Reasoning-Driven Design of Single Atom Catalysts via a Multi-Agent Large Language Model Framework
Large language models (LLMs) are becoming increasingly applied beyond natural language processing, demonstrating strong capabilities in complex scientific tasks that traditionally require human expertise. This progress has extended into materials dis…
Authors: Dong Hyeon Mok, Seoin Back, Victor Fung
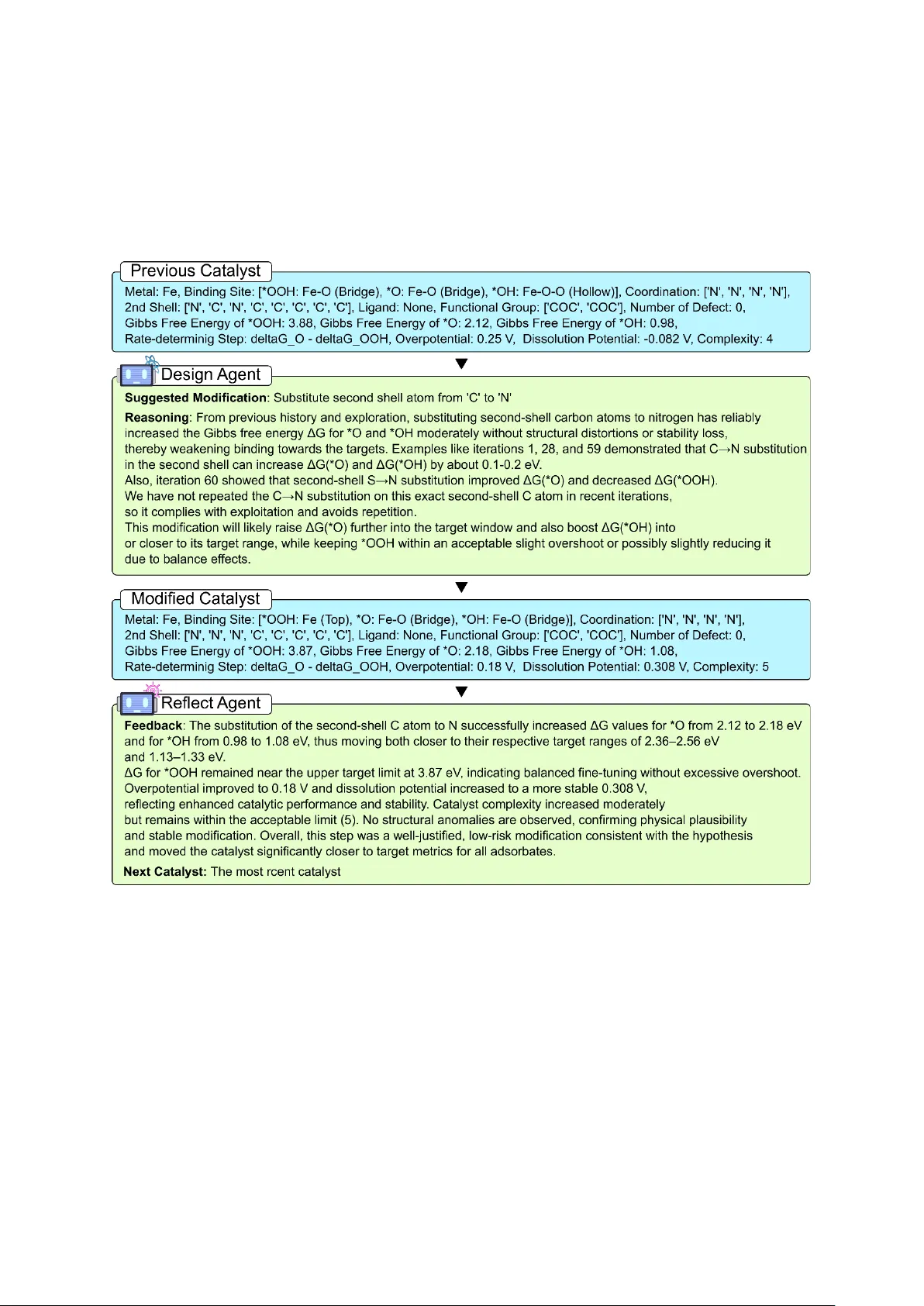
Reasoning-Driven Design of Single Atom Catalysts via a Multi-Agent Large Language Model Framework Dong Hyeon Mok 1 , Seoin Back 2,3,4,* , V ictor Fung 5, * and Guoxiang Hu 6,7, * 1 Department of Chemical and Biomolecular Engineering, Institute of Emerge nt Materials, Sogang University , Seoul 04107, Republic of Korea 2 KU -KIST Graduate School of Conver ging Science and T echnology , Korea University , Seoul 02841, Republic of Korea 3 Department of Integrative Energy Engineering, Korea University , Seoul 02841, Republic of Korea 4 Institute for Multiscale Matter and Systems (IMMS), Ewha W omans University , S eoul 03760, Republic of Korea 5 School of Computation al Science and Enginee ring, Geor gia Institute of T echnology , Atlanta, GA 30332, USA. 6 School of Materials Science and Engineering, Georgia Institute of T echnology , Atlanta, GA 30332, USA. 7 School of Chemistry and Biochemistry , Georgia Institute of T echnology , Atlanta, GA 30332, USA. AUTHOR INFORMATION Corresponding Author E-mail: sback@korea.ac.kr , victorfung@gatech.edu , emma.hu@mse.gatech.edu Keywords Lar ge Language Mode l, Single Atom Catalyst , Oxygen Reduc tion R eaction, Materials Discovery , Scientific Agent Abstract Lar ge language models (LLMs) are becoming increasingly applied beyond natural language processing, d emonstrating strong capabilit ies in complex scientific tasks that traditionally require human expertise. This p rogress has extended into materials discovery , where LLMs introduc e a new paradigm by leveraging reasoning and in -context learning, capabilities absent from conventional machine learning approaches. Here, we present a Multi- Agent-based Electrocatalyst Search Through Reasoning and Optimization (MAESTRO) framework in which m ultiple LLMs with specialized roles collaboratively discover high - performance singl e atom catalysts for the oxygen reduction reaction. W ith in an autonomous design loop, agents iteratively reason, propose modifications, reflect on results a nd accumulate design hist ory . Through in-context learning ena bled by this it erative process, MAESTRO identified design principles not explicitly encoded in the LLMs ’ background knowledge and successfully discover ed catalysts that break conventional scaling relations between reaction intermediates. These results highlight the potential of multi -agent LLM frameworks as a powerful strategy to generate chemical insight and discover promising catalysts. 1. Introduction Identifying new materials that deliver high catalytic activity , selectivity , and long-term stability has remained a significant and ongoing scientific challenge, with major im plications for improving ener gy e fficienc y , lowering indus trial costs, and en abling more sustain able chemical processes. Over time, materials discov ery strategies have evol ved beyond pur ely experimental approaches and first-principles simulations , such as d ensity functional theory (DFT) calculations 1-3 , with machine learning (ML) becoming one of the dominant paradigms in the field 4-9 . Early ML-based approaches primarily relied on high -throughput screening 10 - 13 , in which lar ge d atasets were generated in advance and property pr ediction models were used to filter promisi ng candidates, thereby reducing the computational and experimental cost of subsequent validation. However , as the chemical search space expanded and performance requirements b ecame increasingly demanding, s uch screening-based strategies encountered limitations related to data ava ilability , computational cost, a nd sca lability 14, 15 . T o address these challenges, inverse design strategies were introduced, in which mater ials are designed starting from targe t properties rather than discovered through forward searches 16 - 19 . Approaches based on global optimization 20, 2 1 and generative model s 22 - 24 have demonstrated notable success in the efficient discovery o f promising materials. Despite these advances, data-driven methods remain inherently constrained by their training dataset 25, 26 . While they can discover novel materials within established structure-property relationship s, they often lack explanatory power regarding why a particular mate rial is selected and struggle to identify materials governed by unknown physical mechanisms outside the learned domain. As a result, the discovery of fundamentally new , physics-driven design prin ciples has lar gely remained dependent on human intuition and intervention. Recent adv ances in large language models (LLMs) offer a fundamentally differe nt avenue for inverse design. Or iginally developed for natural language processing, LLMs trained on massive text corpora have demonstrated a capacity for human-like reasoning 27 - 29 . In chemistry and m aterials science, LLMs have begun to move beyond sim ple data mining toward roles involving experimental planning 30 , synthesis guidance 31 , simulation tool orchestration 32 and chemically informed decision-making tasks that were previously exclusive to human experts 33 - 36 . Crucially , LLMs possess the capability for in -context learning, whereby they adapt their reasoning based on prior intera ctions and accumulated context without explicit pa rameter updates 37 . This enables LLMs to acquire task-specific principles and uncover ne w insights not explicitly encoded in their background knowledge, particularly when embedded within iterative decision-making loops 38 . Furthermore, recent research has shift ed from assigning multiple roles to a single LLM toward multi -agent frameworks, whereby several LLM -based agents, each spe cialized for a specific task, interact and collaborate 39, 40 . Suc h systems provide a natural foundation for complex scientific workflows that require hypothesis formulation, reflection and memory . In this work, w e investigate whether an LLM-b ased multi-agent framework can b e successfully applied to heterogeneous electrocatalysis, a relatively narrow yet intrinsically complex domain. While LLMs have re c ently been employed in c atalyst-related studies for task such as data mi ning 41 , synthesis planning 42 , property prediction 43 and catalyst generation 44 , approaches that e xplicitly leverage LLM rea soning to design high-performance catalysts remain scarce compared to other systems. T o address this gap, we develop a Multi-Agent-based Electrocatalyst Search Through Reasoning and Optimization (MAESTRO) fra mework in which multiple LLM agents collectively design active and stable singl e atom ca talysts (SACs) for the oxyg en reduction reac tion (ORR) ( Figure 1 ). W ithin this framework, a gents iteratively propose structural modifications, reflect on outcomes and summarize de sign history , while rapid property evaluation is enabled by a machine learning force field ( MLFF) serving as a surrogate for DFT 45 . Applying the MAESTRO framework, we demonstrate that the agents can autonomously formulate hypotheses regarding catalyst modification strategies and progressively improve both activity and stability through iterative reasoning. Notably , the framework identifies SACs that surpass the theoretical activity limit imposed by conventional scaling relations between reaction intermediates 46 . Detailed analysis revea ls that this enhancement originates from the se lective stabilization of a specific intermediate via hydrogen bonding, a mechanism previously reported in several studies. Importantly , this discovery does not emer ge in the abs ence of in-context learning, providing strong evidence that the proposed framework enables agents to acquire new physics and design p rinciples through accumulated history and iterative reasoning . These results highlight the MAESTRO framework as a promising optimi zation strategy for electrocatalyst discovery , capable of navigating complex design spaces and uncovering nontrivial r elationships between structure and property beyond conventional discovery schemes. 2. Results 2.1 MAESTRO Framework Figure 1. Overview of t he MAESTRO framework and the exploration -exploitation search strategy . Starting from an initial catalyst structure and its associated properties, LLM -based agents and predefined tools iteratively pe rform reasoning, modification, ev aluation, reflection and summarization accor ding to their design ated roles for a fixed number of cycles. The desig n run is divided into two phases, an exploration phase and an exploitation phase. Upon completion of the exploration phase, a summary report is generated by the agent. The MAESTRO framework operates as an iterative design loop compos ed of four nodes, design, calculation, reflection and summary , managed by four agents, Design, Reflect, Summary and Exploration report, along with predefined tools. The primary objective of this loop is to minimize the ORR overpotential ( η ), which serves as a measure of the catalytic activity of the SAC, while simultaneously maximizing the dissolution potential ( U diss ) to ensure electrochemical stability . The design loop initiates by loading an initial SAC structure and e valuating its overpotential and dissolution potential us ing a MLFF surrogate model. The resulting geometric and catalytic data are the n formatted and passed to the design agent withi n the design node. Leveraging structural images and formatted metadata, the design agent formulates a hypothesis to identify specific modi fications, and the underlaying physical reasons, th at could effectively tune binding energies to reduce the overpotential. The design agent is authorized to modify five distinct geometric components of the SAC: the center metal atom, first coordination shell, second shell, axial ligand and functional groups. The available modification types and options are summarized in T able 1 . T able 1. Geometric components of the SAC accessible to the design agent, including modification types and available chemical option s . S pecifically , axial ligands are introduced perpendicular to the surface, positioned beneath the binding site, while functional groups are added to the second coordination shell of the carbon support. Geometric Components Modification T ypes Atom / Molecule Options Center metal atom Substitute Pt , Pd , Ir , Ru , Fe , Co , Mn , Cu , Ni , Cr , V , Ti , Mo , Na , Ta , Ag , Au , Zn , Sn , Bi First coordination shell Substitute, Add, Remove H, C, O, N, S Second coordination shell Substitute C, P , B, S, N Axial ligand Substitute, Add, Remove *O, *OH Functional group Substitute, Add, Remove *COC, *COH, *CO The proposed modification is forwa rded to the modific ation tool, which verifies whether the suggested c hange is applicable to the given SAC. If the modification is deemed unsuitable, the workflow returns to the design agent for a new proposal. Th is includes case in which the agent proposes invalid modifications, such as hallucinated modification type not included in the list or attempts to substitute non-existent elements. If accepted, the modification is applied, and the resulting structure is passe d to the calculation node. Here, the overpotential and dissolution potential of the modified SAC are calculated using the MLF F . If the geometry optimization fails to converge within 100 optimization steps und er a force threshold of 0.05 eV/Å, the failed structure and associated metadata are returned to the de sign agent. Upon successful conver g ence, the calculated properties are forwarded to the reflection node. In the reflection node, the reflection agent evaluates the effec tiveness of the proposed modification by comparing the catalytic activity and stabili ty before and after the change . Based on this comparison, it provides feedback categorizing the modificat ion as successful or unsuccessful. This feedback, along with the proposed modification, the underlying reasoning, and the calculated r esults, is passed to the summary node and formatted as des ign history . While the most recent history is stored in full, previous entries are condensed by t he summary agent to maintain context ef ficiency ( Figure S2 ). Finally , the acc umulated design history , the modified SAC structure and its pe rformance metri cs are fed back to the design agent to initiate the next iteration. T o ensure suffic iently broad exploration, the agents are guided by an exploration - exploitation strategy . During the first half of the process, the explor ation phase, the primary objective is to expand the design space rather tha n minimize the overpotential. Accordingly , the design agent is instruc ted to structurally diverse and distinct types of mo difications without prioritizing im provements in overpotential or dissolution potential. Th e reflect agent provides feedback which prioritizes the discovery of unique structural configurations. At the midpoint of the design a gent, the workflow transitions to the exploitation phase, where optimization proceeds under the original o bjective of minimizing overpotential while maintaining stability . During this tra nsition, an e xploration report ag ent generates a one to two page report summarizing the modifications explored and their corresponding effects ( Figure S6 ). This report dist ills actionable insights to guide the subsequent optimization. Th e exploration report serves as a persistent reference for the design agent th roughout the remainder of the loop. In this study , the primary results were obtained using GPT -4.1-mini as the LLM 29 , Universal Mod els for Atoms (UMA) as the MLF F surrogate model 47 , and F eN 4 as the initial catalyst structure. However , the framework is modular a nd additional experiments using alternative models and st arting materials were also conducted. Detailed agent prompts, catalyst specifications and implementation details are provided in Supplementary Note A . 2.2 V alidation of MLFF and LLM components Figure 2. Pre-validation of MLFF and LLM components. (a-c) Parity plots comparing DFT - calculation and MLFF-prediction results for (a) energies per atom, (b) atomic force s and (c) binding ener gies. (d) An example of the reasoning and modification proposed by the design agent, together with the resulting structure and th e changes in Gibbs free ener gy and d -band center induced by the suggested modification. (e) C orresponding changes in the electronic density of states (DOS) f ollowing the modification. Dashed lines indicate the d-band cent ers of the metal atoms, which shift toward positive values (closer to the Fermi level) upon the substitution of a second-shell carbon atom with nitrogen. Before deploying the M AESTRO framework, we conducted a pre-validation study to ensure the reliability of both the tools and the LLM for the SAC system. Since the UMA employed as the MLF F in this study was not trained on SAC data , and n o publicly available SAC dataset existed for meaningful fine-tuning, we constructed a custom SAC dataset to validate the extrapolation capabilities of UMA. The performance of the pre- trained UMA was evaluated using this out- of - distribution (OOD) dataset, which contains DFT calculated ener gies, forces and bindi ng energies for various structures and their adsorbed int ermediates. As shown in Figures 2 a an d 2b , UMA with ‘OC20’ domain showed Mean Absolute Errors (MAEs) for DFT ene r gies per atom a nd atomic forces of 8.83 meV/atom and 47.73 meV/ Å, respectively . These values are comparable to the MAEs reported by W ood et a l for OOD catalysts 47 ( Figure S3 ). F urthermore, 0.346 eV MAE for bindi ng ener gy was deemed acceptable, especially considering the complete absence of SACs in the pre-training set and the c onsistent directional bias of the prediction error . This systematic beh avior indicates that UMA can reliably capture relative ch anges in binding strength, making it a suitable surrogate model for DFT within the design loop. Detailed in formation regarding the pre -validation datasets and MLFF performance is provided in Supplementary Note B . Regarding the LLM, we validated wheth er the modification proposed b y the model could effec tively steer binding ene r gies in the int ended direction and whe ther the underlying reasoning was physically sound. Th e tar get was to achieve a binding ener gy of 2.46 eV fo r the *O intermediate. As shown in Figure 2d , the LLM successfully suggested a modification that shifted the *O binding ener gy from the ini tial structure toward the desired targe t. Specifically , the LLM hypothesized that N subst itution would decrease the electron density of the center metal atom. This hypothesis was confirmed by d ensity of states (DOS) c alculations, which showed a clear shift of electronic density , qua ntified by the reduction of d -band center position, of the Fe center after substitut ion 48, 49 . While th e correspondence between the intended and realized binding energy changes and their associated reasoning was not perfect in every case , it was correct in the majority of instances. Representative examples of unsuccessful suggestions are provided in Figure S5 . These pre-validation results demonstrate that UMA can function as a practical surrogate for DFT in evaluating the stability and catalytic activity of SACs for ORR without additional fine-tuning. Moreover , the LLM demonstrates a meaningful understanding of structure-property relatio nships of the SACs, enabling it to propose chemically interpretable modifications that systematically guide the ener g ies toward tar get values. 2.3 Performance Evaluation of Overall Framework Figure 3. Performance and behavior of the MAESTRO framework across dif ferent strategies. (a) R epresentative example of the ORR overpotential change during a design run, illustrating the transition from the ex ploration phase to the exploitation after the 50 th modi fication. The red dashed line denotes the running minimum overpotentia l. (b) Corresponding Pareto front obtained from the sam e run, where the red point highlighting the most active catalyst discovered. (c) Evolutio n of overpotential and (d) dissolution potential , averaged ove r 10 independent design runs using “hist ory + exploration” strategy . (e) Comparison of overall design performance across diffe rent strategies. (f) Evolution of catalyst complexity during the design run for each strategy , where higher value indicates a l ar ger number of modifications relative to the initial SAC structure. The objective of the design run is to minimize the ORR overpotential while simultaneously preserving or increasing the disso lution potential. T o systematically evaluate the performance of our framework, we employed four metrics. 1) A verage overpotential : T he mean ORR overpotential obtained across all design runs, excluding the exploration phase. For strategies w ithout an explicit exploration phase, this was computed over the latter half of each design run. 2) Minimum overpotential: The av erage of the low est overpotential achieved during each individual design run. 3) Hypervolume: The ave rage volume of the Pareto front, representing the joint optimization space spanned by limiti ng potential and diss olution potential. A larger volume indicates superior discovery performance in terms of both activity and stability . 4) Active-point volume: The average Pareto volume of the catalyst with the lowest overpotential, representing the combined activity and stability of the most promising candidate discovered. T o benchmark the perfor mance of the proposed “ history + exploration ” strategy , which introduces an exploration phase to prioritiz e a global search be fore exploitation, we defined three baseline strategies: 1) The hist ory strategy performs only exploitation without a preceding exploration phase, thereby isolating the effec t of broadening the design space before focused optimization. 2) The historyless strategy relies solely on the LLM’ s background knowledge and does not incorporate i nformation from previous modification steps, highlighting the impact of in-context learning from accumulated design history . 3) The random strategy applies pure ly random modifications without LLM guidance, serving as a lowe r-bound reference. Figure 3a and 3b present a representative design run, illust rating the gradual changes of overpotential and the corresponding Pareto front of the discovered catalysts. During the exploration phase, the ove rpotential exhibits lar ge fluctuations as the framework bro adly samples the design space. The exploratory behavior was further confirmed by the higher number of unique modifications performed during th is pha se, compared not only to the subsequent exploitation phase, but also to design runs of other strategies without exploration ( Figure S 7 ). In contrast, during the exploitation phase, the overpotential conver ges toward a narrow range, indica ting focused optimization. The progr ession of the minimum overpotential, denoted by the red line and markers, reaches its optimum during th is phase. The Pareto plot highlights the intrinsic trade-off b etween activity a nd stability . Although the most a ctive catalyst identified in this run shows sli ghtly low er stabili ty , it still surpasses the reference F eN 4 catalyst in both metrics. Figure 3c and 3d summa rize the average results over 10 independent design runs. Both graphs reveal a clear trend, where the overpotential gradually decreases while the dissolution potential increa se s following the transition to the exploitation phase ( Figure S8 ). These observations demonstrate that the MAESTRO framework can effectively identify highl y active SACs while maintaining or even improving their electrochemical stability . The origin of this enhanced discovery performance lies in the expansion of th e accessible d esign space through the explora tion phase and in the in-context learning enabled by the design history ( Figur e 3e ). Our strategy exhibits the best performance in terms of both the average overpotential and the minimum overpotential. Notably , even the “ history” strategy consistently outperforms the “ historyless” approach i n average overpotential, underscoring th e im portance of leveraging accumulated design history . In contr ast, the hyp ervolume and active-point volume metrics show comparable values across the different strategies. This behavior arises because these metrics can be disproportionately influenced by high stabili ty values, which are generally easier to achieve than low ove rpotentials. Therefore, maintaining Pareto volume metrics comparable to other strategies while simultaneously achieving lower o verpotentials provides strong evidence that the framework ef fectively mitigates the activity-stability trade-off . F inally , the moderate level of catalyst complexity , defined by the number o f modifications applied to the initial structure, indicates that our strategy maintains a balanced search behavior ( Figur e 3f ). The design agent neither diverges excessiv ely into chemically unrealistic structures nor becomes trapped in a narrow , suboptimal region of the design space. 2.4 Impact of In -context Learning Figure 4. (a) Linear scaling relations among ΔG *OH, ΔG *O and ΔG *OOH . (b) Corr esponding volcano plot using ΔG *OH as the activity descriptor . The red dashed line indicates the theoretical lower limit for overpotential 0.36 V imposed by th e scaling relations. (c) Fre quency of scaling relation violations observed during the design loop, averaged over 10 independent design runs for each strategy . T o elucidate the origin b ehind the good observed performance within the MAESTRO framework, we investigated how in -context learning from design history influences the d esign process. W e c ollected the binding ener gies of 2,017 unique c atalysts fr om a total 4,000 structures generated across four discovery strategies, where e ach strategy was evaluated over 10 independent design runs consisting of 100 modification steps each. W e then analyzed th e linear scaling relations among the binding ene r gies of the three ORR intermediates, *OOH, *O and *OH. Th is analysis confirmed the presence of well-defined linear scaling relations between ΔG *O and ΔG *OH , as well as between ΔG *OOH and ΔG *OH in the S AC system ( Figure 4a ). Furthermore, by deriving a volcano plot using ΔG *OH as the descriptor , we identified a theoretical minimum overpotential of about 0.36 V ( Figure 4b ). This value represents the lowest overpotential achievable by uniformly strengthening or weakening the binding ener gies of all intermediates while remaining on the scaling relations, without selectively modulating individual intermediates ( Figure S9 ). As we confirmed in Figure 2 , the LLM possesses background chemical knowledge that enables it to propose modi fications to control binding strengths as intended. Consistently , in Figure 3 , the historyless strategy , which relies only on this background kno wledge, exhibit ed a minimum overpotential exceeding 0.36 V . This observation indicates that strategies achieving overpotentials below 0.36 V must h ave b een acq uired through leveraging in -context l earning from prior design steps. Specifically , such strategies learn to selectively tune the binding ener gy of specific intermediates, thereby breaking the inherent scaling relations r ather than merely shifting all binding ener gies in tandem 50 . This ef fect can be observed in observing the freque ncy of achieving overpotentials below lower li mit ( Figure 4c ). Across 10 des ign runs, both the random and hist oryless strategies achi eved this va lue fewer than once on average, indicating that breaking the scaling relations in these cases occurs only sporadically by chance. In contrast, strategies with in-context learning from history broke these relations more than three ti mes on average out of 100 modification steps. These results demonstrate that in-c ontext learning from modification history plays a decisive role in overcoming fundamental scaling c onstraint s ( Figure S10 ), despite the higher token consumption compared to strategies without in-context learning ( Figure S 11 ). 2.5 Revealing Catalyst Design Principles Figure 5. (a) Representative modification process to design promising ca talysts by breaking scaling relations a nd lower limit of overpotential. Scaling break and overpotential opti mization occur sequentially . (b) Representation of one of high-performance catalyst structures discovered during the design runs, together with a comparison of MLFF-predicted and DFT - calculated p erformance metrics. Data includes Gibbs free bindi ng energies, overpotential ( η ) and dissolution potentials ( U diss ) for each candidate. Other examples can b e found in Figu re S12 and S13 . T o elucidate the mechanism underlying the observed scaling relation breaking and the successful design of hi gh-performance cat alysts, we analyzed a representative designed catalyst in detail. Figur e 5a shows a n example of a catalyst achieving a n overpotential of 0.31 V , surpassing the scaling-based lower limit of 0.36 V . During the design run, the design agent introduces surface oxygen functional groups (*COC or *COH). These surface oxygen species are observed to form H-bonds with the H atoms of *OH and *OOH intermediates, thereby stabilizing both species. As a result, only th e ΔG *O is selectively increased, leading to a b reak in the scaling relations between ΔG *O H and ΔG *O . After inducing this scaling break, the agent retains the surface oxyg en and subsequently explores first and second shell modi fications, searching for configurati ons that further reduce the overpotential within the altered s caling landscape. Through this sequential process, the MAESTRO framework is able to design catalysts with overpotentials below 0.36 V . Importantly , this scaling break from selective H - bonds wa s confirmed not only at the MLF F bu t also through explicit DFT level. W e performed DFT calculations on 1 1 catalysts with MLFF -predicted overpotentials below 0.36 V , sel ected from the minimum overpotential candidates obtained from each of the 10 design runs starting from FeN4 and CuN4. Among these, six unique catalysts have DFT -calculated overpotentials below 0.36 V , five of which were confirmed to feature surface oxygen forming H -bonds with ORR intermediates ( Figure 5b ). The stabilization of intermediates via selective H- bonding, the resulting scaling break and the associated enhancement in catalytic performance have been reported in multiple prior studies 51 - 55 . Therefore, th e framework does not int roduce a fundamentally ne w design principle. However , as shown in Figure 4c , the agents without in -context learning lack the int rinsic ability to break scaling relations, indicating that these principles were not already pr esent in their background knowl edge, but were gained f rom experimentation. Indeed, through this exploration-exploitation strategy and in-context learning enabled by the design loop, the agents successfully rediscovered a valid design principle that existed outside their prior knowle dge 56 . This result demonstrates that the MAESTRO framework ha s the capacity not only to optimize catalysts but also to potentially uncover novel catalyst design principles bey ond those explicitly provided by human inputs. 3. Discussion 3.1 LLM, Starting State and Hyperparameter T un ing W e have demonstrated t hat the MAESTRO framework operates suc cessfully using GPT -4.1-mini as the LLM and FeN 4 as the initial catalyst. W e further examined how the choice of LLM, the starting SAC structure and key hyperparameters, including the LLM temperature and the number of recent design histories retained in short-term memory , affect the overall performance. First, when the LLM was replaced with GPT -5-mini while all other conditions were kept identical, the agents exhibited slightly more detailed reasoning and feedback. Ho wever , the overall performance remained comparable t o that obtained with G PT -4.1-mini. This observation indicates that GPT -5-mini possesses background knowledge similar to that of GPT - 4.1-mini and that, in both cases, the ability to overc ome scaling relations must be acquired through in-context learning during the design loop rather than being directly encoded in the model ( Figure S14a ). W e also investigated the effect of the starting material by initializing design runs with SACs co ntaining other central metal atoms. In all cases, the average mi nimum overpotential achieved fell below 0.36 V , the limit dictated by the volcano plot. Moreover , each design run broke the scaling r elations more than twice on average, demonstrating that o ur strategy is robust with respect to the choi ce of the initial metal center ( Figure S14b ). Among the starting structures, the design run initiated form PtN 4 exhibited the best over all performance, except for the average overpotential. This behavior can be attributed to the int rinsically high initial overpotential of PtN 4 , which allows for broader exploration to the energy landscape during the exploration phase. Finally , we evaluated the sensitivi ty of the framework to the temperature settings by performing 10 d esign ru ns across a range from 0.2 to 1.8. The r esults indicate that, provid ed the tempera ture remains within a moder ate range, the overall discovery performance is large ly comparable to the default setting of 1.0. In contra st, extreme values, such as 0.2 or 1.8, lead to a noticeable performance degradation ( Figure S15 ). 3.2 Limitations and Future Dir ections The types of SAC modifications employe d in this study focus on fine-tuning the local environment of the active site by element substitution, atom addition/removal, and the introduction of functional groups or li gands. Alth ough this approach expands the a ccessible design space, catalysts identified through such localized modifications may pose challenges for experimental synthesis. In particular , experimentally reproducing an identical local atomic environment remains difficult in prac tice, even when the designed structures satisfy electrochemical stabili ty criteria. As a result, direct experimental v alidation of some discovered catalysts may be non -trivial. In subsequent stu dies, the inclusion of better metrics for synthesizability or even synt hesizability predictions will fac ilitate bridging the gap to experimental validation. Furthermore, this study is limited to the discovery of ORR catalyst within the SAC system. This choice was made to establish a proof-of-concept for agent -based c atalyst discovery , leveraging SACs as a platform in which the active site is sp atially confined and structural complexity is relatively low . Despite these simplifications, the results demonstrate that the framework is capable of selectivity modulating the binding ener gies of mul tiple intermediates while simultaneously managing activity , stability and structural complexity . These c apabilities suggest that the framework can be extende d to more complex systems, such as dual atom catalysts (DACs) or to reactions like CO 2 reduction, where the independent control of intermediate ener gies is even mo re critical. 57 4. Conclusions In this work, we proposed the MAESTRO framework and demonstrated that itera tive interactions among specialized agents within an o ptimization loop can progressively enhance both catalytic activity and stability . Notably , the catalysts identified by the framework b reak the theoretical lower limit of ORR ove rpotential imposed by the conventional scaling relations. These findings indicate that the accumulation of design history , combined with agent-driven reasoning and self-reflection, can reveal n ew phys ical principles not explicitly encoded in the LLM’ s background knowledge. Overall, our results suggest that the MAESTRO framework serves not only as an e f fective catalyst optimization tool but also as a means of autonomously generating nov el chemical insights, significantly reducing the need for human int ervention in the discovery of next-generation catalysts. 5. Methods 5.1 Large Language Models A large language model (LLM) is a transfo rmer -b ased autor egressive generati ve model trained on lar ge-scale text corpora 58 . By learning statistical and semantic patterns in language, an LLM is capable o f performing complex re asoning, planning and dec ision -making tasks that resemble human cognitive processes. In this study , unless otherwise sp ecified, all LLM -based agents we re implemented using OpenAI’ s GPT -4.1-mini model with default temperature and top-p settings. The detailed personas and system prompt tem plates assigned to each agent are provided in Supplementary Note 1 . 5.2 Machine Learning Forc e Field Ma chine learning force field (MLFF) models serve as surrogate models for density functional theory (DFT) calculations. They are trained on large DFT d ataset to predict ener gy and atomic forces bas ed on the geometry and stoichiometry of materials 45 . These models typically interpret materials as graphs, where atoms are tre ated as nod es characterized by elemental features and interatomic bonds are encoded as edges co ntaining geometric information. In this study , we employed the Universal Models for Atoms (U MA) as the MLFF 47 . UMA is based on eSEN 59 , an equivariant graph neural network that represents the atomic environment using spherical harmonic embedding and propagates in formation through multiple message passing layers. UMA is trained as a general-purpose model across diverse material domains and incorporates a Mixture of Linear Expe rt (MoLE) a rchitecture to enhance flexibility and inference efficiency 60 . For geometry optimization and energy prediction o f the SACs, we utilize d the pre trained UMA model corresponding to the ‘OC20’ domain 61 , whic h is specifically weighted toward heterogenous catalyst systems. 5.3 DFT Calc ulations W e performed DFT calculations using the V ienna Ab initi o Simulation Package (V ASP , version 5.4.4) 62, 63 to pre-validate MLFF performance and to evaluate SAC structures discovered by the catalyst design framework. T o en sure consistency , the sa me DFT setting used to generate the UMA training data were adopted. The projected-augmented wave (P A W) pseudopotential method 64 was employed in conjunction with the generalized gradient approximation revised Perdew-Burke-Ernzerhof (GGA-RPBE) exchange-correlation functional 65 . All structures were fully relaxed until the tot al energy and atomic forces conver ged to within 10 -4 eV and 0.0 5 eV/Å, for forces, respectively . A plane wav e kinetic energy cutoff of 350 eV w as applied. Monkhorst-Pack k-point mesh 66 was configured as (3 × 3 × 1). 5.4 Gibbs free ener gy , Overpotential and Dissolution Potential Binding Gibbs free energies were evaluated using the computational hydrogen electrode (CHE) a pproach 67 , in which the chemical potential of a proton-electron pa ir (H + + e - ) is reference d to ½H 2 (g) at 0 V RHE under standard conditions. Baded on this framework, th e Gibbs fr ee ener gies for the adsorption o f *O , *OH and *OOH intermediates were computed as follows: ∆ G O* = E O* − E slab − E H 2 O − E H 2 + ∆(ZPE+ ∫ C p dT -TS) ∆ G OH* = E OH* − E slab − E H 2 O − 1 2 E H 2 + ∆(ZPE+ ∫ C p dT -TS) ∆ G OOH* = E OOH* − E slab +2 E H 2 O − 3 2 E H 2 + ∆(ZPE+ ∫ C p dT - TS ) Here, E * O , E * OH and E * OOH denoted the DFT total ener gi es of the surface with the corresponding adsorbed intermediates, while E H 2 and E H 2 O are DFT energies of gas phase H 2 and H 2 O molecules, respectively . Zero-point ener gies (ZPE) co rrections, enthalpic contribution ( ∫ C p dT ), and entropic contribution terms (TS) were calculated using the Harmonic os cillator approximation for adsorbed s pecies (* O, *OH, *OOH) and the Id eal ga s approximations for gas mol ecules (H 2 , H 2 O), as implemented in Atomic Sim ulation Environment (ASE) 68 . The correction values are given in T able S8 . The theoretical overpotentials of ORR ( η ORR ) was determined from Gibbs free ener gy changes of the four elementary proton-electron transfer steps in the associative ORR pathwa y: ∆ G 1 = ∆ G *OOH − ∆ G O 2 + eU ∆ G 2 = ∆ G O* − ∆ G * OOH + eU ∆ G 3 = ∆ G * OH − ∆ G * O + eU ∆ G 4 = ∆ G H 2 O − ∆ G * OH + eU The limi ting potential ( U L ) is d efined as th e maximum potential at which all r eaction st eps become thermodynamicall y favorable ( ∆ G i ( U L ) ≤ 0 eV). The overpotential is the diffe rence between the standa rd equilibrium potential for ORR (1.23 V) and the limiting potential, calculated as follow s: U L = − max[Δ G 1 , Δ G 2 , Δ G 3 , Δ G 4 ]/ e η ORR = 1.23 − U L The dissolution potential of the SAC ( U d ) is an electroche mical stability , describing the tendency o f the metal center to dissolve under ORR conditions . It is calculated as the dif ference between the b inding energy of the metal atom to the carbon support ( E b ) and the standard dissolution pote ntial of the correspondin g bulk metal ( U 0 ) in aqueous solution (pH = 0), as follows: E b = E SAC-M − E SAC − μ M U d = U 0 − E b / ( e × N e ) where E SAC-M , E SAC are the DFT total en er gies of th e carbon support with and without the metal atom, respectively . μ M is the chemical potential of the metal atom, e is elementary ch ar g e and N e is the number of electrons involved in the metal dissolution process. Code A vailability The code develop ed in this work and relevant information can b e found in GitHub ( https://github.com/ahrehd0506/Catalyst-Design-Agent ) Supporting Information Details of prompt template, Details of pr e-validation for MLF F and LLM, Example of exploration summary report and reasoning of the agents, Additional metrics for d esign framework, Metrics for strategies with various starting materials, parameter and LLM model. Acknowledgements D.H.M. acknowledges the support from Korea Institute for Advancement of T echnology (KIA T) grant funded by the Ministry of T rade, Industry & Energy (MOTIE), Korea Government (RS- 2024-00436106, Human Resource De velopment Program for Industrial Innovation). S.B. acknowledges the support from th e National Research Foundation of Korea (NRF) gr ants funded by the Korea government (MSIT and MOE) (RS-2024-00448287, RS-2025-16063688, RS -2025-00513832, and RS-2025-02214715), and the generous supercomputing time provided by the Korea Institute of Science and T echnolog y Information (KISTI). G.H. acknowledges support from the U.S. Nati onal Science Foundation under Grant # CBET -2442223. This work used NCSA Delta CPU at University of Illino is Urbana-Champaign through allocation MA T250081 from the A dvanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by U.S. National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296. Refer ences (1) Ceder , G.; Chiang, Y . -M.; Sadoway , D.; A ydinol, M.; Jang, Y .-I.; Huang, B. Identification of cathode materials for lithium batteries guided by first -principles calculations. N atur e 1998 , 392 (6677), 694–696. (2) Greeley , J.; Jaramillo, T . F .; Bonde, J.; Chorkendorf f, I.; Nørskov , J. K. C omputational high- throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater . 2006 , 5 (1 1), 909–913. (3) V itos, L.; Korzhavyi, P . A.; Johansson, B. Stainless steel optimization from quantum mechanical calculations. Nat. Mater . 2003 , 2 (1), 25–28. (4) Butler , K. T .; Davi es, D. W .; Cartwright, H.; Isayev , O.; W alsh, A. Machine learning fo r molecular and materials science. Natur e 2018 , 559 (7715), 547–555. (5) Ong, S. P . Accelerating materials science with high-throughput computations and machine learning. Comp. Mater . S ci. 2019 , 161 , 143–150. (6) Choudhary , K.; DeCost, B.; Chen, C.; Jain, A.; T avazza, F .; Cohn, R.; Park, C. W .; Choudhary , A.; Agrawal, A.; Billinge, S. J. Recent advances and applications of deep learning methods in materials science. npj Comp. Mater . 2022 , 8 (1), 59. (7) Esterhuizen, J. A.; Go ldsmith, B. R.; Linic, S. Interpre table machine learning for knowledge generation in heterogeneous catalysis. Nat. Catal. 2022 , 5 (3), 175 – 184. (8) Back, S.; Aspuru-Guzik, A.; Ceriotti, M.; Gryn'ova, G.; G rzybowski, B.; Gu, G. H.; Hein, J.; Hippalgaonkar , K.; Hormázabal, R.; Jung, Y . Accelerated chemica l science with AI. Digital Discovery 2024 , 3 (1), 23–33. (9) Alberi, K.; Nardelli, M. B.; Zakutayev , A.; Mitas, L.; Curtarolo, S.; Jain, A.; Fornari, M.; Marzari, N.; T akeuc hi, I.; Green, M. L. The 2019 materials by design roadmap. J. Phys. D Appl. Phys. 2018 , 52 (1), 013001. (10) Ludwig, A. Disco very of new materials using combinatorial synt hesis and high - throughput characterization of thin -film materials libraries combined with computational methods. npj Comp. Mater . 2019 , 5 (1), 70. (1 1) A ykol, M.; Kim, S.; Hegde, V . I.; Snydacker , D.; Lu, Z.; Hao, S.; Kirklin, S.; Morgan, D.; W olverton, C. High-throughput computational design of cathode coatings for Li -ion batteries. Nat. Commun. 2016 , 7 (1), 13779. (12) Y ohannes, A. G.; Lee, C.; T alebi, P .; Mok, D. H.; Karamad, M.; Back, S.; S iahrostami, S. Combined high-throughput DFT and ML screening of transition metal nitrides for electrochemical CO2 reduction. ACS Catal. 2023 , 13 (13), 9007–9017. (13) Pyzer -Kn app, E. O.; Suh, C.; Gómez-Bombarelli, R.; Aguilera-Iparraguirre, J.; Aspuru - Guzik, A. What is high -throughput virtual screening? A perspective fro m organic materials discovery . Annu. Rev . Mater . Res. 2015 , 45 (1), 195–216. (14) Elton, D. C.; Boukouvalas, Z.; Fuge, M. D.; Chung, P . W . Deep learning for molecular design—a review of the state of the art. Mol. Syst. Des. Eng. 2019 , 4 (4), 828–849. (15) Peng, J.; Schwalbe-Koda, D.; Akkiraju, K.; Xie, T .; Giordano, L.; Y u, Y .; Eom, C. J.; Lunger , J. R .; Zheng, D. J.; R ao, R. R. Human-and machine-centred designs of molecules and materials for sustainability and decarboniza tion. Nat. Rev . Mater . 2022 , 7 (1 2), 991–1009. (16) Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018 , 361 (6400), 360–365. (17) Noh, J.; Gu, G. H.; Kim, S .; Jung, Y . Machine-enabled inverse d esign of inorga nic solid materials: promises and challenges. Chem. Sci. 2020 , 11 (19), 4871–4881. (18) Zunger , A. Inverse design in search of materials with target functionaliti es. Nat. Rev . Chem. 2018 , 2 (4), 0121. (19) Fung, V .; Zhang, J.; Hu, G.; Gane sh, P .; Sumpter , B. G. Inverse design of two-dimensional materials with invertible neural networks. npj Comp. Mater . 2021 , 7 (1), 200. (20) Oganov , A. R.; Pickard, C. J.; Zhu, Q.; Needs, R. J. Structure predicti on drives materials discovery . Nat. Rev . Mater . 2019 , 4 (5), 331–348. (21) Doll, K.; Schön, J.; Jansen, M. Structure prediction based on ab initio simulated annealing for boron nitride . Ph ysical Review B—Condensed Matter and Materials Physics 2008 , 78 (14), 1441 10. (22) Ze ni, C.; Pinsler , R.; Zügner , D.; Fowle r , A.; Horton, M.; Fu, X.; W ang, Z.; Shysheya, A.; Crabbé, J.; Ueda, S. A generative mod el for in orga nic materials design. Nature 2025 , 639 (8055), 624–632. (23) Jumper , J.; Evans, R.; Pritzel, A.; Green, T .; Figurnov , M.; Ronneber ger , O.; T unyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFol d. natur e 2021 , 596 (7873), 583–589. (24) Joshi, C. K.; Fu, X.; Liao, Y .-L.; Gharakhanyan, V .; Miller , B. K.; Sriram, A.; Ulissi, Z. W . All-atom diffusion trans formers: Unified generative modelling of mol ecules and m aterials. arXiv pr eprint arXiv:2503.03965 2025 . (25) Li, Q.; Miklaucic, N.; Hu, J. Out-of-Distribution Material Property Prediction Using Adversarial Learning. J. Phys. Chem. C 2025 , 129 (13), 6372–6385. (26) Li, K.; Rubungo, A. N.; Lei, X.; Pers aud, D. ; Choudhary , K.; D eCost, B.; Dieng, A. B.; Hattrick-Simpers, J. P robing out-of-distribution generalization in machine learning for materials. Commun. Mater . 2025 , 6 (1), 9. (27) W ei, J.; W ang, X.; Schuurmans, D.; Bosma, M.; Xia, F .; Chi, E.; Le, Q. V .; Zhou, D. Chain - of-thought prompting elicits reasoning in large language models. Adv . Neural Inf. Pr ocess. Syst. 2022 , 35 , 24824–24837. (28) Huang, J.; Chang, K. C.-C. T owards reason ing in large language m odels: A survey . In Findings of the association for computational linguistics: ACL 2023 , 2023; pp 1049–1065. (29) Achiam, J.; Adler , S.; Agarwal, S.; Ahmad, L.; Akkay a, I.; Aleman, F . L.; Almeida, D.; Altenschmidt, J.; Altman, S.; Anadk at, S. Gpt-4 technical report. arXiv pr eprint arXiv:2303.08774 2023 . (30) Y oshikawa, N.; Skreta, M.; Darvish, K.; Arellano-Rubach, S.; Ji, Z.; Bj ørn Kristensen, L.; Li, A. Z.; Zhao, Y .; Xu, H.; Kuramshin, A. Large language models for chemistry robotics. Autonomous Robots 2023 , 47 (8), 1057–1086. (31) M. Bran, A.; Cox, S. ; Schilter , O.; Baldassari, C.; White, A. D.; S chwaller , P . Augmenting lar ge language models with chemistry tools. Nat. Mach. Intell. 2024 , 6 (5), 525–535. (32) Chaudhari, A.; Ock, J.; Barati F arimani, A. Modular lar ge languag e model agents for multi-task computational materials science. 2025 . (33) Jablonka, K. M.; Ai, Q.; Al-Feghali, A.; Badhwar , S.; Bocarsly , J. D.; Bran, A. M.; Bringuier , S.; Brinson, L. C.; Choudhary , K.; Circi, D. 14 e xamples of how LLMs can transform materials science and chemistry: a reflection on a l ar ge languag e model h ackathon. Digital Discovery 2023 , 2 (5), 1233–1250. (34) Zhang, D.; Jia, X.; Tra n, H. B.; Jang, S. H.; Zhang, L.; Sato, R.; Hashimoto, Y .; Sato, T .; Konno, K.; O rimo, S.-i. “DIVE” into hydrogen storage materials discovery with AI agents. Chem. Sci. 2026 . (35) Xin, H.; Kitchin, J. R.; López, N.; Schweitzer , N. M.; A rtrith, N.; Che, F .; Grabow , L. C.; Gunasooriya, G. K. K.; Kulik, H. J.; Laino, T . Roadmap for transforming heterogeneous catalysis with artificial intelligence. Nat. Catal. 2026 , 1–10. (36) T akahara, I.; Mizogu chi, T .; Liu, B. Accelerated inorganic materials design with generative AI agents. Cell Reports Physical Science 2025 , 6 (12). (37) Chiang, Y .; Hsieh, E.; Chou, C. -H.; Riebesell, J. LLaMP: Lar ge language model made powerful for high-fidelity materials knowledge retrieval and distillation. arXiv pr eprint arXiv:2401.17244 2024 . (38) Jia, S.; Zhang, C.; Fung, V . LLMatDesign: A utonomous Materials Discovery with Large Language Models. arXiv pr eprint arXiv:2406.13163 2024 . (39) Zhang, Y .; Sun, R.; Chen, Y .; Pfister , T .; Zhang, R.; Arik, S . Chain of agents: La r ge language models c ollaborating on long-context tasks. Adv . Neural Inf. Pr ocess. Syst. 2024 , 37 , 132208–132237. (40) Liang, T .; He, Z.; Jiao, W .; W ang, X.; W ang, Y .; W ang, R.; Y ang, Y .; Shi, S.; Tu, Z. Encouraging div er gent thinking in large langua ge models through multi -agent deb ate. In Pr oceedings of the 2024 confer ence on empirical methods in natural language pr ocessing , 2024; pp 17889–17904. (41) W ei, C.; Shi, Y .; M u, W .; Zhang, H.; Qin, R .; Y in, Y .; Y u, G.; Mu, T . Large Language Models Assisted Materials Development: Case of Predictive Analytics for Oxygen Evolution Reaction Catalysts of (Oxy) hydroxides. ACS Sustainable Chemistry & Engineering 2025 , 13 (14), 5368–5380. (42) Lin, J.; Zhao, D.; Lu, S.; Li, R.; Xu, X.; W ang, Z.; Li, W .; Ji, Y .; Zhang, C.; Shi, L. Conversational Large-Language-Model Artificial Intelligence Ag ent fo r A ccelerated Synthesis of Metal–Or g anic Frameworks Catalysts in Olefin Hydrogena tion. ACS Nano 2025 . (43) Ock, J.; Guntuboina, C .; Barati Farimani, A. Catalyst ener gy prediction with CatBER T a: unveiling feature exploration strategies through large languag e models. ACS Catal . 2023 , 13 (24), 16032–16044. (44) Mok, D. H.; Back, S. Generative pretrained transformer for heterogeneous catalysts. J. Am. Chem. Soc. 2024 , 146 (49), 33712–33722. (45) Unke, O. T .; Chmiela, S.; Sauceda, H. E.; Gastegger , M.; Poltavsky , I.; Schutt, K. T .; Tkatchenko, A.; Muller , K.-R. Machine learning force fields. Chemical Reviews 2021 , 121 (16 ), 10142–10186. (46) Kulkarni, A.; Siahrostami, S.; Patel, A.; Nørskov , J. K. Understanding catalytic activity trends in the oxygen reduction reac tion. Chemical r eviews 2018 , 1 18 (5), 2302–2312. (47) W ood, B. M.; Dzamba, M.; Fu, X.; Gao, M.; Shuaibi, M.; Barroso -Luque, L.; Abdelmaqsoud, K.; Gharakhanyan, V .; Kitchin, J. R.; Levine, D. S. UMA: A Family of Universal Models for At oms. arXiv pr eprint arXiv:2506.23971 2025 . (48) Nørskov , J. K.; Abild-Pedersen, F .; Studt, F .; Bligaard, T . Density functional theory in surface chemistry and c atalysis. Pr oceedings of th e N ational Academy of S ciences 201 1 , 108 (3), 937–943. (49) Jiao, S.; Fu, X.; Huang, H. Descriptors for the evaluation of electrocatalytic reactions: d‐ band theory and beyond. Advanced Functional Materials 2022 , 32 (4), 2107651. (50) Huang, Z.-F .; Song, J.; Dou, S.; Li, X.; W ang, J.; W ang, X. Strategies to bre ak the scaling relation toward enhanced oxygen electrocatalysis. Matter 2019 , 1 (6), 1494–1518. (51) Craig, M. J.; Coult er , G.; Dolan, E.; S oriano-López, J.; Mates-T o rres, E.; Schmitt, W .; García-Melchor , M. Universal scaling r elations for the rational design of molecular water oxidation catalysts with near - zero overpotential. Nat. Commun. 2019 , 10 (1), 4993. (52) Matheu, R.; Ertem, M. Z.; Benet-Buchholz, J.; Coronado, E.; Batista, V . S.; Sala, X.; Llobet, A. Intramolecular proton transfer boosts water oxidation catalyzed by a Ru c omplex. J. Am. Chem. Soc. 2015 , 137 (33), 10786–10795. (53) Baran, J. D.; Gronbeck, H.; Hellman, A. Analysis of Porphyrine s as Catalysts for Electrochemical Reduction of O2 and Oxidation of H2O. J. Am. Chem. Soc. 2014 , 136 (4), 1320–1326. (54) Y ang, L.; Zhang, Y .; Huang, Y .; Deng, L.; Luo, Q.; Li, X.; Jiang, J. Promoting Oxygen Reduction Reaction on Carbon‐Based Materials by Selective Hydrogen Bonding. ChemSusChem 2023 , 16 (16), e202300082. (55) Zou, H.; Shu, S. ; Y ang, W .; Chu, Y . -c.; C heng, M.; Dong, H.; Liu, H.; Li, F .; Hu, J.; W ang, Z. Steering acidic oxygen reduction sel ectivity of single -atom catalysts through the second sphere ef f ect. Nat. Commun. 2024 , 15 (1), 10818. (56) Pérez-Ramírez, J.; López, N. Strategies to b reak linear scaling relationships. Nat. C atal. 2019 , 2 (1 1), 971–976. (57) Pedersen, A.; Barrio, J.; Li, A.; Jervis, R.; Brett, D. J.; T itirici, M. M.; Stephens, I. E. Dual‐ metal atom electrocatalysts: theory , synthesis, charac terization, and applications. Advanced Ener gy Materials 2022 , 12 (3), 2102715. (58) V aswani, A.; S hazeer , N.; Parmar , N.; Uszkoreit, J.; Jones, L.; Gomez, A. N.; Kaiser , Ł.; Polosukhin, I. Attention is all you nee d. Adv . Neural Inf. Proc ess. Syst. 2017 , 30 , 6000– 6010. (59) Fu, X.; W ood, B. M.; Barr oso-Luque, L.; Levi ne, D. S.; Gao, M.; Dzamba, M.; Zitnick, C. L. Learning smoo th and expressive inter atomic potentials for physical property predi ction. arXiv pr eprint arXiv:2502.12147 2025 . (60) Jacobs, R. A.; Jordan, M. I.; Barto, A. G. T ask decomposition through competition in a modular connectionist architecture: The what and where vision tasks. Cognit ive science 1991 , 15 (2), 219–250. (61) Chanussot, L.; Das, A.; Goyal, S.; Lavril, T .; Shuaibi, M.; Riviere, M.; T ran, K.; H eras - Domingo, J.; Ho, C.; Hu, W . Open catalyst 2020 (OC20) dataset and community challenges. ACS Catal. 2021 , 11 (10), 6059–6072. (62) Kresse, G.; Furthmüller , J. Ef ficiency of ab-initio t otal ener gy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater . Sci. 1996 , 6 (1), 15–50. (63) Kresse, G.; Hafner , J. Ab initio molecular dynamics for open-shell tra nsition metals. Phys. Rev . B 1993 , 48 (17), 131 15. (64) Hammer , B .; Hansen, L. B.; Nørskov , J. K. Improve d adsorption ener ge tics within density- functional theory using revised Perdew -Burke-Ernzerhof functionals. Phys. Rev . B 1999 , 59 (1 1), 7413. (65) Kresse, G.; Joubert, D. From ultrasoft ps eudopotentials to the projector augmented -wave method. Phys. Rev . B 199 9 , 59 (3), 1758. (66) Monkhorst, H. J.; P ack, J. D. S pecial points for Brillouin -zone integrations. Phys. Rev . B 1976 , 13 (12), 5188. (67) Nørskov , J. K.; Rossmeisl , J.; Logadottir , A.; Lindqvist, L.; Kitchin, J. R.; Bligaard, T .; Jonsson, H. Origin of the overpotential for oxyg en reduction at a fuel -cell cathode. J. Phys. Chem. B 2004 , 108 (46), 17886–17892. (68) Larsen, A. H.; Mort ensen, J. J.; Blomqvist, J.; Castelli, I. E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M. N.; Hammer , B .; Hargus, C. The atomic simulation environment — a Python library for working with atoms. J. Phys. Condens. Matter 2017 , 29 (27), 273002. Supplementary Information Supplementary Note A. Details of Prompt T emplate A.1 System Pr ompt Four discovery strategies were implemented withi n Multi-Agent-based Electrocatalyst Search Through Reasoning and Optimization (MAESTRO) framework: history + exploration , history , historyless and random . Except random strategy , which relies on stochastic modification selection, other strategies utilized LLM -based agents. For the LLM-based strategies, both the design and reflect agents operate under strategy specific configurations. In particular , their system prompts, which govern overall behavior throughou t the entire design loop, dif fer depending on the selected str ategy ( T able S1 and S2 ). The summary and exploration report agent do not employ strategy specific prompt variations at the system or input prompt level. Instead, their participation in the framework is determined solely by the chose strategy . Specifically , the summary agent is deactivated for strategies that do not utilize design history , while the exploration report agent is deactivated for strategies that do not include exploration phase ( T able S3 ). A.2 Output Format The design and reflect a gents employ fixed and structured output formats to ensure robust interac tion throughout the design loop. These output formats re main identical ac ross all discovery strategies. If an agent generates a response that deviates fr om the prescribed for mat, for example due to hallucinated or malformed output s, the response is returned to the agent together with explicit feedback describing the formatting error . When an agent repeatedly produces invalid outputs beyond a predefined threshold, the corresponding de si gn run is automatically terminated. The output of the design agent consists of the selected modification type, the associated parameters and the reasoning underlying the proposed change. The reflect agent produces structured feedback that evaluates both the proposed modification and the resulting performance. Based on this reflection, the agent recommends the next catalyst to be modified by selecting fr om a se t of recently modified catalysts stored in the d esign history . This recommendation m echanism functions as an “undo” operation, allowing the framework to revert to a previous d esign state when the modi fied catalyst diverges excessively from th e ta r get region or becomes trapped in repetitive modification cycles. A. 3 Scientific Rule T o ensure consistent physical interpretation across agents, all agents were instructed in the electrochemica l convection for binding Gibbs free ener gy . Unlike general chemical context, where high energy typically indicates strong interactions, binding in electrochemistry is characterized by stron ger binding at lower (more n egative) energy val ue. Because LLM background knowledge is lar gely domi nated by general chemistry conventions, omission of this distinction can lead to systematic re asoning errors. T o miti gate this issu e, we incorporated few-shot examples and terminology to enforce the correct interpretation of binding ener gy ( T able S4 ) T able S1. System prompt templets of design agent Design Agent Sy stem Pr ompt Base Y ou are an expert Computational Chemist Designer specialized in Single Atom Catalysts (SACs). {strategy} {scientific_rules} {output_format} {strategy} history + hxplorat ion Y our task: - T o p ropose one well-reasoned modification for the current catalyst to reach the target property . - T o k eep the catalyst stable. Th e higher dissolution potential indicates higher stability . - T o k eep the catalyst from being too complex. Y ou will be given: - The current catalyst description - T arget type/value or target range s - Recent modification history - Self-reflection of previous modification - A d iscovery strategy (exploration or exploitati on) Y ou MUST : - Propose exactly ONE modification in the 'modifications' list. - W rite a hypothesis (8 to 1 1 sentences) explaining WHY this modification should move the system toward the tar get. - If strategy is 'exploration', DO NOT use same modifications in history . - If strategy is 'exploitation', MUST choose modification based on history . historyless Y our task: - T o p ropose one well-reasoned modification for the current catalyst to reach the target property . - T o k eep the catalyst stable. Th e higher dissolution potential indicates higher stability . - T o k eep the catalyst from being too complex. Y ou will be given: - The current catalyst description - T arget type/value or target range s Y ou MUST : - Propose exactly ONE modification in the 'modifications' list. - W rite a hypothesis (8 to 1 1 sentences) explaining WHY this modification should move the system toward the tar get. Others Y our task: - T o p ropose one well-reasoned modification for the current catalyst to reach the target property . - T o k eep the catalyst stable. Th e higher dissolution potential indicates higher stability . - T o k eep the catalyst from being too complex. Y ou will be given: - The current catalyst description - T arget type/value or target range s - Recent modification history - Self-reflection of previous modification Y ou MUST : - Propose exactly ONE modification in the 'modifications' list. - W rite a hypothesis (8 to 1 1 sentences) explaining WHY this modification should move the system toward the tar get. {output format} {"modifications": [{ "modification_type": "$TYPE", "parameters": ["$PROPER TY_1", "$PROPER TY_2"], "reasoning": "$HYPOTHESIS", }]} $HYPOTHESIS: Y our scientific reasoning. Explain why chosen modification will mo ve the SAC toward the target value. $TYPE: One of the allowed modification types. $PROPER TY_1: The first parameter (e.g., element to remove). If t h e type does not require a parameter , use "None". $PROPER TY_2: The second parame ter (e.g., new element). If the type only requires one parameter , use "None". T able S2. System prompt templets of reflect agent Reflect Agent Sy stem Pr ompt Base Y ou are an expert Computational Chemist Criticism specialized in Single Atom Catalysts (SACs). {strategy} Y our task 1) Reflection (<= 7 sentences): - State whether the modification moved the catalyst toward the tar get o r away from it, - If the result looks unphysical or failed, provide reflectio n on the modification why it was failed. 2) Choose the NEXT starting ca talyst (undo mechanism): Y ou MUST choose exactly one source : - next_catalyst_type = 'recent' > continue from one of RECENT candidates - next_catalyst_type = 'best' > revert to BEST (global best so far) 3) If you choose NEXT starting ca talyst as 'recent', choose index[0..N-1] (0=oldest, N-1=most recent) Decision guidelines: - Choose the MOST RECENT catalyst (RECENT[N -1]) if it is physically reasonable and not worse than other options. - Choose an earlier RECENT index if the most recent result is clearly unphysical, ov erly complex, or significantly further from the target than a previous re cent catalyst. - Choose 'best' if: (a) RECENT candidates show repeated unphysical/divergent behavior (ex, all target value of recent candidates is far from the best), OR (b) BEST is clearly th e most reliable an d closest- to -target state among BEST vs RECENT , especially when the most recent step degraded the result. {output_format} {scientific_rules} {strategy} history + explorat ion Y ou will be given: - The tar get type/value - The modification + hypothesis p roposed by the DesignAgent - Catalyst description and evaluated values before/after the modification - Optional images of optimized structures, - Current catalyst complexity (higher = more complex), - A li st of the N most recent catal ysts (RECENT , chronological: 0=oldest, N-1=most recent), - The global best catalyst so far (BEST) with its property a nd complexity . - A d iscovery strategy (exploration or exploitati on). Strategy-dependent decision guidelines 1) Exploration: - Prefer continuing from RECENT catalyst that has potential to expand catalyst space and avoid cho osing catalysts already used. - Never use BEST . 2) Exploitation - Prefer reverting to BEST or RE CENT ca talyst with good performance when the most recent step degraded performa nce or increased complexity without benefit. - Continue from RECENT only if the new state is reliable and comparable to BEST (or clearly impr oving toward target). Others Y ou will be given: - The tar get type/value - The modification + hypothesis p roposed by the DesignAgent - Catalyst description and evaluated values before/after the modification - Optional images of optimized structures, - Current catalyst complexity (higher = more complex), - A li st of the N most recent catal ysts (RECENT , chronological: 0=oldest, N-1=most recent), - The global best catalyst so far (BEST) with its property a nd complexity . {output format} { "reflection": "", "next_catalyst_type": <'recent' or 'best'> "next_catalyst_index": , "next_catalyst_reason": "<4~5 se ntences explaining why this catalyst is the best starting point for the next iteration and why didn't choose another option>" } T able S3. System prompt templets of summary and exploration report agent Summary Agen t System Pr ompt Base Y ou are a Historian of a Computational Chemist experiment specialized in Single Atom Catalysts (SACs) Y ou will be given a list of modification steps (modification, results) performed by catalyst design agent. Y our task is to summarize the progress into a concise paragraph. Present the summary while keeping the following points in mind. - Which modification ind uced an increase or decrease in Gibbs free energ y (Delta G). - Which modification was succe ssful or unsuccessful in achieving the target. - Which modification was the most critica l. Keep it under 200 words. This summ ary will be read by the Designer to decide the next step. {scientific_rules} Exploration Repor t Agent System Pr ompt Base Y ou are ReportAgent, an expert Computational Chemist analyst specialized in Single Atom Catalysts (SACs). Y ou are invoked once after iterations of EXPLORA TION and immediately before switching to EXPLOIT A TION. Y ou will be given: - The full exploration history: each it eration's starting catalyst, applied modification (type + parameters), DesignAgent hypothesis, evaluator results (before/after values), ReflectionAgent asse ssment, and complexity . Y our task: W rit e a co ncise, 1-p age report that summarizes explo ration outcomes in a way that directly impro ves exploitation decisions. This is not a narrative; it is a decision-ready technical brief. {scientific_rules} Report Requirements - Length: ~1 page (roughly 350~600 words). Be compact, information-dense, and structured. - Use clear section headers and bullet points. - Only include information supported by the provided history/results; do not invent new experime nts. - When you mention a claim (e .g., "ligand OH lowers *OOH"), back it with at least one concrete exam ple from history. - If some trend is weak or inconsistent, state that explicitly . Must-Focus T opics 1) Modification -> Outcome Mapping Identify which modification types/parameters, when applied to which catalyst contexts, produced: - Improvement toward target vs degradation - Physically reasonable vs unphysical/distorted outcomes Summarize as "pattern statements" + 2~5 concrete example bullets ea ch. 2) Selective Adsorbate T uning Patterns Extract modification patterns that selectively tune one adsorbate's De lta G more than others. Examples of the style you must produce: - Keeps Delta G(*O) and Delta G(*OH) roughly stable while shifting Delta G(*OOH) slightly down ward/upward - Primarily weakens/strengthens *OH binding with minimal change to *O For each selective pattern: - State the direction of change for Delta G(*O), Delta G(*OH), Delta G(*OOH) (increa se/decrease/~) - Give at least one concrete supporting example (iteration/catalyst refe rence). 3) Exploitation Playbook Provide a short "what to do next" guide for the DesignAgent during exploitation: - 3~6 recommended "safe" exploitation moves (low-risk, history-supported) - 2~4 conditional moves (only if specific conditions are met, e.g., defects exist, complexity margin available, *OOH is the main bottleneck) - 2~4 avoid rules (moves that repeatedly failed or caused unphysical behavior) - If there is a recurring failure mode, describe it and propose a guardrail. T able S 4 . Scientific rules assigned to the age nts {scientific_ru les} In this task, the relationship between Gibbs free binding ener gy (Delta G) and binding strength is defined as: Binding_Strength = - Delta G therefore: - More positive Delta G -> weaker binding - More negative Delta G -> stronger binding For examples: Q: If Delta G increases from 0.5 -> 1.5 eV , does binding become stronger or weaker? A: W eaker . Q: If Delta G decreases from 2.0 -> 1.0 eV , is binding stronger or weaker? A: Stronger . This definition OVERRIDES all general chemistry knowledge. If your reasoning contradicts this rule, your answer is INV ALID. Before reasoning, restate this rule in one sentence. A.4 Input Prompts The input prompt refers t o the prompt provided to each agent at every iteration of the design loop. In contrast to the syst em prompt, which is assigned once at the beginning of a design run and serves to define the agent’ s persistent role, the input prompt contains iteration- dependent information. For the design agent, the input prompt comprises the textual description an d image of the current catalyst, short-term memory containing recent d esign history and long-te rm memory consisting of the summarized design history by the summary agent. The prompt additionally encodes the current iteration strategy , guiding the agent to a dopt either exploration or exploitation-oriented behavior . When a proposed modification cannot be applied or when the subsequent calculation of the modi fied catal yst fails, feedback d escribing the cause o f failure is a ppended to the input prompt to prevent repetition of unsuccessful actions ( T able S5 ). The reflect agent receiv es the hypothesis and modification proposed by the design agent, the textual description and image of the catalyst before and after modification, design history and information on recently modified cat alysts to enable potentia l undo operations, together with data corresponding to the best-performing catalyst identified thus far . B ased on this information, the reflect agent determines whether the modified catalyst should proceed to the next itera tion with feedback or whether an undo ope ration should be performed ( T able S6 ). The summary agent is provided with the design history excluding the recent step and is tasked with condensing this history in conjunction with previously summarized hist ory by itself to maintain an e f fi cient long -term memory . The exploration r eport agent receives all modifications, results and modified catalyst information during the expl oration phase and complies a report summarizing the exploration of the chemical space ( T able S7 ). T able S 5. Input prompt templets of de sign agent Design Agent Inpu t Pro mpt Base {strategy} {feedback} Propose modifications to tune its Gibbs free ener gy ( ΔG) of *O, *OH, *OOH adsorbates to a tar get value to reduce ORR overpotential based on given information. T arget Gibbs free ener gy of *O: {2.46 - threshold} ~ {2.46 + threshold} eV T arget Gibbs free ener gy of *OH: {1.23 - threshold} ~ {1.23 + thre shold} eV T arget Gibbs free ener gy of *OOH: {3.69 - threshold} ~ {3.69 + threshold} eV Current state of catalyst is {textual description} The {num_recent_history} recent modificaitons, reasonings, feedbacks and self-reflections are following: {formatted_history}. The summary of modification history is {summarized_history }. The simplified history of previous modifications is {simplified_history}. {strategy} Exploration Phase Exploitation Pha se STRA TEGY : EXPLORA TION Y our primary objective is to explore uncertain but plausible regions of the catalyst space Rule: - Y ou MUST not suggest the mod ifications alreday used in history - Just make sure the complexity doesn't exceed the maximum, do not care about it below that. Below is the list of modifications that alreday used: {modification_list} STRA TEGY : EXPLOIT A TION Y our primary objective is to explore uncertain but plausible regions of the catalyst space Rule: - Choose proper modifications to reach the tar get by referring to the Gibbs free energy change in the previous history and report. - T ry to maintain complexity of catalyst moderate - Never suggest the failed combination of the modification + current catalysts already used in history . Below is the report that summarized the exploration phase: {exploration_report} Y ou must still aim toward the targe t, but information gain has priority over immediate best performance. Performance toward the target ha s priority over novelty . {feedback} If recent modif ication succeeds If recent modifica tion fails - Y our recent modifications {previous_ modifications} are failed. The reason of the most recent failure is {failed_reason}. Please re-propose the modification based on the given format and information. NEVER suggest same modification with previous failed modifications T able S 6. Input prompt templets of re flect agent Reflect Agent Input Pro mpt Base {strategy} Design agent suggested following hypothesis and modification. Hypothesis: {reasoning} Modification: {modification} After completing the modification, we obtained the following catalyst Before modification: {previous_catalyst_textual_description} After modification: {current_catalyst_textual_description} Please write a brief post-action reflection on the modification in less than five sentences, \ explaining how successful it was in achieving {2.46 - threshold} ~ {2.46 + threshold} eV f or Gibbs free energy of *O {1.23 - threshold} ~ {1.23 + threshold} for Gibbs free energ y of *OH and {3.69 - threshold} ~ {3.69 + threshold} for Gibbs free energ y of *OOH, and the reasons for its success or failure.\ The {num_recent_history} recent modificaitons, reasonings and feedbacks is following: {formatted_history}. The summary of history is {summarized_history} The simplified history of previous modifications is {simplified_history}. Recommended maximum value of complexity is {max_complexity}. Global best catalyst found so far is {best_catalyst_textual_description }. Recent catalyst list is following (oldest to newest): {recent_catalysts_list} If all target value of recent catalys ts is fall apart from the best, choose 'best' {strategy} Exploration Phase Exploitation Phase STRA TEGY : EXPLORA TION Y our priority is information gain and coverage of the catalyst space. - Prefer continuing from RECENT catalyst that has potential to expand catalyst space and avoid choose catalysts already used. - MUST explore as many catalyst + modification possibilities as possible. STRA TEGY : EXPLOIT A TION Y our priority is fast convergence toward the tar g et with reliable, low-risk steps. - Recommends you judge to be the most suitable catalyst for the designer to achieve the tar get.. - Balance the use of RECENT an d BEST . DO NOT stick to the BEST and select ca talysts with the potential to achieve new BEST . T able S 7. Input prompt templets of summary and exploration re port agent. Summary Agen t Input Pr ompt Base Following is recent summarized history by yourself: {summarized_history} And following is recent detailed history: {formatted_history} Please summarize history based on given summarize d and detailed history \ Exploration Repor t Agent Input Pr ompt Base Following is detailed history: {formatted_history} Please write the report based on history A.5 Formatting T o communicate catalyst information to LLM-b ased agents, the catalyst structure must be expressed in a form compatible with natur al language. However , accurately d escribing geometric details such as interatomic bonding and atomic coordinate using text alone remains challenging. Although se veral a pproaches fo r representing materials in nat ural language have been propo sed 1 , this study focuses on the relatively simple single atom catalyst (SAC) system. Accordingly , we designe d a textual description that encodes o nly the k ey structural features characteristic of SACs and energy-related properties as shown in Figure S1a . In addition, all LLMs employed in this work are multimodal and capable of processing image inputs. Since two orthogonal perspectives are suf ficient to capture the full geometry of SACs, both top view and side view images of each catalyst and its bindi ng configurations were provided to th e agents alongside the textual description ( Figure S1b ). The design history was also formed to facilitate efficient int erpretation by the LLMs. T wo distinct history formatting methods were employed. The first format retains comprehensive information, including the modification type, reasoning from the de sign agent, and the fe edback from the reflect agent. This formatted history serves as short-term memory . The second format is a condensed representation that records only the applied modi fication and their corresponding energy changes, excluding agent intervention. This sim plified history functions as long-term memory preserving only objective state transitions. Figure S1. ( a) Examples of textual description of SAC. (b) Examples of SAC image provided to the agents. Figure S2. Schematic of the formatted and simplified history representation derived from the information stored at each design step. Supplementary Note B. Details of Pr e -validation for MLFF an d LLM B.1 Dataset In this study , we employed pre-trained UMA as MLFF surrogate for DFT . Because SAC syst em was not included in the training data of UMA, it was necessary to assess its optimization reliability for SACs prior to application within the design framework. Moreover , as no publicly available SAC benchmark d ataset su itable for this purpose exists, we constructed a dedicate validation dataset by performing DFT calculations on structure representative of those expected to emer g e during the catalyst design loop. For this validation dataset, geometry opti mizations were carried out for M -N 4 SACs with nine diffe rent transitions metals (Co, Cu, Fe, Ir , Mn, Ni, Pd, Pt, Ru) as the center atom, considering three intermediates (*OOH, *O, *OH) as well as configurations in which an additional axial ligand (OH) binds simultaneously with the adsorbate. T his resulted in a datase t comprising 3,107 optimiza tion image s, c orresponding 3,107 DFT total ener gies, 579,057 atomic forces and 54 binding ener gies. T o furthe r evaluate prediction accuracy under variations in the local environment of the binding site, ad ditional datasets were generated for CoN 4 systems featuring COC or COH functional groups on the s econd shell of carbon support, as well as structures in whi ch first shell N atom we re subst ituted with hydrogen or removed to form defects. This data c onstruction yielded an additional 420 DFT total energies and 76,098 atomic forces. B.2 Pre-validation of MLLFF V alidation was primarily performed using the UMA with the ‘OC20’ domain, which is weighted toward heterogenous alloy catalysis systems and was also employed in the actual design framework. F or comparison, validation was additionally conducted using UMA with the ‘OMat’ domain, which is biased toward crystalline bulk mate rials. The results revealed that UMA with the ‘OMat’ domain e xhibited substantially inferior prediction pe rformance ( Figure S3 ) compared to the model weighted on ‘OC20’. In contrast, the UMA with ‘OC20’ maintained high auccracy even for SACs with modified local environments ( Figure S4 ) Notable, the validation datasets include systems that were absent from the UMA training data, such as tw o-dimensional materials with functional groups or defects, octahedral geometrics involving simultaneous ligand and adsorbate binding and *OOH adsorbate. Considering these out -of-distribution characteristics, the performance demonstrated by UMA in this study is particularly significant, supporting its suitability as a surrogate model. Figure S3. Parity plots between MLF F-predicted and DFT -calculated ener gies per atom , atomic forces and binding ener gies of pristine M- N 4 SAC system. In this prediction, UMA with OMA T task is used as M LFF . Figure S4. Parity plots between MLFF -predicted and DFT -calculated energies per atom and atomic forces of modified M-N 4 SAC system. In this pre diction, UMA with OC20 task is used as MLFF . B.3 Pre-validation of L LM In most cases, the modifications proposed by the LLM induced changes in binding ener gies consistent with both the intended direction and the underlying reasoning provided by the model. Nevertheless, a limited number of dis crepancies were observed. The first type of inconsistency arises fro m structural distortions driven by local environment ef fects, as illustrated in Figure S5a . In thi s case, a functional group located in the second shell migrated toward the m etal center during MLFF -optimization. This migration destabilized the bare catalyst structur e, inc reasing its DFT total en er gy and consequently leading to a reduction in the binding ener gy . Although the LLM correctly anticipated the change in electron density , it failed to predict binding ener gy tr end due to the unforeseen structural rearrangement. A second sou rce of incon sistency origina tes from i ncorrect analogies to prior c ases, a s shown in Figure S5b . Even when identical modifications are applied, the resulting binding ener gy tr ends can differ depending on the current condition of catalyst. In such instances, the LLM neglected this contextual dependence, leading to erroneous predictio ns for both binding ener gy and electronic density changes. T o miti gate these limitations within the proposed f ramework, we introduced additional mechanisms to inform the LLM when substantial structural rearrangements occur after geometry optimization, such as changes in binding c onfigurations. Furthermore, modi fications were recorded in the design history together with updated textural des criptions of the catalyst structures. This approach enables LLM to associate binding energy trends with both the catalyst environment and the applied modification, ther eby improving it s contextual reasoning in subsequent design iterations. Figure S 5. Examples of discrepancies b etween re asoning of th e design agent and calculated results. (a) Case of binding site change due to movement of functional group. (b) Case of incorrect reference to previous observation. Figure S6. An example of e xploration summary report from exploration report agent. Figure S7. Number of the unique modifications proposed during the 50 design steps, a veraged over 10 independent desi gn runs. The labels “Exp loration” and “Exploitation” correspond to the exploration and exploitation phase of history + exploration strategy , respectively , Figure S8. Evolution of overpotential and dissolut ion potential averaged over 10 independent design runs using (a) history , (b) historyless and (c) random strategy . Figure S9. Evolution of the Gibbs free binding energies of *OOH, *O a nd *OH over 100 design steps from a design run that failed to break the scaling relations. S haded regions denote the optimal ener gy windows required to a chieve and overpotential below 0.1 V . Because the binding ener gies increase and de crease concurrently according to the scaling relations, the three intermediates cannot simultaneously fall within their respective optimal ranges. Figure S10. An example of design and self-reflection workflow lea ding to a minimum overpotential (0.18 V). The design agent suggests a modification by leveraging accumulated design history to selectively tune binding energies. The r eflect agent subsequently evaluate s modification and calculation results, confirms its e f fectiveness and provide feedback to guide the next design step. Figure S1 1. Input and output t oken usage changes by p rogress of d esign run. The historyless strategy maintains nearly constant input token usage comparable to the initi al value, wherea s the history and history + exploration strategies exhibit a gradual increase in input token usage as design history accumulates. Figure S 12. V arious representative modification process to design pro mising catalysts by breaking scaling relations and lower limit of overpotential. Figure S13. Representation of one of high -performance catalyst structures discovered during the design runs, together with a comparison of MLFF-predicted and DFT -calculated performance metrics. Data includes Gibbs free bindi ng energies, overpotential ( η ) and dissolution potentials ( U diss ) for each candidate. Figure S14. Comparison of overall design per formance among (a) different LLMs and (b) dif ferent starting SACs. The dashed line indicates lower limi t of overpotential defined by the scaling relations. Figure S15. Minimum overpotentials averaged over 10 design runs with each tempe rature setting. The dashed line indicates lower limit of overpotential defined by the scaling relations. T able S8. The Gibbs free energy correction value s for adsorbates and gaseous molecules. All units are in eV . Adsorbates ZPE ∫C p dT - TS Molecules ZPE ∫C p dT - TS O* 0.08 0.03 -0.06 H 2 0.28 0.09 -0.41 OH* 0.37 0.04 -0.07 H 2 O 0.57 0.10 -0.67 OOH* 0.44 0.05 -0.09 Refer ence (1) Jia, S.; V arma, A.; Manivannan, P .; Chayapathy , D.; Fung, V . Benchmarking T ext Representations for Crystal Structure Generation with Lar ge Language Models. In AI for Accelerated Materials Design-ICLR 2025 .
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment