e v an der w aals ready?, J. Ph ys. Con- dens. Matter. 24 , 424218 (2012) . [7] S. A. T awfik, T. Gould, C. Stampfl, and M. J. F ord , Ev al- uation of v an der waals density functionals for lay ered materials, Phys. Rev. Mater. 2 , 034005 (2018) . [8] L. Barroso-Luque, M. Sh uaibi, X. F u, B. M. W o o d, M. Dzamba, M. Gao, A. Rizvi, C. L. Zitnick, and Z. W. Ulissi, Op en materia ls 2024 (OMat24 ) inorganic materi - als dataset and mo dels, arXiv preprint (2024) . [9] D. S. Levine, M. Shuaibi, E. W. C. Sp otte-Smith, M. G. T aylor, M. R. Ha syim, K. Michel, I. Batatia, G. Cs´ anyi, M. Dzamba, P . Eastman, et al. , The op en molecules 2025 (OMol25) d ataset, ev aluations, and mo dels, arXiv preprint arXiv:2505.08762 (2025) . [10] R. J. Maurer, C. F reysoldt, A. M. Reilly , J. G. Bran- denburg, O. T. Hofmann, T. Bj¨ orkman, S. Leb` egue, and A. Tk atchenko, Adv ances in d ensit y-functional calcula- tions for materials modeling , Annu. Rev. Mater. Res. 49 , 1 (2019) . [11] O. T. Unke, S. Chmiela, H. E. Sauceda, M. G astegger, I. P oltavsky , K. T. Sch¨ utt, A. Tk atchenk o, and K.-R. M ¨ uller, Machine learning force fields, Chem. Rev. 121 , 10142 (202 1) . [12] A. Kab ylda, J. T. F rank , S. Su´ arez-Dou, A. Khabibrakhmanov, L. Medrano Sandonas, O. T. Unke, S. Chmiela, K.-R. M ¨ uller, and A. Tk atchenk o, Molecular simulations with a pretrained neura l netw ork and universal pairwise force fields, J. Am. Chem. So c. 147 , 33723 (2025) . [13] S. Grimme, A. Hansen, J. G. Brandenburg, and C. Ban- n warth, Disp ersion-corrected mean-field electronic struc- ture metho ds, Chem. Rev. 116 , 5105 (2016) . [14] S. Grimme, J. Anton y , S. Ehrlich, and H. Krieg, A con- sistent an d accurate ab initio parametrization of density functional disp ersion correction (DFT- D) for the 94 ele- ments H-Pu, J. Chem. Phys. 132 , 15 4104 (2010) . [15] A. Tk atchenk o and M. Scheffler, Acc urate molecular v an der waals interactions from ground-state electron den- sity and free-atom reference data, Phys. Rev. Lett. 102 , 073005 (20 09) . [16] N. F erri, R. A. DiStasio Jr, A. Ambrosetti, R. Car, and A. Tk atchenk o, Electronic prop erties of molecules and surfaces with a self-consistent interatomic v an der waals density functi onal, Phys. Rev. Lett. 114 , 176802 (2015 ) . [17] A. D. Beck e and E. R. John son, Exchange-hole d ipole moment and the disp ersion interaction revisited, J. Chem. Phys. 127 , 154108 (2007) . [18] E. R. Johnson, Ch apter 5 - the exchange-hole dip ole mo- ment disp ersion mo del, in Non-Covalent Interactions in Quantum Chemistry and Phys ics , edited by A. Otero de la Roza and G. A. DiLabio (Elsevier, 2017) pp. 169–194. [19] A. Tk atchenk o, R. A. DiStasi o Jr, R. Car, a nd M. Schef- fler, Accurate and efficient metho d for many-bo dy v an der waals interactions, Phys. Rev. Let t. 10 8 , 23 6402 (2012) . [20] A. Tk atc henko, A. Ambrosetti, and R. A. DiStasio, In- teratomic metho ds for the di spersion energy derived from the adiabatic connection fluctuation-dissipa tion theorem, J. Chem. Phys. 138 , 074106 (2013) . [21] A. Ambrosetti, A. M. Reilly , R. A. DiStasio, and A. Tk atc henko, Long-range correlation energy calculated from coupled atomic resp onse functions, J. Chem. Phys. 140 , 18A508 (2014) . [22] R. A. DiStasio, V. V. Gobre, and A. Tk atchenk o, Man y- b ody v an der waals interactions in molecules and con- densed matter, J. Phys. Condens. Matter 26 , 213202 (2014) . [23] J. Hermann , D. Alfe, and A. Tk atc henko, Nanoscale π – π stack ed molecules are b ound by collective charge fluctu- ations, Nat . Commun. 8 , 1 4052 (2017) . [24] M. Kim, W. J. Kim, T. Gould, E. K. Lee, S. Leb ` egue, and H. Kim, umbd: A materials-ready disp ersion correction that uniformly treats metallic, ionic, and v an der waals b onding, J. Am. Chem. So c. 142 , 2346 (2020) . [25] J. Hermann and A. Tk atchenk o, Density functional mo del for v an der waals interactions: Unifying many- b ody atomic approaches with nonlocal functionals, Phys. Rev. Lett. 124 , 146401 (2020) . [26] T. Gould, S. Leb egue, J. G. An gy an, and T. Buˇ cko, A fractionally ionic approach to p olarizabilit y and v an der w aals many-bo dy disp ersion calculations, J. Chem. The- ory Comput. 12 , 5920 (2016) . [27] A. Khabibrakhmanov, M. Gori, C. M¨ uller, and A. Tk atchenko, Nonco v alent in teractions in density func- tional the ory: All the charge density w e do not see, J. Am. Chem. So c. 147 , 40763 (2025) . [28] P . P . Paul, V. S. Thakur, and S. Ban erjee, DFT-D3 for solid-state materials: A data-driven persp ective on accu- racy , Adv. Theory Simul. 9 , e01072 (202 5) . [29] R. Sedlak and J. Rezac, Empirical D3 disp ersion as a re- placement for ab initio disp ersion terms in density func- tional theory-based symmetry-adapted p erturbation the- ory , J. Chem. Theory Comput. 13 , 1638 ( 2017) . [30] J. Propp e, S . Gugler, and M. Reiher, Gaussian pro cess- based refinement of disp ersion correct ions, J. Chem. The- ory Comput. 15 , 6046 (2019) . [31] E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Ban n w arth, and S. Grimme, A gener- ally ap plicable atomic-charge dep endent london disp er- sion correctio n, J. Chem. Phys. 150 , 154122 (2019) . 12 [32] J. Witte, N. Mardirossian, J. B. Neaton, and M. Head- Gordon, Assessing DFT-D3 damping functions across widely used density functionals: Can we do b etter?, J. Chem. Theory Comput. 13 , 2043 (2017) . [33] N. V. Tk achenk o and M. Head-Gordon, Smo other semi- classical disp ersion for density functional theory via d3s: Understanding and addressing unphysical minima in the d3 d ispersion correction mo del, J. Che m. Theory Com- put. 20 , 9741 (2024) . [34] L. Briccolani-Bandini, B. Gnnanap areddy , F. Labat, E. Br´ emond, J. C. Sancho-Garc ´ ıa, A. Otero-de-la Roza, G. Scalmani, M. J. F risch, G. Card ini, G. A. DiLabio, et al. , W eak noncov alent interactions in nonequilibrium structures: How go od are the disp ersion corrections?, J. Phys. Chem. Lett. 17 , 703 (2026) . [35] J. Hermann, M. St¨ oh r, S. G´ oger, S. Chaudhuri, B. Aradi, R. J. Maurer, and A. Tk atchenk o, libmbd: A general- purp ose pack age for scalable qu an tum many-bo dy d is- p ersion calculations, J. Chem. Phys. 159 , 174802 (2023) . [36] P . P . Poier, T. Jaffrelot Inizan, O. Adjoua, L. La- gardere, and J.-P . Piquemal, Accurate deep learning- aided den sit y-free strategy for man y-b ody disp ersion- corrected density functional theory , J. Phys. Chem. Lett. 13 , 4381 (2022) . [37] K. R. Bry enton and E. R. Johnson, Many-bo dy dis- p ersion in mo del systems and t he sensitivity of self- consistent screening, J. Chem. Ph ys. 158 , 204110 (2023) . [38] S. Gansc ha, O. T. Unk e, D. Ahlin , H. Maennel, S. Kash u - bin, and K.-R. M ¨ uller, The QCML dataset, quantum c hemistry reference data from 33.5 M DFT and 14.7 B semi-empirical c alculations, Sci. Data 12 , 406 (2025) . [39] A. G. Donchev, A. G. T aub e, E. Decolvenaere, C. Har- gus, R. T. M cGibbon, K.-H. Law, B . A. Gregersen, J.-L. Li, K. Palmo, K. Siv a, et a l. , Quantum chemical b ench- mark databases of gold-standard dimer interaction ener- gies, Sci. Data 8 , 55 (2021) . [40] V. Gharakhany an, L. B arroso-Luque, Y. Y ang, M. Sh uaibi, K. Michel, D. S. Levine, M. Dzamba, X. F u, M. Ga o, X. Liu, et al. , Op en molecular crystals 2025 (OMC25 ) dataset and mo dels, Sci. Data (2026) . [41] A. P . Jones, J. Crain , V. P . Sokhan, T. W. Whitfield, an d G. J. M art yna, Quan tum drude oscillator model of atoms and molecules: Many-bo dy p olarization and disp ersion interactions for atomistic simulation, Phys. Rev. B 87 , 144103 (20 13) . [42] A. Khabibrakhmanov, D. V. F edorov, A. Ambrosetti, J. Crain, K. L. Hunt, E. R. Johnson, K. D. Jordan, S. G´ oger, M. Gori , M. R. Karimpour, et al. , Accu- rate noncov alent in teractions in atomistic systems via quantum drude oscillators, J. Chem. Phys. 163 , 055701 (2025) . [43] O. A. Vydrov and T. V an V o orhis, Disp ersion interac- tions fro m a lo cal p olarizability mo del, Phys. R ev. A. 81 , 062708 (2010) . [44] J. T. F rank, O. T. Unke, K.-R. M ¨ uller, and S. Chmiela, A euclidean transformer for fast an d stable mac hine learned force fields, Nat. Commun. 15 , 6539 (2024) . [45] C. J. N ic k erson, K. R. Bryen ton, A . J. Price, and E. R. Johnson, Compariso n of densi t y-functional theory dis- p ersion co rrections for the DES15K data base, J. Phys. Chem. A. 127 , 8712 (2023) . [46] H. Zhao, B. L˝ orincz, T. Henkes, D. Berta, P . Nagy , A. Tk atc henko, and S. V uc ko vic, Accurate density fu nc- tional theory for non-co v alen t in teract ions in c harged sys- tems, ChemRxiv (2025) . [47] A. Kabylda, S. Su´ arez-Dou, N. Dav oine, F. N. Br ¨ unig, and A. Tk atchenk o, Qcell: Comprehensive quantum- mechanical dataset spanning diverse biomolecular frag- ments, arXiv preprint arXiv:2510.0 9939 (2025) . [48] S. Grimme, S. Ehrlic h, and L. Go erigk, Effect of the damping function in disp ersion corrected density func - tional theo ry , J. Comp ut. Chem. 32 , 14 56 (2011) . [49] E. Caldeweyher, C. Bannw arth, and S. Grimme, Exten- sion of the d3 disp ersion co efficient mo del, J. Chem. Phys. 147 , 034112 (2017) . [50] P . Hauseux, A. Am brosetti, S. P . Bordas, and A. Tk atc henko, Colossal enhancement of atomic force re- sp onse in v an der w aals materials a rising from many- b ody electronic correlations, Phys. Rev. Lett. 128 , 106101 (20 22) . [51] M. Pulev a, L. Medrano Sandonas, B. D. L˝ orincz, J. Charry , D. M. Rogers, P . R. Nagy , and A. Tk a tc henko, Extending quantum-mec hanical b enchmark accuracy to biological ligand-p ock et interactions, Nat. Commun. 16 , 8583 (2025 ) . [52] R. I. Sosa, M. Ga lan te, and A . Tk atchenk o, Po wer of the many-bo dy force: M agnitudes and angles of atomic v an der waals disp ersion forces in extended molecular systems, Small Struct . 6 , 2500226 (2025) . [53] J. P . Perdew, K. Bu rk e, and M. Ernzerhof, Generalized gradient approximation mad e simple, Phys. Rev. Lett. 77 , 3865 (1996) . [54] J. Potticary , L. R. T erry , C. Bell, A. N. Papanik olopou- los, P . C. Christianen, H. Engelk amp, A. M. Collins, C. F ontanesi, G. Ko ciok-K¨ ohn, S. Crampin, et al. , An unforeseen p olymorph of coronene by the application of magnetic fields du ring crystal growth, Nat. Com m un. 7 , 11555 (201 6) . [55] J. Nyman and G. M. Day , Static and lattice vibration al energy differences betw een p olymorphs, CrystEngComm 17 , 5154 (2015) . [56] A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. F riis, M. N. Grov es, B. Hammer, C. Hargus, et al. , The atomic sim- ulation environmen t—a python library for working with atoms, J. Phys. Condens. Matter. 29 , 273002 (2017) . [57] S. P . Ong, W. D. Ric hards, A. Jain, G. Hautier, M. Ko cher, S. Ch olia, D. Gunter, V. L. Chevrier, K. A. P ersson, and G. Ceder, Python materials genomics (py- matgen): A robust, op en-source python library for ma- terials analys is, Comput. Mat er. Sci. 68 , 31 4 (2013) . Supplemen tary Information for: MBD-ML: Man y-b o dy disp ersion from mac hine learning for molecules and molecular crystals S1. MBD-ML TRAINING AND P ARAMETERS The MBD-ML mo del is based on the SO3LR [1] ar- c hitecture, with its long-range contributions disabled, so that the remaining mo del is equiv alent to SO3krates. F or the purp ose of th e M BD -M L mo del, the SO3LR architec- ture was extended to also predict C r 6 and α r 0 ratios. The training was conducted on the QCML data set for which a combined loss function including th ese ratios with equal w eighting was employ ed, using a robust loss function with α = 1. The learning rate w as adapted exp onen tially us- ing the AMSGr ad optimizer with a learning rate of 0 . 001 and a deca y rate of 0 . 9 applied every 1M steps. Com- pared to the original SO3LR mo del, MBD-ML was kept relatively ligh t weigh t, with a feature dimension of 64, 4 heads, a maximal degree of 4 an d 32 rad ial basis func- tions. Th us, with long-range contributions disabled and a short-range cutoff radius of 4 . 0 ˚ A, MBD-ML exhibits an effective receptive field of 8 . 0 ˚ A. S2. SELECTION OF DA T A SUBSETS DES370k W e used the dimer structures of the DES370k set, which w ere r ecom pu t ed on the PBE0+MBD-NL level of theory as part of the recently published QCel l biomolecular data set [2]. OMol25 F or the constructi on of the OMol25 sub set used in this stud y , the structures of t h e SPICE and the PDB protein p o c ket fragments s ub se t as the union of those tw o contains molecules ranging from 3 to 350 atoms. T o ac hieve a un i for m selection of system sizes, the molecules of t he se tw o sets were binned by the num- b er of atoms, using bins of size 10 (1 − 10 atoms, 11 − 20 etc.) and a total of 883 mol ecu l es w as randomly pick ed in a uniform fashion. OMC25 T o ensure a maximally div erse subset for the present work, each of the 200 molecular crystals that were pic ked featur e a unique molecule, i.e there are no tw o p olymorphs of the same mol e cu lar crystal. Other than that the structures w ere chosen randomly without addi- tional constraints. OMat24 Starting from the OMat24 v alidation set, 6 materials categories w ere defined based on the band gap and the num ber of atoms in the unit cell: 1. small-metal 2. small-insulator 3. intermediate-metal 4. intermediate-insulator 5. large-metal 6. large-insulator , where metal and insulator structures were defined b y the PBE ban d gap of the original OMat24 en try to b e less t h an 0 . 5 eV and ≥ 0 . 5 eV, resp ectiv ely , while small , intermediate and large structures were made up of at most 50 atoms, at most 100 atoms and o ver 100 atoms, resp ectiv ely . F or each of these 6 categories, up to 50 ma- terials were randomly selected and recomputed on the HSE06+MBD-NL level of theory , of which 203 calcula- tions w ere completed succesfully and used to b enc hmark the MBD-ML mo del in this work. S3. AB INITIO CALCULA TIONS All DFT re-calculations of the OMol25, DES370k, OMC25 and OMat24 data sets w ere p erformed using the FHI-aims electronic structure pack age [ 3, 4] (v ersion 231212.1). All molecules and molecular crystal struc- tures, including the subsets of the OMol25, DES370k and OMC25 data s et s were computed on the PBE0+MBD- NL level of theory , while the inorganic material struc- tures from the OMat24 data set were computed on the HSE06+MBD-NL level. F or the conv ergence of the SCF - cycle the default accuracy settings in com bination with default tight basis sets were used. Relativistic effects w ere included using the scalar atomic ZORA me t ho d. F or b oth hybrid functionals a MBD damping parame- ter β of 0 . 83 was used. F or all molecular crystals, k- meshes with a spacing of 0 . 03 ˚ A − 1 w ere employ ed after finding that for a random subset of 50 OMC25 molec- ular crystal structure s this yielded forces con v erged to b elo w 1 meV/ ˚ A. F or the inorganic mat er i al s, the k-mesh w as chosen based on the PBE band gap provided i n the original OMat24 data set. F or materials with a PBE band gap of less than 0 . 5 eV, a very fine k-mesh spac- ing of 0 . 0125 ˚ A − 1 w as employ ed, while materials with larger ban d gaps were recomputed on the HSE06+MBD- NL level of theory w it h a coarser k-spacing of 0 . 02 ˚ A − 1 w as used. 2 S4. EX CLUSION OF ANIONS FR OM QCML S4.1. Basis set conv ergence for anions F or the QCML subset of n egat i vely charged molecules, w e iden tified outliers in the ratios α r 0 and C r 6 in the orig- inal data. These out l ie r s corresp ond to atoms with un- usually large v alues, reaching 20–60, whereas b oth ra- tios typically lie withi n the 0–2 range. In most cases, the anomalous v alues o ccur for h ydrogen atoms, which is particularly u ne x p ected. A detailed analysis of the underlying ab initio calcu- lations revealed that, for anions, α r 0 and C r 6 are h i ghly sensitive to the cut pot parameter in FHI-aims. This parameter defines the onset of a st e ep l y increasing con- fining p otential v cut ( r ) used to lo calize numerical atom- cen tered radial functions. F or more details on v cut ( r ) and its role in constructing basis functions, we refer the reader to Section 3 of the original FHI-aims pap er [3]. In practical terms, cut pot , together with t h e width parameter w (kept fixed here), determines the spatial s up- p ort of a basi s function. The default tight settings for organic elements ( cut pot = 4 ˚ A, w = 2 ˚ A) yield a total radial extent of 6 ˚ A, which is generally sufficient to ob- tain meV-accurate total energies. How ever, the accurate description of diffuse electron density in anions requires explicit conv ergence tests with resp ect to cut pot . W e randomly selected several an ion s exh i bit i ng anoma- lous ratios and p erformed calculations v ar y in g cut pot b et w een 3 and 8 ˚ A, w hil e keeping all other computational parameters consistent with QCML settings [5]. The total energies of the anions sho w stron g sensitivity to cut pot , v arying by up to ± 1 . 5 eV relative to the default 4 ˚ A set- ting (F i gure S1a). Conv ergence is slow, ind ic at in g signifi- can t contributions from slowly decaying density tails. By con trast, neutral molec ul es of comparable size (b enzene, p en tane) exhi b it v ariations of only ab out 2 meV betw een cut pot = 4 ˚ A and 8 ˚ A. Fig. S1. Con vergence of ( a ) total energies and ( b ) MBD-NL disp ersion energies with resp ect to the cut pot parameter for tw o example anions. T otal en er gi es are plotted as the differences relative to the default 4 ˚ A set- ting (marked by a vertical dashed line).. The corresp onding MBD-NL disp ersion energies are lik ewise uncon verged and increase unphysically as the ba- sis b ecomes more extended (Figure S 1b) . This b ehavior is further illustrated b y plotting α r 0 and C r 6 for hydrogen atoms versus cut pot , whic h reveals a dramati c increase in b oth ratios with i n cr eas i ng basis extent (Figu r e S2). Hydrogen at oms are emphasized b ecause they are t ypi- cally lo cated at the molecul ar p eriphery and thus most sensitive to density tails, although similar trends are ob- serv ed for some heavier atoms. Ph ysically , this divergence arises b ecause a more ex- tended basis perm it s slow er deca y of the electron densit y , and the VV p ol ar i zab il i ty functional is highly sensitive to density gradients i n the tail region [6]. The p erformance of VV an d MBD-NL for anions was not assessed in the original work, and this issue has not previ ou sl y b een re- p orted. F urther analysis shows that the problematic sp ecies are actually thermo dynamically unstable, as indicated b y their comput ed electron affinities (T able S1). F or an N -electron syst e m, the electron affinity is defined as EA( N ) = E ( N ) − E ( N + 1), wit h p ositive v alues 3 indicating stability of the ( N + 1)-electron state. W e ev aluated EAs b y sequentially removing electrons from the c harge states present in QCML. Calculations e m- plo yed the long-range corrected LC- ω PBEh functional ( ω = 0 . 2 Bohr − 1 ) [7] as implemented in Q-Chem [ 8] , to- gether with aug-cc-pVQZ basis sets to ens ur e adequate diffuseness [9]. As summarized in T able S1, only singly charged an- ions are stable in all four cases; hi gh er charge states are clearly unstable. Consequen tly , suc h pathological en tries m ust b e recomputed with physically meaning- ful charge states to form a reliable subset of negatively c harged molecules within QCML. T o mitigate similar is- sues in future datasets, additional screening could b e applied, for example using HOMO energies as a pro xy for stability . As shown in T able S1, HOMO v alues ob- tained with a long-range corrected functional cor r el at e w ell with EAs, consistent with the ionization p otential theorem applied to the ( N + 1)-electron system, wh ich yields EA( N ) = IP( N + 1) = − HOMO ( N + 1) for the exact functional [10]. A more compr eh en si ve inv estigation, including system- atic b enc hmarking of basis sets and exchange–correlation functionals, is warran ted but lies b eyond the scop e of the present work. Until such v alidation and filtering are p erformed, w e do not recommend using QCML data for negatively c harged molecules, particularly di sp ersion en- ergies and derived ratios. T ABLE S1. T otal energies, electron affinities and HOMO levels (all in eV) for v arious charged state s of the four considered molecules. The first row in each case corresp onds to the charged state included in QCML.. C 4 H 7 C 2 NH 4 Q E tot EA HOMO Q E tot EA HOMO -2 -4252.28 -2.81 2.79 -2 -3621.08 -3.36 3.29 -1 -4255.09 0.96 -1.00 -1 -3624.44 1.61 -1.58 0 -4254.13 -9.15 0 -3622.84 -9.07 C 5 N 2 H 9 C 6 O 2 H 9 Q E tot EA HOMO Q E tot EA HOMO -3 -8290.99 -4.16 4.76 -3 -10441.5 -4.74 4.75 -2 -8295.15 -2.86 2.42 -2 -10446.2 -2.62 2.71 -1 -8298.02 1 .67 -1.72 -1 -10448.9 1.3 0 -1.82 0 -8296.35 -7.04 0 -10447.6 -8.77 4 Fig. S2. Boxplots of α r 0 (left) and C r 6 (right) with v arying cut pot for hydrogen at oms in the tw o negatively charged molecules: ( a ) [C 4 H 7 ] 2 − and ( b ) [C 5 N 2 H 9 ] 3 − (the structures shown in the insets). The inset in the upp er left panel illustrates t he α r 0 ratios in p entane molecu l e for cut pot = 4 ˚ A and 8 ˚ A.. Fig. S3. The same as Figure S2 but for ( a ) [C 2 NH 4 ] 2 − and ( b ) [C 6 O 2 H 9 ] 3 − (the structur es shown in the insets ). . 5 S4.2. P erformance of MBD-ML with anions included If these pathological anions from the QC ML data set are included in the accuracy meas ur em ent of the MBD- ML mo del, one finds numerous outliers in the MBD ratio prediction (see Figures S4) , which is plausible due to the insufficient computational settings for negativ ely charged molecules outlin ed in the previous subsection. (a) 0 20 40 MBD -NL C r 6 0 10 20 30 40 MBD -ML C r 6 RMSE = 0.102 MAE = 0.019 Q < 0 ( 14. 3%) Q = 0 ( 57. 5%) Q > 0 ( 28. 2%) (b) 0 1 2 3 C r 6 ab s. e r r or 10 − 5 10 − 3 10 − 1 Relative Frequency Median = 0.011 Mean = 0.019 95th %ile = 0.054 Q < 0 Q = 0 Q > 0 (c) 0 5 10 MBD -NL α r 0 0.0 2.5 5.0 7.5 10.0 12.5 MBD -ML α r 0 RMSE = 0.067 MAE = 0.018 (d) 0 1 2 3 α r 0 ab s. e rr or 10 − 5 10 − 3 10 − 1 Relative Frequency Median = 0.011 Mean = 0.018 95th %ile = 0.049 Q < 0 Q = 0 Q > 0 Fig. S4. P erformance of PBE0+MBD-ML in predic ti n g the C 6 and α 0 ratios of the QCML test set including all negatively charged molecules. W e find, how ev er, that the resulti ng MBD energy and force contributions are basical l y unaffected from these outliers (see Figure S5). S5. MBD-ML VS MBD-NL PERFORMANCE F OR OMOL25 AND DE S370K SUBSETS While the main analysis of the MBD-ML p erformance on the OMol25 and DES370k subsets excluded alk ali and alk aline earth elements, here we additionally sho w the ac- curacy of the mo del if those elements are included relative to the ab ini tio MBD -NL refe re nc e. Figure S6 and S7 il- lustrate the p erformance of the MBD-ML mo del on the OMol25 subset with and without alk ali and alk ali earth elemen ts. Figure S8 and S9 do so analogously for the (a) -40 -20 0 E MBD − NL ( me V/at om) -40 -30 -20 -10 0 E MBD − ML ( me V/at om) RMSE = 0.286 MAE = 0.178 (b) 0 25 50 75 F MBD − NL ( me V/ Å /at om) 0 20 40 60 80 100 F MBD − ML ( me V/ Å /at om) RMSE = 0.477 MAE = 0.322 Q < 0 Q = 0 Q > 0 Fig. S5. P erformance of PBE0+MBD-ML in predic ti n g the MBD contribution to th e total energy and atomic forces of the QCML test set including all negat ively c harged molecules. (a) 0.5 1.0 1.5 MBD -NL C r 6 0.5 1.0 1.5 MBD -ML C r 6 RMSE = 0.029 MAE = 0.020 10 0 10 1 10 2 10 3 (b) 0.5 1.0 1.5 MBD -NL α r 0 0.5 1.0 1.5 2.0 MBD -ML α r 0 RMSE = 0.033 MAE = 0.022 10 0 10 1 10 2 10 3 (c) -40 -20 E MBD − NL ( me V/at om) -40 -30 -20 -10 E MBD − ML ( me V/at om) RMSE = 0.373 MAE = 0.252 10 0 10 1 (d) 25 50 75 F MBD − NL ( me V/ Å /at om) 50 100 150 F MBD − ML ( me V/ Å /at om) RMSE = 1.383 MAE = 0.654 10 0 10 1 10 2 10 3 Fig. S6. P erformance of PBE0+MBD-ML in predic ti n g the C 6 and α 0 ratios and the MBD contribution to the total energy and at om ic forces of the OMol25 test set including alk ali an d alk aline earth elements. DES370k test set. 6 (a) 0.5 1.0 1.5 MBD -NL C r 6 0.5 1.0 1.5 MBD -ML C r 6 RMSE = 0.030 MAE = 0.021 10 0 10 1 10 2 10 3 (b) 0.5 1.0 1.5 MBD -NL α r 0 0.5 1.0 1.5 2.0 MBD -ML α r 0 RMSE = 0.033 MAE = 0.023 10 0 10 1 10 2 10 3 (c) -40 -20 E MBD − NL ( me V/at om) -40 -30 -20 -10 E MBD − ML ( me V/at om) RMSE = 0.226 MAE = 0.198 10 0 10 1 (d) 25 50 F MBD − NL ( me V/ Å /at om) 20 40 60 F MBD − ML ( me V/ Å /at om) RMSE = 0.707 MAE = 0.564 10 0 10 1 10 2 10 3 Fig. S7. P erformance of PBE0+MBD-ML in predic ti n g the C 6 and α 0 ratios and the MBD contribution to the total energy and at om ic forces of the OMol25 test set excluding alk ali and alk aline earth elements. (a) 1 2 MBD -NL C r 6 2 4 6 8 10 MBD -ML C r 6 RMSE = 0.034 MAE = 0.015 10 0 10 1 10 2 10 3 10 4 10 5 (b) 1 2 MBD -NL α r 0 1 2 3 4 5 MBD -ML α r 0 RMSE = 0.037 MAE = 0.016 10 0 10 1 10 2 10 3 10 4 10 5 (c) -40 -20 E MBD − NL ( me V/at om) -150 -100 -50 E MBD − ML ( me V/at om) RMSE = 0.836 MAE = 0.204 10 0 10 1 10 2 10 3 10 4 (d) 100 200 F MBD − NL ( me V/ Å /at om) 100 200 300 F MBD − ML ( me V/ Å /at om) RMSE = 2.648 MAE = 0.461 10 0 10 2 10 4 10 6 Fig. S8. P erformance of PBE0+MBD-ML in predic ti n g the C 6 and α 0 ratios and the MBD contribution to the total energy and atomic forces of the DE S370k test set including alk ali an d alk aline earth elements. (a) 1 2 MBD -NL C r 6 1 2 3 MBD -ML C r 6 RMSE = 0.022 MAE = 0.013 10 0 10 1 10 2 10 3 10 4 10 5 (b) 1 2 MBD -NL α r 0 0.5 1.0 1.5 2.0 2.5 3.0 MBD -ML α r 0 RMSE = 0.025 MAE = 0.014 10 0 10 1 10 2 10 3 10 4 10 5 (c) -30 -20 -10 E MBD − NL ( me V/at om) -30 -20 -10 E MBD − ML ( me V/at om) RMSE = 0.165 MAE = 0.117 10 0 10 1 10 2 10 3 (d) 20 40 F MBD − NL ( me V/ Å /at om) 10 20 30 40 50 F MBD − ML ( me V/ Å /at om) RMSE = 0.387 MAE = 0.266 10 0 10 1 10 2 10 3 10 4 10 5 Fig. S9. P erformance of PBE0+MBD-ML in predic ti n g the C 6 and α 0 ratios and the MBD contribution to the total energy and atomic forces of the DE S370k test set excluding alk ali and alk aline earth elements. 7 S6. SENSITIVITY ANAL YSIS OF MBD-ML The present study demonstrates excellent accurac y of the MBD-ML mo del for nearly all target quantities, in- cluding relative ener gi es, atomic forces, and stress tensor comp onen ts. How ever, as noted in the main text and evident from Fig. 4, the total MBD energy predicted by MBD-ML can deviate from the ab initio reference b y more than 100 , meV for l ar ge systems containing ov er one hundred atoms. As shown in T able I II, this discrep- ancy becomes negligible when computing physically mea- surable energy differences. Nevertheless, understanding the origin of this residual disagree me nt is instructive. In principle, exact predicti on of the α r 0 and C r 6 ratios would eliminate this error entirely . T o assess the sensitivity of the MBD en er gy t o thes e inputs, we p erformed a sensitivit y analysis for p olymorph I of coronene. The MBD-ML predicted vdW energy for coronene I differs from the MBD-NL reference b y − 61 , meV ( − 0 . 85 , meV / atom), consistent with the MAE of the MBD-ML mo del on the OMC25 subset (T able I). W e first examined the impact of ran dom, non- systematic noise in α r 0 and C r 6 on the total MBD en- ergy . Starti ng from the PBE0+MBD-NL reference ratios of coronene I, uniform zero-mean noise with a v ariable range b etw een 0 . 0 and 0 . 04 was applied indep enden tly to b oth ratios. F or each noise level combination, the MBD- NL energy was recomputed 100 times, and the resulting standard deviation was recorded (Fig. S10). The cho- sen range reflects the a verage prediction errors for these ratios on the OMC25 subset (T able I). P erturbations within this range pro duce standard de- viations in the total MBD energy of up to ± 38 , meV , sub- stan tially sm al le r than the − 61 , meV deviation observed for MBD-ML on coronene I. Based on Fig. S10 and the rep orted MAEs, the non-sy st e matic contribution to the MBD energy error is estimated to b e ab out 20 , meV. The analysis also indicat e s that the MBD energy is slightly more sensitive to noise in C r 6 than in α r 0 , as evidenced by consistently larger deviat i ons in the low er-left region of the heat map, although the difference amounts to only a few meV. In addition to random errors, systematic bias m ust b e considered. F or the 200 molecular c ry s t als in the OMC25 dataset, the mean residuals (serving as a proxy for bias) in Fig. 4 are − 0 . 02 for α r 0 and − 0 . 01 for C r 6 . T o quantify the effect of such bias, the ratios for coronene I were indep enden tly shifted b y v alu es b etw een − 0 . 04 and 0 . 04 (Fig. S11). The resulting energy errors reveal a muc h stronger sensitiv i ty to systematic shifts, with deviations reac hing up to 700 , meV and − 778 , meV . Again, shifts in C r 6 exert a larger influence on the vdW energy than comparable shifts in α r 0 . In terestingly , energy errors are reduced when shifts in b oth ratios are of similar magn i t ude. P oints along the 0.0 0.01 0.02 0.03 0.04 Noi se i n α r 0 0.0 0.01 0.02 0.03 0.04 Noi se i n C r 6 0.00 5.67 10.81 14.55 23.80 6.83 9.60 11.79 18.22 24.27 14.15 16.65 19.53 20.68 26.70 23.88 24.99 23.87 27.80 32.28 28.32 31.46 29.11 38.72 35.38 0 10 20 30 S t d . d e v . E MBD − NL ( me V) Fig. S10. Sensit i v i ty of the MBD-NL energy to the in tro duction of uniform random noise to a r 0 and C r 6 . -0.04 -0.02 0.00 0.02 0.04 Bi as of α r 0 -0.04 -0.02 0.00 0.02 0.04 Bi as of C r 6 140 217 292 365 436 505 572 637 700 24 104 180 255 327 397 465 532 596 -90 -10 68 144 218 289 359 427 492 -205 -123 -44 34 109 182 253 322 389 -320 -236 -155 -76 0 74 147 217 285 -435 -349 -267 -187 -109 -33 40 112 182 -549 -462 -378 -297 -217 -140 -66 7 78 -664 -575 -490 -407 -326 -248 -172 -97 -25 -778 -688 -601 -517 -435 -355 -277 -202 -129 -750 -500 -250 0 250 500 E r r or of E MBD − NL ( me V) Fig. S11. Sensit i v i ty of the MBD-NL energy to the in tro duction of a systematic bias to a r 0 and C r 6 . diagonal of Fig. S11, where biases ar e balanced, exhibit smaller deviations. The observed biases of − 0 . 02 and − 0 . 01 ((T able I) corresp ond to an estimated MBD energy error of ab out − 44 , meV . Combining this systematic con- tribution with the estimated non-systematic u nc er t aint y yields a total error estimate of − 44 ± 20 , meV, consistent with the observed − 61 , meV deviation for coronene I. Bey ond rationalizing residual errors, this analysis pro- vides guidance on the accuracy requirements for future mo dels. Sensitivity estimates of this type can b e used to determine the level of precision in predicted vdW ratios necessary to achiev e a desired accu rac y in the resulting MBD energies. 8 S7. DET AILS ON FOR CE DEVIA TION DISTRIBUTION FOR MBD-ML, D3 AND D4 10 − 3 10 0 10 2 Mag. dev. (%) MBD-ML D3 D4 10 − 1 10 0 10 1 10 2 An g. d e v . ( ° ) Fig. S12. Distribution s of vdW force deviations from the MBD-ML, D3 and D4 metho d compared to MBD- NL. The distributions are represented b y violi n plots. Blac k c ros se s corresp ond to the mean v alue, the red line to the median. The vertical edges of the b o xes are the quartiles, i.e the 25-th and 75-th p ercen tile of the devia- tion distribution. The whiskers corresp ond to the mini- m um and the 95% deviation. Here we provide the quan titative statistical data on the distribution of the forc e magnitudes and angles of the MBD-ML, the D 3 and the D4 metho d relativ e to the ab ini tio MBD-NL m et ho d. D3 and D4 vdW force con tributions were computed using the dftd3 ( https:// github.com/dftd3/simple- dftd3 ) and dftd4 ( https: //github.com/dftd4/dftd4 ) pack ages. Both correc- tions were computed us i ng the default PBE0 parameters, included Beck e-Johnson (BJ) damp in g and a Axilro d- T eller-Muto three-b o dy term and w ere compared to PBE0-based MBD-NL forces with a damping parame- ter β = 0 . 83. The same β was used for the MBD-ML calculations. The di st r i bu t i ons are illustrated via violin plots in Figure S12, exact statistical v alues are given in T able S2. S8. GEOMETR Y OPTIMIZA TION OF MOLECULAR CR YST ALS F or the structure relaxation of the molecular crys- tals the Aims calculat or of the Atomic Simulation En vironment (ASE) was employ ed. F or geometry optimizations that inv olv ed the MBD-ML mo del, a new T ABLE S2. Mean, median, and 95 th p ercen tile devia- tions for relative magnitude and angular force predi ct i ons of MBD-ML, D3 and D4 c omp are d to MBD-NL on the OMol25 test set. MBD-ML D3 D4 Magnitude Deviation (%) Mean 2.6 28.2 39.5 Median 1.8 18.2 35.4 95 th P ercentile 7.7 60.7 80.8 Angular Deviation ( ° ) Mean 1.7 17.4 19.8 Median 1.1 12.6 15.2 95 th P ercentile 5.1 46.6 50.8 Molecule P olymorphs interaction type aspirin I (ACSALA07) I I (ACSALA17) mixed maleic acid I (MALIAC12) I I (MALIAC13) h b 2-amino-5-nitropyrimidine I (PUPBAD01) I I (PUPBAD02) I II (PUPBAD) h b maleic hydrazide I (MALEHY10 ) I I (MALEHY01) I II (MALEHY12) h b p en tacene I (PENCEN) I I (PENCEN04) vdW coronene I (CORONE04 a ) I I (N/A a ) vdW o-diio dobenzene I (ZZZPRO06) I I (ZZZPRO07) vdW a Structures di rec tl y taken from Ref 11. T ABLE S3. Molecular crystal p olymorphs from which geometry optimizations w ere p erformed. Along with the roman numeral denoting the p olymorph the C SD ref- erence co de and the type of interaction predominantly found in the system is pr ovided: mixed, v an-der-W aals (vdW) or hydrogen-bonded (hb). ASE calculator was impl em ented and combined with the Aims calculator as a SumCalculator , so that the final calculator ob j ect yielded total energies, atomic forces and stress tensors whi ch were the sum of the pure FHI-aims output and the MBD- ML con tributions to these prop erties. An example script to perform a PBE0+MBD-ML geometry relaxation can b e found in the libmbd rep ository under https://github.com/ em819/libmbd/tree/master/examples/mbd- ml- ase/ aspirin- structure- relaxation . All geometry opti- mizations of mol ec ul ar crystals were p erformed with the ASE implementation of the BFGS algor i th m and 9 in volv ed tw o steps: Starting from the CSD reference structure, in the first step only the atomic positions w ere relaxed with a force threshold fmax of 0 . 005 eV/ ˚ A. The resulting structure w as relaxed again, including b oth the atomic p ositions and the unit cell using the FrechetCellFilter without any constraints to a fmax of 0 . 005 eV/ ˚ A. In this second step, the initial guess for the Hessian in the ASE BF GS algorit h m, alpha was c hosen as 150 . 0 instead of the default 70 . 0 to counteract o vershooting due to the notoriously sh al l ow and rugged p oten tial energy surface of molecular crystals, leading to more stable c onv ergence. While the geometry op- timizations were p erformed with the PBE DF A with MBD-NL, MBD-ML and without and vdW metho d, the final optimized structure was recompute d with th e PBE0 functional instead. F or PBE-based calcu l at ion s that were combined with a the MBD metho d (MBD-NL or MBD-M L) , a damping parameter β of 0.81 was used, while for the final PBE0 recalculations, a v alue of 0.83 w as set. [1] A. Kabylda, J. T. F rank, S. Su´ arez-Dou, A. Khabibrakhmanov, L. Medrano Sandonas, O. T. Unke, S. Chmiela, K.-R. M ¨ uller, and A. Tk atchenk o, Molecular simulations with a pretrained neura l n e tw ork and universal pairwise force fields, J. Am. Chem. So c. 147 , 33723 (2025). [2] A. Kabylda, S. Su´ arez-Dou, N. Dav oine, F. N. Br ¨ unig, and A. Tk atchenk o, Qcell: Comprehensive quantum- mechanical dataset spanning diverse biomolecular frag- ments, arXiv preprint arXiv:2510.09939 (2025). [3] V. Blum, R. Gehrk e, F. Hanke, P . Ha vu, V. Havu, X. Ren , K. Reuter, and M. Sc heffler, Ab in i ti o molecular simulations with numeric atom-centered orbital s, Com- put. Phys. Commun. 180 , 2175 (2009). [4] J. W. Abb ott, C. M. Acosta, A. Akkoush, A. Am brosetti, V. Atalla, A. Bagrets, J. Beh ler, D. Berger, B. Bieniek, J. Bj¨ ork, et al. , Roadmap on adv ancements of the fhi- aims softw are pack age, arXiv (2025). [5] S. Ganscha, O. T. Unke, D. Ahlin, H. Maennel, S . Kash u- bin, and K.-R. M ¨ uller, The QCML dataset, quantum c hemistry reference data from 33.5 M DFT and 14.7 B semi-empirical c a l cu l a t io n s, Sci. Data 12 , 406 (2025). [6] J. Hermann and A. Tk atchenk o, Density functional mo del for v an der waals interactions: Unifying many- b ody atomic approaches with nonlo cal func ti o n a ls , Phys. Rev. Lett. 124 , 146401 (2020). [7] M. A. Rohrdanz, K. M. Martins, and J. M. Herb ert, A long-rang e-c o rrec t ed density functional that p erforms w ell for b oth ground-state prop erties and time-dep endent density func ti o n a l theory excitation energies, includ- ing charge-transfer exci t ed states, J. Chem. Phys. 130 , 054112 (20 0 9 ) . [8] E. Epifanovsky , A . T. Gilb ert, X. F eng, J. Lee, Y. Mao, N. Mardiros sia n , P . Pokhilk o, A. F. White, M. P . Co ons, A. L. Dempw olff, et a l. , Softw are for the fro ntiers of quan- tum chemistry: An ov erview of developmen ts in the q- c hem 5 pack ag e, J. Chem. Phys. 155 , 08 4 8 0 1 (2021). [9] R. A. Kendall, T. H. Dunning, and R. J. Harrison, Elec- tron affinities of the first-row atoms revisited. systematic basis sets and wa v e functions, J. Chem. Phys. 96 , 6796 (1992). [10] J. P . Perdew, R. G. Parr, M. Levy , an d J. L. Balduz, Density-functional theory for fra c t io n a l particle num ber: Deriv ative discontinuities of the energ y , Phys. Rev. Lett. 49 , 1691 (1982). [11] J. Potticary , L. R. T erry , C. Bell, A. N. Papanik olopou- los, P . C. Christianen, H. Engelk amp, A. M. Collins, C. F ontanesi, G. Ko ciok-K¨ ohn, S. Crampin, et al. , An unforeseen p olymorph of coronene by the application of magnetic fields du rin g crystal growth, Nat. Commun. 7 , 11555 (201 6 ) . 10 T ABLE S4. Structural comparison of p olymorphs relaxed wit h PBE and PBE+MBD-ML relative to the PBE+MBD-NL-relaxed reference structures. RMSD and maximum atomic displacemen t (Max. Disp.) quantify the deviation of atomic positi on s, while ∆ V gives the relative change in unit cell volume. Cases where the PBE geometry optimizat i on did not conv erge are marked wi th “–”. ANP refers to 2-amino-5-nitropyrimidine. RMSD ( ˚ A) Max. Disp. ( ˚ A) ∆ V (%) Molecule Polymorph PBE PBE+MBD-ML PBE PBE+MBD-ML PBE PBE+M B D- M L Mixe d Inter actions Aspirin I 0.177 0.002 0.248 0.003 26.00 0.09 I I – 0.007 – 0.018 – 0.22 Hydr o gen Bonde d Maleic acid I 0.035 0.002 0.060 0.003 24.16 0.29 I I 0.040 0.001 0.077 0.002 27.19 0.19 ANP I 0.222 0.004 0.385 0.006 28.32 0.22 I I 0.401 0.002 1.046 0.004 37.47 -0.18 I II 0.062 0.001 0.149 0.001 13.22 0.05 Maleic hydrazide I 0.055 0.001 0.078 0.002 28.13 0.05 I I 0.170 0.003 0.196 0.007 30.76 -0.11 I II 0.042 0.001 0.068 0.002 23.23 0.00 van der Waals P entacene I 0. 1 7 6 0.004 0.293 0.008 36.1 8 -0.46 I I – 0.007 – 0.014 – -0.55 Coronene I – 0.004 – 0.007 – -0.26 I I 0.277 0.001 0.499 0.002 37.42 -0.18 o -Diio dob enzene I 0 .1 1 9 0.005 0.179 0.008 29.8 6 -0.50 I I 0.085 0.038 0.119 0.056 26.65 0.12
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment