Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction
Graph self-supervised learning (GSSL) has demonstrated strong potential for generating expressive graph embeddings without the need for human annotations, making it particularly valuable in domains with high labeling costs such as molecular graph ana…
Authors: Jiele Wu, Haozhe Ma, Zhihan Guo
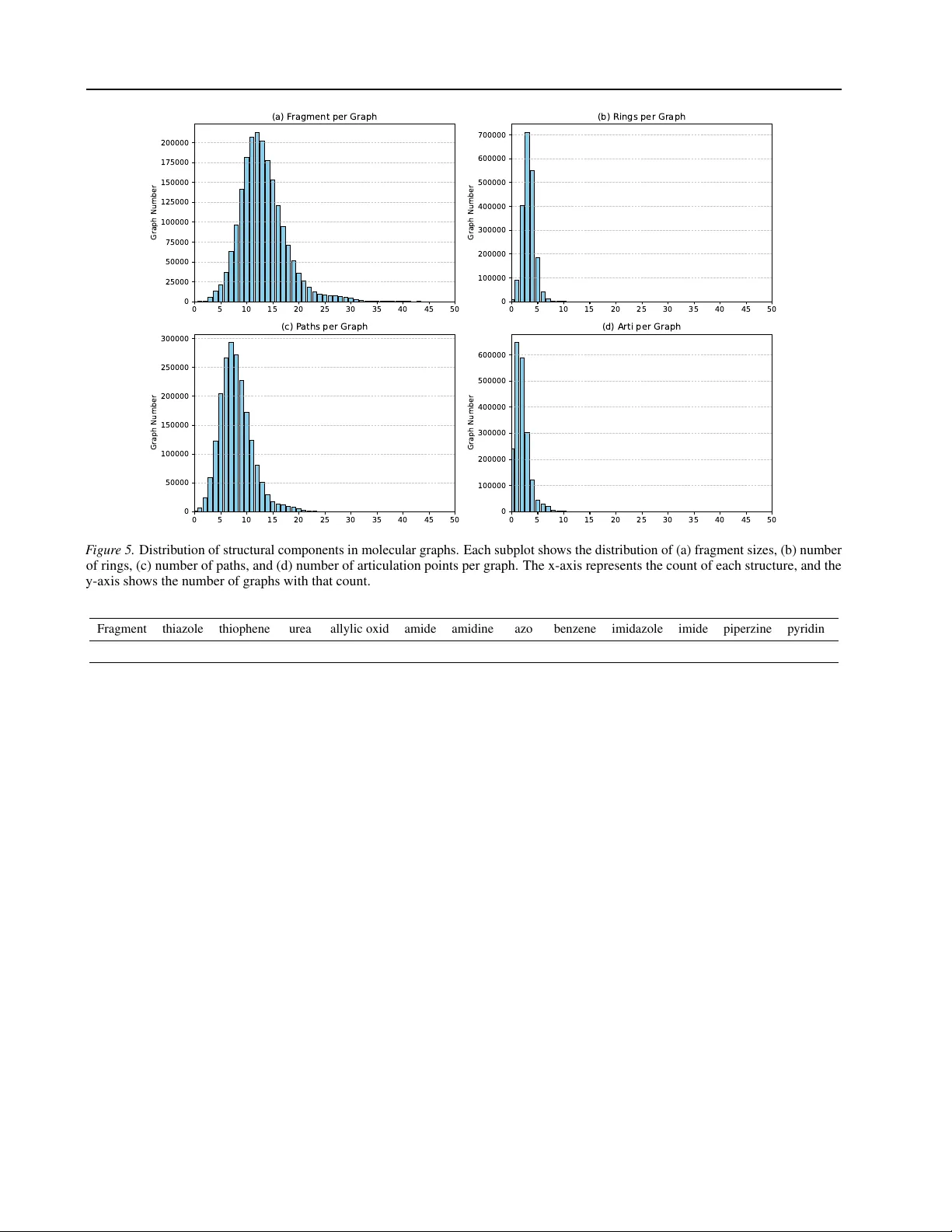
Hierar chical Molecular Repr esentation Learning via Fragment-Based Self-Supervised Embedding Pr ediction Jiele W u 1 Haozhe Ma 1 Zhihan Guo 2 Thanh V inh V o 1 Tze Y un Leong 1 Abstract Graph self-supervised learning (GSSL) has demonstrated strong potential for generating e x- pressiv e graph embeddings without the need for human annotations, making it particularly valu- able in domains with high labeling costs such as molecular graph analysis. Howe v er , exist- ing GSSL methods mostly focus on node- or edge-le vel information, often ignoring chemically relev ant substructures which strongly influence molecular properties. In this work, we propose Gra ph S emantic P redicti ve Net work (GraSPNet), a hierarchical self-supervised frame work that ex- plicitly models both atomic-lev el and fragment- lev el semantics. GraSPNet decomposes molecu- lar graphs into chemically meaningful fragments without predefined vocab ularies and learns node- and fragment-lev el representations through multi- lev el message passing with masked semantic pre- diction at both levels. This hierarchical seman- tic supervision enables GraSPNet to learn multi- resolution structural information that is both ex- pressiv e and transferable. Extensiv e experiments on multiple molecular property prediction bench- marks demonstrate that GraSPNet learns chemi- cally meaningful representations and consistently outperforms state-of-the-art GSSL methods in transfer learning settings. The code will be re- leased upon acceptance. 1. Introduction Graphs are a po werful tool for representing structured and complex non-Euclidean data in the real world, as they nat- urally capture relationships and dependencies between en- tities ( Ma & T ang , 2021 ; Bacciu et al. , 2020 ). T echniques such as Graph Neural Networks (GNNs) ha ve demonstrated 1 School of Computing, National Univ ersity of Singapore, Sin- gapore 2 Computer Science and Engineering, The Chinese Univer - sity of Hong Kong, China. Correspondence to: Tze Y un Leong < leongty@nus.edu.sg > . notable success in this context, enabling models to cap- ture both local and global structural information ( Corso et al. , 2024 ). Molecules are inherently graph-structured data, where atoms and chemical bonds are represented as nodes and edges, respecti vely . Molecular representation learning focuses on deriving meaningful embeddings of molecular graphs, forming the foundation for a wide range of applica- tions, including molecular property prediction ( Y ou et al. , 2020 ), drug discov ery ( Gilmer et al. , 2017 ), and retrosyn- thesis ( Y an et al. , 2020 ). Howe ver , the costly process of la- beling molecular properties and the scarcity of task-specific annotations underscore the need for self-supervised learning approaches in molecular representation. In self-supervised learning, supervisory signals are deri ved directly from the input data, typically following either in variance-based or generati ve-based paradigms ( Liu et al. , 2021 ). These approaches aim to learn mutual information between dif ferent views of a graph by applying node-le vel and edge-le vel augmentations ( Zhang et al. , 2021a ), or to re- construct certain characteristics of the graph from original or corrupted inputs ( Kipf & W elling , 2016b ; Hou et al. , 2022 ). Howe ver , for graph classification tasks such as molecu- lar property prediction, chemically meaningful substruc- tures—such as functional groups—often play a decisive role in determining molecular behavior ( Ju et al. , 2023 ). While reconstructing atoms (nodes) and bonds (edges) helps capture local structure, it may fail to encode higher-le vel semantics that are critical for downstream tasks requiring a deeper understanding of molecular graphs. The main challenge lies in preserving different lev els of semantic information within graph data. Figure 1 illus- trates the hierarchy of representation learning in molecular graph analysis, progressing from chemical representations to graph-le vel embeddings and ultimately to downstream tasks. In other domains, such as natural language process- ing and computer vision, tokenizer is commonly used to divide input data into smaller units—such as words or pixel patches—that encapsulate semantically meaningful infor- mation for representation learning ( V aswani et al. , 2017 ; Dosovitskiy et al. , 2021 ). Methods that adapti vely adjust the size and position of patches to capture richer semantic regions from images further underscore the importance of 1 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction C1 0 H1 0 O 4 SMI L ES: C(O )(= O)C= CC 1 =C C= C(O )C(OC )=C1 Structure Formula No de - lev e l Chemical Group Fr agm e n t - lev e l Graph Represen t ation Molecule- le vel … … Downstream Ta s k s Phy s io lo gy Bio ph y s i c s Phy s ic al Chem istry Qua ntu m Mec hanic s …… F igure 1. An example of hierarchical representation learning on molecular graph. The molecule is represented as a string-based notations (SMILES) and encoded at three semantic levels—node (atoms), fragment (e.g., functional groups), and graph to support various downstream tasks. semantic preservation in data ( Chen et al. , 2021 ). In graphs composed of nodes with relational connections, clusters of interconnected nodes often carry significant information and serve as key indicators of the graph’ s ov erall charac- teristics ( Milo et al. , 2002 ). Therefore, the exploration of fragment-lev el semantic information in graphs is of great importance. Although fragment-based representation learn- ing techniques ha ve been proposed, they often rely on dis- crete fragment counting ( Kashtan et al. , 2004 ), which may lack chemical validity , or on generativ e processes that are computationally intensiv e ( T ang et al. , 2020 ; Zhang et al. , 2021b ). Consequently , a more efficient and generalized method for capturing rich semantic information in graphs remains largely une xplored. In this work, we focus on the problem of extracting seman- tically rich representations from lar ge pre-trained molecular structure datasets. W e aim to address the follo wing ques- tion: Ho w can we better utilize the hierarchical semantic information in large unlabeled molecule graph databases and generalize to various downstream tasks? W e propose Gra ph S emantic P redictiv e Net work ( GraSPNet ), a hierar- chical pretraining method based on predicting the representa- tions of nodes and fragments without relying on pre-defined chemical vocab ularies. In contrast to pre vious approaches that focus on reconstructing node-level inputs or predict- ing node representations, GraSPNet utilizes both nodes and fragments representation as prediction targets, benefiting from information at multiple resolutions. By incorporat- ing a hierarchical conte xt and target encoder , the model is guided to learn representations that capture multiple lev- els of semantic information, including fine-grained node features and structures, node–fragment dependencies, and coarse-grained fragment patterns. 2. Related W ork Pretraining on Graphs. GNNs are a class of deep learn- ing models designed to capture complex relationships by aggregating the features of the local neighbors of the node through neural networks ( Hamilton et al. , 2017 ; Kipf & W elling , 2016a ). T o alle viate the generalization problem of graph-based learning, graph pretraining has been activ ely explored to benefit from large databases of unlabeled graph data. Pretraining methods on graphs can be categorized as contrastiv e methods and generativ e methods. Graph con- trasti ve learning methods ( Y ou et al. , 2020 ; Zhu et al. , 2021 ; Y ou et al. , 2021 ; Zhao et al. , 2021 ; Y u et al. , 2022 ) learn in variant representations under graph augmentations, while generativ e methods like Graph Autoencoders ( Hinton & Zemel , 1993 ; Pan et al. , 2018 ; W ang et al. , 2017 ; P ark et al. , 2019 ; Salehi & Davulcu , 2020 ) rely on reconstruction objec- tiv es from the input graph. Recently , masked autoencoder framew orks ( He et al. , 2022 ) including GraphMAE ( Hou et al. , 2022 ), S2GAE ( T an et al. , 2023 ), MaskGAE ( Li et al. , 2023 ) where certain node or edge attrib utes are perturbed and encoders and decoders are trained to reconstruct them with the remaining graph. Fragment-based GNN. Representation learning on molecules has made use of hand-crafted representation in- cluding molecular descriptors, string-based notations, and image ( Zeng et al. , 2022 ). Besides the node- and graph-lev el methods which represent atoms as nodes and bonds as edges, fragment-based approaches explicitly modeling molecular substructures to learn higher-order semantics. Existing methods mostly generate fragments based on pre-defined knowledge or purely geometric structure and learn fragment representations through autoregressi ve processes ( Zhang et al. , 2021b ; Jin et al. , 2020 ; Rong et al. , 2020 ). These methods treat fragments merely as graph tokenizer ( Liu et al. , 2024 ), decomposing molecules into various of recon- 2 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction struction units without fully incorporating fragment-level semantics into the learned representation. 3. Preliminaries Giv en a graph G = V , A, X , where V is the set of N nodes (atoms), A ∈ R N × N is the adjacency matrix, and X = [ x 1 , x 2 , · · · , x N ] ⊤ ∈ R N × D is the node feature ma- trix. Each entry A ij ∈ 0 , 1 indicates the presence ( A ij = 1 ) or absence ( A ij = 0 ) of a chemical bond between atoms i and j . Each node feature x i represents an atom’ s properties, encoding its individual characteristics. 3.1. Graph Neural Network Graph Neural Network relies on message passing to learn useful representations for node based on their neighbors. Giv en an input graph G = { V , A, X } , the node embedding is calculated by: H k = M ( A, H ( k − 1) ; W ( k ) ) = ReLU ( ω ( e A ) H ( k − 1) W ( k − 1) ) , (1) where e A = A + I , e D = P j e A ij and W ( k ) ∈ R d × d is a trainable weight matrix. ω ( e A ) is the normalization oper- ator that represents the GNN’ s aggregation function. An additional readout function aggregates the final node em- beddings into a graph-lev el representation H G , defined as: H G = READOUT( H k v | v ∈ V ) , (2) where V is the node set and k denotes the layer index. The R E A D O U T operation is a permutation-in variant pooling function, such as summation. The resulting graph repre- sentation H G captures global structural and node-le vel se- mantic information of the graph and can be used for v arious downstream tasks. 3.2. Masked graph modeling Masked Graph Modeling (MGM) aims to pre-train a graph encoder using component masking for downstream appli- cations. Specifically , it masks out some components (e.g., atoms, bonds, and fragments) of the molecules and then trains the model to predict them gi ven the remaining compo- nents. Giv en a graph G = { V , A, X } , the general learning objectiv e of MGM can be formulated as: L MGM = − E V m ∈ V " X v ∈ V m log p ( f V m | V \ V m ) # , (3) where V m denotes the masked components of graph V and V \ V m are the remaining components. 4. Methodology The fundamental architecture of the GraSPNet is illustrated in Figure 3 . In general, the architecture is designed to WL = Frag- WL ≠ G 2 G 1 F 1 F 2 Nod e - lev e l Fra g - lev e l Articula tion P oint Atom R ings Atom Paths Frag e dge s Node - > F rag Frag -> Nod e F igure 2. Illustration of our graph fragmentation process. The left figure sho ws graph G 1 and G 2 which can not be distinguished by WL test while higher -level fragment graph F 1 and F 2 exhibit different connections that can be distinguished by WL. The right figure sho ws an example of our graph fragmentation. The ring is first selected, follo wed by the extraction of multiple paths. The articulation points are designated as unique fragment types to prev ent cycles in the fragment graph. predict the representations of multiple semantic target by lev eraging the learned representations of a context graph with missing information. In this section, we detail the design and implementation choices for each component of the architecture. 4.1. Graph Fragmentation Understanding data semantics is often considered essential in machine learning ( Fei et al. , 2023 ). Graph fragmentation methods aim to decompose an entire graph into structurally informativ e subgraphs that are closely related to the graph’ s properties. This hierarchical structure is crucial for enhanc- ing the expressi veness of GNNs. The widely used WL-test ( Leman & W eisfeiler , 1968 ) which captures structural differences between graphs by repeat- edly updating node labels based on their neighbors had been prov ed to be the expressi veness upper bound of message passing network ( Xu et al. , 2019 ). Higher-le vel fragmenta- tion graph with nodes and fragments representation together with the interaction between them is more expressi veness and can be more powerful than 2-WL test in distinguish graph isomorphisms. As illustrated in Figure 2 , WL-test fails to distinguish between G 1 and G 2 where both graphs exhibit symmetric structures at the atom lev el, leading to 3 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction identical label refinement through WL iterations. Ho wev er , as for their corresponding fragment graphs, F 1 forms a sim- ple two-node graph, while F 2 includes a three-node chain with a distinct central ‘path’ fragment. This structural asym- metry is detectable by the WL test on the fragment le vel, enabling better discrimination of molecular graphs. Existing fragmentation methods are typically rule-based procedures, including BRICS ( Degen et al. , 2008 ) and RE- CAP ( Lewell et al. , 1998 ) which follow chemical heuris- tics but often generate large, sparse vocab ularies with low- frequency or unique fragments, limiting generalization. Oth- ers, like METIS ( Karypis & Kumar , 1998 ), use graph parti- tioning algorithm to minimize edge cuts may disrupt chemi- cally meaningful structures and produce non-deterministic, molecule-specific fragments. T o construct a fragmented graph with high e xpressiveness and strong generalization ability , the fragmentation method should both capture key structural fragments and general- ize across diverse molecular graphs. W e propose a frag- mentation strategy that decomposes each molecule into rings, paths and articulation points, forming a higher -level graph that supports learning both fragment representations and their structural relationships. Specifically , given the SMILES of a molecule, we transform it into graph with G = { V , A, X } using RDKIT where V and A are atoms (nodes) and bonds (edges) respecti vely , X is the corre- sponding atom feature. V is then divided into subgraphs V 1 , . . . , V m where m is the number of generated fragments using the follo wing three steps. Ring extraction. W e first identify all minimal rings in the molecular graph and form the first type of subgraphs, each corresponding to a ring. Path extraction. For the remaining nodes and edges, we extract paths in which all intermediate nodes ha ve degree of 2, while the endpoints may hav e other degrees. These form the second type of subgraphs. Articulation point extrac- tion. Finally , any remaining nodes with degree greater than 3 are selected as individual articulation-point subgraphs. This process results in a set of overlapping subgraphs, where each node is assigned to e xactly one subgraph, except for connector nodes that bridge tw o adjacent substructures. Fig- ure 2 illustrates an example of our fragmentation method. This design preserves topological information and prev ent most circles in the fragmentation graph, enabling ef fectiv e fragment-lev el representation learning. All fragments induce a ne w fragment node set V f , with an associated adjacency matrix A f ∈ [0 , 1] m × m representing the connectivity between fragments and A nf ∈ [0 , 1] n × m mapping the original node to fragments. Specifically , ∀ ( i, j ) , A f ij = 0 ⇒ V f i ∩ V f j = ∅ , A f ij = 1 ⇒ V f i ∩ V f j = ∅ , indicating whether two fragments share o verlapping nodes. The size of the overlapping corresponds to the minimum cut value between the tw o fragment in the original graph. 4.2. Model Based on the giv en graph with fragmentation, we proposed GraSPNet which is a predictive architecture focused on learning fragmentation level information. The model is constructed by nodes and fragmentation encoding, conte xt encoder , target encoder , and predictor which is shown in Figure 3 . 4 . 2 . 1 . F R AG M E N T E N C O D I N G W e first introduce the graph information encoding which includes both node encoding and fragmentation encoding. Previous w ork generate fragment embeddings by aggregat- ing nodes or one hot encoder . T o achieve better general- ization and ensure that similar fragmentations share similar initial features, we follow the ( W ollschl ¨ ager et al. , 2024 ) by incorporating the fragment class and size into the em- bedding. Formally , the fragment embeddings are generated as: h f = W 1 · X ( f ) ∥ α · ( W 2 · X ( f )) , (4) where h f is the fragment embedding, W 1 , W 2 are tw o learn- able encoding matrix, X ( f ) is the one-hot initial vector representing fragment type, scaling factor α is equals to the fragment size. 4 . 2 . 2 . C O N T E X T E N C O D E R W e design our context encoder to lev erage hierarchical rep- resentation learning. T o train the encoder , a destructi ve data augmentation is applied by randomly masking nodes and fragments in the input graph. The masking process is modeled as a Bernoulli distrib ution applied independently to each node and fragment which is defined formally as follows: V m ∼ B ernoul li ( p ) , where p < 1 is the masking ratio. W e denote the masked node set as V m and the remaining node set as V − V m . The fragment masked set are define as V F m with the same p . The input node features X n ∈ R d n × 1 are projected into d - dimensional embedding through a linear embedding layer: h n i = W n · X n i + b, where W n ∈ R d × d n and b ∈ R d are learnable parameters. h f is generated from Equation 4 . Giv en the initial node and fragment embeddings, we ap- ply a series of L message-passing layers, where both node and fragment representations are iterativ ely updated using a 4 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction Frag mentati on CC 1= CC =C(C =C1 ) CC C(= O) C Ran do m Mas k Emb ed di ng Ext ract io n Mes sa ge Pas sing Fragm ent Emb ed di ng Message Passing No de Emb ed ding No de - Frag M P s Fra g -l e vel M Ps Fra g - node MPs No de - fr agme nt in ter ac tio n Message Passi ng N- la yer No de - le ve l MP s M- la y er No de - le ve l MP s \\ St op grad i ent 𝒁 𝒇 𝒁 𝒏 𝒁 𝒇 𝒁 𝒏 No de Fr a g ment De coder Message Passing MLP Proj ect or 𝑳 𝒏 𝑳 𝒇 + Minimizin g Distance T arget Enc od e r Conte xt En c o der F igure 3. Overvie w of the Graph Semantic Predicti ve Network (GraSPNet) frame work. The original molecule is fragmented to form a higher-le vel fragment graph. Masked node and fragment graphs are input into the context encoder , while the target encoder processes the original unmasked graphs. The predictor uses context representations to predict node and fragment embeddings, and the loss minimizes the distance between the prediction and the target encoder’ s representations. graph neural netw ork. The message-passing procedure in the encoder consists of four components: M n → n , M n → f , M f → f , and M f → n , which denote message passing from nodes to nodes, nodes to fragments, fragments to fragments, and fragments to nodes, respectively . Here, M n → n corre- sponds to standard message passing o ver the original atom nodes, while the remaining components collecti vely form the fragment-lev el message passing layer . For each layer , the node representations are first updated through node-le vel message passing. If a fragment layer is applied, the updated node features are then aggregated into the fragments to which each node belongs. Subsequently , higher-le vel fragment message passing is performed to cap- ture structural information based on fragment representa- tions and their connecti vity . Finally , the updated fragment features are injected back into the corresponding node rep- resentations. The message passing between nodes and frag- ments can be represented as: H ( l ) node = f ( l ) node ( A node , H ( l − 1) node ) , W ( l ) 0 , H ( l ) frag = f node-frag ( A node-frag , H ( l − 1) frag , H ( l ) node , W ( l ) 1 , H ( l ) frag = f frag ( A frag , p o ol( H ( l ) frag )) , W ( l ) 2 , H ( l ) node = f ( l ) frag-node ( A frag-node , H ( l ) node , H ( l ) frag ) , W ( l ) 3 , where l is the layer index, W 0 , W 1 , W 2 , W 3 ∈ R d × d are learnable weight matrices of the l th message passing layer with feature dimension d . Let A node ∈ R n × n denote the original adjacency matrix of the molecular graph. The matri- ces A node-frag , A frag-node ∈ R n × m and A frag ∈ R m × m repre- sent the node-to-fragment mapping, fragment-to-node map- ping, and the adjacency between fragments, respectively , where n is the number of nodes and m is the number of frag- ments. Function f ( l ) node and f ( l ) frag denotes the l -th node-le vel and fragment-lev el message-passing layer , which can be any standard GNN architecture (e.g., GCN, GIN). f frag-node denote a fragment-le vel message passing, implemented us- ing an MLP and guided by the fragment connecti vity and node-fragment associations. The aggre gation from node fea- tures to fragment features is defined via a pooling function: h p = 1 | V p | P i ∈ V p h l i,p ∈ R d where 1 ≤ p ≤ m and V p is the set of nodes assigned to fragment p . 4 . 2 . 3 . T A R G E T R E P R E S E N TA T I O N The target encoder adopts the same architecture as the con- text encoder , incorporating both node and fragment encod- ing along with k layers of message passing with fragment layer to learn representations of the original input graph enriched with higher-lev el semantic information. T o pre- vent representation collapse, we use a lighter version of the context encoder with fe wer layers and update its parameters using an Exponential Moving A verage (EMA) of the context encoder parameters. 4.3. Predictor and Loss Giv en the learned node representations Z n and fragment representations Z f from the context encoder , the objective is to capture richer semantic information beyond individual node embeddings. Our prediction model consists of two 5 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction components: node representation prediction and fragment representation prediction. For node prediction, we first remo ve the representations of masked nodes from the context output. The remaining node embeddings are passed through a k -layer message passing network to predict the target node representations. Unlike a simple MLP decoder , this approach encourages the encoder to capture topological dependencies rather than merely re- constructing indi vidual node features. The prediction is defined as: ˆ Z n = g ( A, Z context n ; W ) , where g denotes the message passing function and A is the node adjacency ma- trix. For fragment prediction, a k -layer MLP is used to predict the target fragment embeddings from the context output: ˆ Z f = M LP ( Z context f ; W ) . The loss function is a commonly used MSE loss to measure the distance between the target encoder fragment represen- tation and the predicted fragment representation, which is formally written as: L = α 1 | V n m | | V n m | X i =1 D ( ˆ Z i n , Z i n )+(1 − α ) 1 | V f m | | V f m | X i =1 D ( ˆ Z i f , Z i f ) , where V n m and V f m denote the masked node and fragment sets, respectiv ely; | V n m | and | V f m | represent their cardinality , D ( · ) denotes the Euclidean distance between two v ectors and α is hyperparameter . 5. Experiments In this section, we conduct extensi ve experiments to ev aluate the performance of GraSPNet across various benchmark datasets, aiming to assess the model’ s e xpressiveness and generalization ability . W e also conduct additional studies on the impact of fragment layer position on performance. 5.1. Settings. W e use the open-source RDKit package to pre-process SMILES strings and perform fragmentation for various datasets. For pretraining GraSPNet and all baseline models, we leverage 2 million unlabeled molecules from the ZINC15 database ( Sterling & Irwin , 2015 ). During downstream pre- diction, only the pre-trained context encoder is used and fine-tuned on each benchmark dataset. Final predictions are obtained by applying mean pooling over all node repre- sentations in a graph, follo wed by a linear projection layer . Detailed configurations are provided in the Appendix. 5.2. Evaluation and Metrics The ev aluation focuses on molecular property prediction tasks using benchmark datasets from MoleculeNet ( W u et al. , 2018 ), a collection compiled from multiple pub- lic databases. Specifically , we select eight classification datasets related to physiological and biophysical property prediction: BBBP , T ox21, T oxCast, SIDER, ClinT ox, MUV , HIV , and B ACE. These datasets cover a div erse range of molecular properties, including blood–brain barrier perme- ability (BBBP), toxicity prediction (T ox21, T oxCast, Clin- T ox), adverse drug reactions (SIDER), bio-acti vity against HIV (HIV), and binding affinity to drug targets (BA CE). W e also conduct experiment on regression task datasets including FreeSolv , ESOL and Lipophilicity which focus on physical chemistry . Detailed descriptions are pro vided in the Appendix. For do wnstream tasks, datasets are split 80/10/10 % into training, v alidation, and test sets using the scaffold split protocol, in line with prior w orks. 5.3. Baselines. As experimental baselines, we include representati ve SSL methods from dif ferent categories: contrasti ve-based methods including Infomax ( V eli ˇ ckovi ´ c et al. , 2018 ), ADGCL ( P an et al. , 2018 ), GraphCL ( Y ou et al. , 2020 ), GraphLoG ( Xu et al. , 2021 ) and JOA O ( Y ou et al. , 2021 ); generative-based methods including MAE ( Kipf & W elling , 2016b ), ContextPred ( Hu et al. , 2020b ), S2gae ( T an et al. , 2023 ), GraphMAE ( Hou et al. , 2022 ) and GraphMAE2 ( Hou et al. , 2023 ); predictive methods like Attribute Mask- ing ( Y ou et al. , 2020 ), BGRL ( Thakoor et al. , 2021 ), Mole- BER T( Xia et al. , 2023 ) and SimSGT ( Liu et al. , 2024 ); as well as fragment-based method, MGSSL ( Zhang et al. , 2021b ), HiMOL ( Zang et al. , 2023 ) and S-CGIB ( Lee et al. , 2025 ). W e compare all baselines with our proposed method. 5.4. Molecule Property Prediction. The molecule property prediction tasks follo ws the setting and we use the same node encoder structure with 5 layers of graph isomorphism network (GIN) along with batch normal- ization for each layer . The higher-le vel fragment message passing layer is constructed with 2 layers of GIN to avoid ov er-squashing in smaller fragment graph. T able 1 reports the R OC-A UC (%) scores for eight molecular property pre- diction benchmarks using different self-supervised pretrain- ing strategies. Overall, GraSPNet consistently achie ves the best or second-best performance across most tasks, demon- strating its strong generalization ability . Across most datasets, pretraining helps significantly . Com- pared with the baseline without pretraining, all self- supervised methods improve R OC-A UC, highlighting the benefit of pretraining on molecular graphs. GraSPNet out- performs all competing methods on BBBP (74.4%), T ox21 (77.3%), T oxCast (65.5%), Sider (62.5%), Bace (82.9%), and Clintox (84.1%), indicating superior transferability to div erse biochemical endpoints. On MUV , SimSGT achie ves the highest R OC-A UC (79.0%), follo wed closely by GraSP- Net (78.5%). For HIV , MGSSL leads with 79.5%, while 6 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction T able 1. Ev aluation on molecular property prediction tasks. For each do wnstream dataset, we report the mean and standard deviation of the R OC-A UC (%) scores over three random scaf fold splits. The best and second best scores are marked bold and underline, respectively . BBBP T ox21 T oxCast Sider MUV HIV Bace Clintox No Pre-train 67.8 ± 1.4 73.9 ± 0.8 62.4 ± 0.4 58.3 ± 1.8 73.4 ± 2.5 76.3 ± 1.2 76.8 ± 2.6 62.6 ± 4.4 MAE 68.7 ± 1.3 75.5 ± 0.5 62.0 ± 0.8 58.0 ± 1.0 69.7 ± 1.5 74.2 ± 2.2 77.2 ± 1.6 70.1 ± 3.2 Infomax 69.2 ± 0.7 74.6 ± 0.5 61.8 ± 0.8 60.1 ± 0.7 74.8 ± 1.5 75.0 ± 1.3 76.3 ± 1.8 71.2 ± 2.5 Attr mask 65.6 ± 0.9 77.2 ± 0.4 63.3 ± 0.8 59.6 ± 0.7 74.7 ± 1.9 77.9 ± 1.2 78.3 ± 1.1 77.5 ± 3.1 CP 72.1 ± 1.5 74.3 ± 0.5 63.5 ± 0.3 60.2 ± 1.2 70.2 ± 2.8 74.4 ± 0.8 79.2 ± 0.9 70.2 ± 2.6 ADGCL 70.5 ± 1.8 74.5 ± 0.4 63.0 ± 0.5 59.1 ± 0.9 71.5 ± 2.2 75.9 ± 1.8 74.2 ± 2.4 78.5 ± 3.7 GraphCL 71.4 ± 1.1 74.5 ± 1.0 63.1 ± 0.4 59.4 ± 1.3 73.8 ± 2.0 75.6 ± 0.9 78.3 ± 1.1 75.5 ± 2.4 JO A O 71.8 ± 1.0 74.1 ± 0.8 63.9 ± 0.4 60.8 ± 0.6 74.2 ± 1.2 76.2 ± 1.8 77.2 ± 1.7 79.6 ± 1.4 BGRL 72.5 ± 0.9 75.8 ± 1.0 62.1 ± 0.5 60.4 ± 1.2 76.7 ± 2.8 77.1 ± 1.2 74.7 ± 2.6 65.5 ± 2.3 GraphMAE 71.7 ± 0.8 76.0 ± 0.9 64.8 ± 0.6 60.0 ± 1.0 76.3 ± 1.9 75.9 ± 1.8 81.7 ± 1.6 80.5 ± 2.0 MGSSL 69.7 ± 0.9 76.5 ± 0.3 64.1 ± 0.7 61.5 ± 1.0 76.3 ± 1.9 79.5 ± 1.8 79.7 ± 1.6 80.7 ± 2.1 SimSGT 72.8 ± 0.5 76.8 ± 0.9 65.2 ± 0.8 60.6 ± 0.8 79.0 ± 1.4 77.6 ± 1.9 81.5 ± 1.0 82.0 ± 2.6 GraphLOG 67.2 ± 1.3 76.0 ± 0.8 63.6 ± 0.7 59.8 ± 2.1 72.8 ± 1.8 72.5 ± 1.6 82.8 ± 0.9 76.9 ± 1.9 S2GAE 67.6 ± 2.0 69.6 ± 1.3 58.7 ± 0.8 55.4 ± 1.3 60.1 ± 2.4 68.0 ± 3.8 68.6 ± 2.1 59.6 ± 1.1 Mole-BER T 70.8 ± 0.5 76.6 ± 0.7 63.7 ± 0.5 59.2 ± 1.1 77.2 ± 1.1 76.5 ± 0.8 82.8 ± 1.4 77.2 ± 1.4 GraphMAE2 71.6 ± 1.6 75.9 ± 0.8 65.4 ± 0.3 59.6 ± 0.6 78.5 ± 1.1 76.1 ± 2.2 81.0 ± 1.4 78.8 ± 3.0 GraSPNet 74.4 ± 1.5 77.3 ± 0.8 65.5 ± 0.5 62.5 ± 1.1 78.5 ± 1.3 78.0 ± 0.8 82.9 ± 3.1 84.1 ± 2.1 GraSPNet is second-best (78.0%). Compared to earlier con- trastiv e learning approaches such as GraphCL, JO A O, and ADGCL, GraSPNet yields clear impro vements (e.g., +3–5% on BBBP and Clintox). These results suggest that GraSPNet better captures transferable molecular semantics, especially for small datasets lik e BBBP and Clintox where pretrain- ing benefits are most pronounced. Furthermore, its robust performance across both toxicity-related (T ox21, Sider) and bioactivity-related (HIV , Bace) benchmarks indicates its adaptability across heterogeneous molecular tasks. W e further compare our method with other fragment-based approaches, as reported in T able 3 . Our method achieves the best performance on Clintox (82.5) and MUV (78.5), and ranks second on HIV and BBBP . Compared with S- CGIB, which relies on aggregating one-hop subgraphs of each node for pre-training and fine-tuning, our approach gen- erates meaningful fragmentations that enable more ef fectiv e representation learning while av oiding the high memory ov erhead of subgraph enumeration. In contrast to MGSSL, which constructs fragments dynamically during training and thus incurs substantial computational cost, our method per- forms fragmentation in a more ef ficient manner, leading to faster training without sacrificing predicti ve accuracy . Apart from classification tasks, we also reports fine-tuning performance on three regression benchmarks from physical chemistry as shown in T able 4 , measured by RSE (lower is better). Our method achie ves the best results on all three tasks. These results highlight GraSPNet’ s ability to cap- ture higher-le vel molecular semantics that are critical for T able 2. Ablation study on molecular prediction tasks. w/o F: without fragment information. w/o H-MP: without higher-lev el MP . T ype Clintox BBBP Sider Bace GINE 71.8 ± 1.3 70.2 ± 1.5 59.6 ± 1.5 76.9 ± 2.3 w/o F 78.9 ± 1.4 71.3 ± 1.4 60.0 ± 1.0 80.7 ± 2.2 w/o H-MP 81.9 ± 1.3 72.8 ± 1.3 61.3 ± 1.1 81.2 ± 2.5 Full 84.1 ± 1.5 74.2 ± 1.6 62.5 ± 1.2 82.9 ± 2.7 T able 3. Comparison with other fragment-based methods on four molecular property prediction benchmarks. Clintox MUV HIV BBBP MGSSL 80.7 ± 1.2 76.3 ± 1.1 79.5 ± 0.8 69.7 ± 1.2 S-CGIB 74.6 ± 1.5 74.1 ± 1.0 77.3 ± 0.9 85.4 ± 0.7 HiMOL 80.8 ± 0.9 76.3 ± 1.4 77.1 ± 1.2 70.5 ± 1.3 Ours 82.5 ± 1.7 78.5 ± 1.3 78.0 ± 1.1 74.4 ± 1.2 modeling fundamental physical chemistry properties such as solubility and lipophilicity . Overall, these results demonstrate that our approach con- sistently improv es predictiv e performance across multiple datasets, highlighting the strength of incorporating fragment- lev el message passing, while also identifying areas for future enhancement on specific molecular tasks. 5.5. Ablation Study . T able 2 presents an ablation study on four molecular prop- erty prediction datasets: clintox, bbbp, sider, and bace. The 7 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction T able 4. Performance comparison on regression tasks in terms of RMSE( ↓ ). Methods FreeSolv ESOL Lipophilicity ContextPred 3 . 195 ± 0 . 058 2 . 190 ± 0 . 026 1 . 053 ± 0 . 048 AttrMasking 4 . 023 ± 0 . 039 2 . 954 ± 0 . 087 0 . 982 ± 0 . 052 EdgePred 3 . 192 ± 0 . 023 2 . 368 ± 0 . 070 1 . 085 ± 0 . 061 Infomax 3 . 033 ± 0 . 026 2 . 953 ± 0 . 049 0 . 970 ± 0 . 023 JO AO 3 . 282 ± 0 . 002 1 . 978 ± 0 . 029 1 . 093 ± 0 . 097 GraphCL 3 . 166 ± 0 . 027 1 . 390 ± 0 . 363 1 . 014 ± 0 . 018 GraphFP 2 . 528 ± 0 . 016 2 . 136 ± 0 . 096 1 . 371 ± 0 . 058 MGSSL 2 . 940 ± 0 . 051 2 . 936 ± 0 . 071 1 . 106 ± 0 . 077 GR OVE 2 . 712 ± 0 . 327 1 . 237 ± 0 . 403 0 . 823 ± 0 . 027 SimSGT 1 . 953 ± 0 . 038 1 . 213 ± 0 . 032 0 . 835 ± 0 . 037 GraSPNet 1 . 232 ± 0 . 05 1 . 161 ± 0 . 037 0 . 813 ± 0 . 052 SIDER ClinT o x BBBP BA CE Dataset 55 60 65 70 75 80 85 A ccuracy (%) 59.6 74.3 70.5 80.5 59.9 74.5 70.8 80.1 60.2 74.9 71.0 81.2 61.6 72.8 72.3 79.3 61.5 70.2 72.1 78.6 GINE Layer Layer 1 Layer 2 Layer 3 Layer 4 Layer 5 F igure 4. Performance of incorporating fragment information after different GINE layers in the conte xt encoder . baseline GINE model performs moderately across all tasks. Removing fragment information during masked node re- construction (“w/o F”) results in noticeable performance drops on most datasets, highlighting the contribution of chemically meaningful fragments to representation learning. Excluding higher -level message passing (“w/o Higher -MP”) further de grades performance, demonstrating the impor - tance of multi-lev el interaction modeling. The full model, which incorporates both fragment-aware masking and hi- erarchical message passing, consistently achie ves the best results across all tasks, indicating that both components are essential for effecti ve molecular representation learning. Moreov er, we conduct an ablation study on the use of artic- ulation points in fragment construction, as sho wn in T able 7 in the Appendix. Incorporating articulation points leads to more robust performance across datasets. 5.6. Fragment Layer Analysis. Since the fragmentation layer can be placed after any node- lev el message passing layer , we conduct experiments to test the influence of node-fragment interaction after each layer . The result is shown in Figure 4 . W e conduct pretraining on 20,000 molecules on ZINC and fine-tune it on 4 do wn- stream tasks: clintox, bbbp, sider, and bace. W e use 5-layer of GINE as context encoder and add the node-fragment in- teraction layer and higher -level fragment layer after each message passing layer . The result shows that as the depth increases from layer 1 to layer 3, the performance gradually improv es on all datasets, indicating that early layer hierar - chical message passing enriches node representations with meaningful local semantical information, which benefits multiple downstream performance. Placing the fragment layer after layer 4 provides marginal gains for BBBP and SIDER but be gins to degrade performance on ClinT ox and B ACE, and adding the fragment layer after layer 5 consis- tently results in performance deterioration across all tasks. This shows that late layer node and fragment information interaction may enlarge the influence of ov er-smoothing and be harmful to downstream performance. Overall, these results demonstrate that positioning the fragment layer at an intermediate depth achie ves the best trade-of f between enriched semantics and a voiding ov er-smoothing, thereby enhancing fragment-aware molecular representations. 5.7. Chemical Representati veness. W e also conduct e xperiments to further e xplore the chem- ical representativ eness of GraSPNet generated molecule representation. we perform a probing task to predict frag- ment counts for the 23 common RDKit fragments using the frozen GraSPNet representation and report the accurac y for each fragment. The results are shown in Appendix T able 6 . GraSPNet achie ves high accuracy in detecting chemically valid fragments, indicating that the learned representati ons capture both local motifs and global compositional structure without pre-defined vocab ulary . 6. Conclusion In this work, we proposed a fragment-based pretraining framew ork for molecular graph representation learning that jointly predicts node and fragment embeddings to capture semantically meaningful graph representations. By intro- ducing fragmentation based on both geometric and chemical structure, we construct higher -lev el graph abstractions that are expressiv e yet maintain a lower v ocabulary size, pro- moting better generalization. The representation learned through hierarchical message passing and embedding pre- diction at both node and fragment levels, enabling the model to capture semantic information across multiple resolutions. The model demonstrate competiti ve performance across se v- eral benchmark datasets under transfer learning settings. A limitation of our method is that the fragmentation design is tailored to molecular graphs and may not generalize well to node classification tasks, such as social networks. In future work, we plan to incorporate multimodal signals and natural language annotations to further explore the benefits of fragmentation in graph learning. 8 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction 7. Impact Statement This paper presents work whose goal is to advance the field of machine learning. There are many potential societal consequences of our work, none of which we feel must be specifically highlighted here. References Assran, M., Duv al, Q., Misra, I., Bojano wski, P ., V incent, P ., Rabbat, M., LeCun, Y ., and Ballas, N. Self-supervised learning from images with a joint-embedding predicti ve architecture. In Proceedings of the IEEE/CVF Confer ence on Computer V ision and P attern Recognition , pp. 15619– 15629, 2023. Bacciu, D., Errica, F ., Micheli, A., and Podda, M. A gentle introduction to deep learning for graphs. Neural Net- works , 2020. Bardes, A., Garrido, Q., Ponce, J., Chen, X., Rabbat, M., LeCun, Y ., Assran, M., and Ballas, N. V -jepa: Latent video prediction for visual representation learning. 2023. Chen, Z., Zhu, Y ., Zhao, C., Hu, G., Zeng, W ., W ang, J., and T ang, M. Dpt: Deformable patch-based transformer for visual recognition. In Pr oceedings of the 29th A CM international confer ence on multimedia , pp. 2899–2907, 2021. Corso, G., Stark, H., Jegelka, S., Jaakkola, T ., and Barzilay , R. Graph neural networks. Natur e Reviews Methods Primers , 4(1):17, 2024. Degen, J., W egscheid-Gerlach, C., Zaliani, A., and Rarey , M. On the art of compiling and using’ drug-like’chemical fragment spaces. ChemMedChem , 3(10):1503, 2008. Dosovitskiy , A., Beyer , L., Kolesniko v , A., W eissenborn, D., Zhai, X., Unterthiner , T ., Dehghani, M., Minderer , M., Heigold, G., Gelly , S., Uszkoreit, J., and Houlsby , N. An image is worth 16x16 words: T ransformers for image recognition at scale. ICLR , 2021. Fei, Z., F an, M., and Huang, J. A-jepa: Joint-embedding predictiv e architecture can listen. arXiv pr eprint arXiv:2311.15830 , 2023. Garrido, Q., Assran, M., Ballas, N., Bardes, A., Najman, L., and LeCun, Y . Learning and le veraging world mod- els in visual representation learning. arXiv preprint arXiv:2403.00504 , 2024. Gilmer , J., Schoenholz, S. S., Riley , P . F ., V inyals, O., and Dahl, G. E. Neural message passing for quantum chem- istry . In International conference on machine learning , pp. 1263–1272. PMLR, 2017. Guo, Z., Guo, K., Nan, B., T ian, Y ., Iyer , R. G., Ma, Y ., W iest, O., Zhang, X., W ang, W ., Zhang, C., et al. Graph- based molecular representation learning. In Pr oceedings of the Thirty-Second International J oint Confer ence on Artificial Intelligence , pp. 6638–6646, 2023. Hamilton, W ., Y ing, Z., and Leskovec, J. Inducti ve repre- sentation learning on large graphs. NeurIPS , 2017. He, K., Chen, X., Xie, S., Li, Y ., Doll ´ ar , P ., and Girshick, R. Masked autoencoders are scalable vision learners. In Pr oceedings of the IEEE/CVF confer ence on computer vision and pattern r ecognition , pp. 16000–16009, 2022. Hinton, G. E. and Zemel, R. Autoencoders, minimum de- scription length and helmholtz free energy . Advances in neural information pr ocessing systems , 6, 1993. Hou, Z., Liu, X., Cen, Y ., Dong, Y ., Y ang, H., W ang, C., and T ang, J. Graphmae: Self-supervised mask ed graph autoencoders. In Pr oceedings of the 28th ACM SIGKDD Confer ence on Knowledge Discovery and Data Mining , 2022. Hou, Z., He, Y ., Cen, Y ., Liu, X., Dong, Y ., Kharlamov , E., and T ang, J. Graphmae2: A decoding-enhanced masked self-supervised graph learner . In Pr oceedings of the A CM web confer ence 2023 , pp. 737–746, 2023. Hu, W ., Fey , M., Zitnik, M., Dong, Y ., Ren, H., Liu, B., Catasta, M., and Leskovec, J. Open graph benchmark: Datasets for machine learning on graphs. Advances in neural information pr ocessing systems , 33:22118–22133, 2020a. Hu, W ., Liu, B., Gomes, J., Zitnik, M., Liang, P ., Pande, V ., and Lesko vec, J. Strategies for pre-training graph neu- ral networks. In International Confer ence on Learning Repr esentations (ICLR) , 2020b. Jin, W ., Barzilay , R., and Jaakkola, T . Hierarchical gen- eration of molecular graphs using structural motifs. In International confer ence on machine learning , pp. 4839– 4848, 2020. Ju, W ., Liu, Z., Qin, Y ., Feng, B., W ang, C., Guo, Z., Luo, X., and Zhang, M. Fe w-shot molecular property prediction via hierarchically structured learning on relation graphs. Neural Networks , 163:122–131, 2023. Karypis, G. and Kumar , V . A f ast and high quality multilevel scheme for partitioning irre gular graphs. SIAM J ournal on scientific Computing , 20(1):359–392, 1998. Kashtan, N., Itzko vitz, S., Milo, R., and Alon, U. Efficient sampling algorithm for estimating subgraph concentra- tions and detecting network motifs. Bioinformatics , 20 (11):1746–1758, 2004. 9 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction Kipf, T . N. and W elling, M. Semi-supervised classifica- tion with graph con volutional networks. arXiv pr eprint arXiv:1609.02907 , 2016a. Kipf, T . N. and W elling, M. V ariational graph auto-encoders. arXiv pr eprint arXiv:1611.07308 , 2016b. LeCun, Y . A path to wards autonomous machine intelligence version 0.9. 2, 2022-06-27. Open Review , 62(1):1–62, 2022. Lee, O.-J. et al. Pre-training graph neural networks on molecules by using subgraph-conditioned graph informa- tion bottleneck. In Pr oceedings of the AAAI Confer ence on Artificial Intelligence , volume 39, pp. 17204–17213, 2025. Leman, A. and W eisfeiler, B. A reduction of a graph to a canonical form and an algebra arising during this reduc- tion. Nauchno-T echnicheskaya Informatsiya , 2(9):12–16, 1968. Lewell, X. Q., Judd, D. B., W atson, S. P ., and Hann, M. M. Recap retrosynthetic combinatorial analysis procedure: a powerful ne w technique for identifying privile ged molec- ular fragments with useful applications in combinatorial chemistry . Journal of chemical information and computer sciences , 38(3):511–522, 1998. Li, J., W u, R., Sun, W ., Chen, L., Tian, S., Zhu, L., Meng, C., Zheng, Z., and W ang, W . What’ s behind the mask: Under- standing masked graph modeling for graph autoencoders. In Pr oceedings of the 29th A CM SIGKDD Conference on Knowledge Discovery and Data Mining , pp. 1268–1279, 2023. Liu, S., Guo, H., and T ang, J. Molecular geometry pretrain- ing with se (3)-inv ariant denoising distance matching. In The Eleventh International Confer ence on Learning Repr esentations , 2022. Liu, X., Zhang, F ., Hou, Z., Mian, L., W ang, Z., Zhang, J., and T ang, J. Self-supervised learning: Generativ e or contrastiv e. IEEE transactions on knowledg e and data engineering , 35(1):857–876, 2021. Liu, Z., Shi, Y ., Zhang, A., Zhang, E., Ka waguchi, K., W ang, X., and Chua, T .-S. Rethinking tokenizer and decoder in masked graph modeling for molecules. Advances in Neural Information Pr ocessing Systems , 36, 2024. Ma, Y . and T ang, J. Deep learning on gr aphs . Cambridge Univ ersity Press, 2021. Milo, R., Shen-Orr , S., Itzkovitz, S., Kashtan, N., Chklovskii, D., and Alon, U. Network motifs: simple building blocks of complex networks. Science , 298(5594): 824–827, 2002. Pan, S., Hu, R., Long, G., Jiang, J., Y ao, L., and Zhang, C. Adversarially regularized graph autoencoder for graph embedding. In IJCAI International J oint Confer ence on Artificial Intelligence , 2018. Park, J., Lee, M., Chang, H. J., Lee, K., and Choi, J. Y . Symmetric graph con volutional autoencoder for unsuper- vised graph representation learning. In Pr oceedings of the IEEE/CVF international confer ence on computer vision , pp. 6519–6528, 2019. Rong, Y ., Bian, Y ., Xu, T ., Xie, W ., W ei, Y ., Huang, W ., and Huang, J. Self-supervised graph transformer on large- scale molecular data. Advances in Neural Information Pr ocessing Systems , 33:12559–12571, 2020. Salehi, A. and Da vulcu, H. Graph attention auto-encoders. In 32nd IEEE International Conference on T ools with Artificial Intelligence , ICT AI 2020 , pp. 989–996. IEEE Computer Society , 2020. Sterling, T . and Irwin, J. J. Zinc 15–ligand disco very for e veryone. Journal of c hemical information and modeling , 55(11):2324–2337, 2015. Suresh, S., Li, P ., Hao, C., and Neville, J. Adversarial graph augmentation to improve graph contrasti ve learning. Advances in Neural Information Pr ocessing Systems , 34: 15920–15933, 2021. T an, Q., Liu, N., Huang, X., Choi, S.-H., Li, L., Chen, R., and Hu, X. S2gae: Self-supervised graph autoencoders are generalizable learners with graph masking. In Pr o- ceedings of the Sixteenth A CM International Confer ence on W eb Searc h and Data Mining , pp. 787–795, 2023. T ang, B., Kramer , S. T ., Fang, M., Qiu, Y ., W u, Z., and Xu, D. A self-attention based message passing neural net- work for predicting molecular lipophilicity and aqueous solubility . Journal of cheminformatics , 12:1–9, 2020. Thakoor , S., T allec, C., Azar, M. G., Munos, R., V eli ˇ ckovi ´ c, P ., and V alko, M. Bootstrapped representation learning on graphs. In ICLR 2021 W orkshop on Geometrical and T opological Repr esentation Learning , 2021. V aswani, A., Shazeer , N., Parmar , N., Uszkoreit, J., Jones, L., Gomez, A. N., Kaiser , Ł ., and Polosukhin, I. Attention is all you need. NeurIPS , 2017. V eli ˇ ckovi ´ c, P ., Fedus, W ., Hamilton, W . L., Li ` o, P ., Bengio, Y ., and Hjelm, R. D. Deep graph infomax. In Interna- tional Confer ence on Learning Representations , 2018. W ang, C., P an, S., Long, G., Zhu, X., and Jiang, J. Mgae: Marginalized graph autoencoder for graph clustering. In Pr oceedings of the 2017 A CM on Conference on Informa- tion and Knowledge Manag ement , pp. 889–898, 2017. 10 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction W ollschl ¨ ager , T ., Kemper , N., Hetzel, L., Sommer , J., and G ¨ unnemann, S. Expressi vity and generalization: fragment-biases for molecular gnns. In Pr oceedings of the 41st International Confer ence on Machine Learning , pp. 53113–53139, 2024. W u, Z., Ramsundar , B., Feinber g, E. N., Gomes, J., Ge- niesse, C., Pappu, A. S., Leswing, K., and Pande, V . Moleculenet: a benchmark for molecular machine learn- ing. Chemical science , 9(2):513–530, 2018. Xia, J., Zhao, C., Hu, B., Gao, Z., T an, C., Liu, Y ., Li, S., and Li, S. Z. Mole-bert: Rethinking pre-training graph neural networks for molecules. In The Eleventh International Confer ence on Learning Representations , 2023. Xu, K., Hu, W ., Lesko vec, J., and Je gelka, S. Ho w powerful are graph neural networks? In International Confer ence on Learning Repr esentations , 2019. Xu, M., W ang, H., Ni, B., Guo, H., and T ang, J. Self- supervised graph-lev el representation learning with local and global structure. In International Confer ence on Machine Learning , pp. 11548–11558. PMLR, 2021. Y an, C., Ding, Q., Zhao, P ., Zheng, S., Y ang, J., Y u, Y ., and Huang, J. Retroxpert: Decompose retrosynthesis prediction like a chemist. Advances in Neural Information Pr ocessing Systems , 33:11248–11258, 2020. Y ou, Y ., Chen, T ., Sui, Y ., Chen, T ., W ang, Z., and Shen, Y . Graph contrastiv e learning with augmentations. NeurIPS , 2020. Y ou, Y ., Chen, T ., Shen, Y ., and W ang, Z. Graph contrasti ve learning automated. In ICML , 2021. Y u, L., Pei, S., Ding, L., Zhou, J., Li, L., Zhang, C., and Zhang, X. Sail: Self-augmented graph contrastiv e learn- ing. In AAAI , 2022. Zaidi, S., Schaarschmidt, M., Martens, J., Kim, H., T eh, Y . W ., Sanchez-Gonzalez, A., Battaglia, P ., Pascanu, R., and Godwin, J. Pre-training via denoising for molecu- lar property prediction. In The Eleventh International Confer ence on Learning Representations , 2022. Zang, X., Zhao, X., and T ang, B. Hierarchical molecular graph self-supervised learning for property prediction. Communications Chemistry , 6(1):34, 2023. Zeng, X., Xiang, H., Y u, L., W ang, J., Li, K., Nussinov , R., and Cheng, F . Accurate prediction of molecular properties and drug targets using a self-supervised image represen- tation learning framew ork. Nature Mac hine Intelligence , 4(11):1004–1016, 2022. Zhang, H., W u, Q., Y an, J., W ipf, D., and Y u, P . S. From canonical correlation analysis to self-supervised graph neural networks. Advances in Neural Information Pro- cessing Systems , 34:76–89, 2021a. Zhang, Z., Liu, Q., W ang, H., Lu, C., and Lee, C.-K. Motif- based graph self-supervised learning for molecular prop- erty prediction. Advances in Neur al Information Pr ocess- ing Systems , 34:15870–15882, 2021b. Zhao, J., W en, Q., Sun, S., Y e, Y ., and Zhang, C. Multi- vie w self-supervised heterogeneous graph embedding. In ECML/PKDD , 2021. Zhu, J., Rossi, R. A., Rao, A., Mai, T ., Lipka, N., Ahmed, N. K., and Koutra, D. Graph neural networks with het- erophily . In Proceedings of the AAAI confer ence on arti- ficial intelligence , pp. 11168–11176, 2021. 11 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction A. Datasets Description The benchmark datasets used for downstream prediction tasks are summarized in T able 5 , including their descriptions, number of graphs, and number of prediction tasks. T able 5. Summary of molecular property prediction benchmarks. Dataset Description Number of Graphs Number of T asks BBBP Blood-brain barrier permeability 2,039 1 T ox21 T oxicology on 12 biological targets 7,831 12 T oxCast T oxicology via high-throughput screening 8,575 617 SIDER Adverse drug reactions of marketed medicines 1,427 27 ClinT ox Clinical trial failures due to toxicity 1,478 2 MUV V alidation of virtual screening techniques 93,087 17 HIV Ability to inhibit HIV replication 41,127 1 B A CE Inhibitors of human β -secretase 1 binding results 1,513 1 B. Distribution of Fragments The distribution of fragment size and number of each fragments including rings, paths and articulation per graph in pretraining ZINC dataset are shown in Figure 5 . The analysis demonstrates that the proposed heuristic rules produce fragment distrib utions consistent with established chemical principles. The extracted ring structures are predominantly 5- and 6-membered rings, corresponding to common stable chemical motifs such as benzene and pyridine. Like wise, the distribution of “Path” fragments peaks at lengths of 2–4 atoms, which align with typical aliphatic link ers (e.g., ethyl and propyl chains) that influence molecular fle xibility . These observations indicate that the proposed method systematically recov ers chemically meaningful and representative substructures, rather than arbitrary graph partitions, and is well aligned with known re gularities in molecular structures. C. Additional Experiments In this section we list the additional experimental result which is mentioned in the main paper . The fragements prediction accuracy is shown in T able 6 , the training time is shown in T able 8 and the articulation point ablation study is sho wn in T able 7 . D. Higher -level Graph Expressi veness W e consider a general 3-node cycle distinguishing problem, as sho wn in Figure 7 , illustrated by two non-isomorphic graphs G 1 and G 2 , each containing 8 nodes and 12 edges. Although structurally different, G 1 and G 2 cannot be distinguished by the 1-WL test, since nodes sharing the same features also have identical neighborhood structures. As a result, all nodes receiv e identical label refinements across WL iterations, pro viding no discriminativ e information. In contrast, the corresponding higher-le vel fragment graphs, which incorporate both fragment-le vel nodes and fragment-le vel edges, introduce additional structural information. Specifically , in G 1 , the 3-cycle fragment is connected to a single 4-c ycle fragment, whereas in G 2 , the 3-cycle fragment is connected to another 3-c ycle fragment as well as a 4-cycle fragment. This difference in fragment-le vel connectivity yields distinct higher -lev el representations, enabling the two graphs to be distinguished. T able 6. RDKiT fragments prediction accurac y of GraSPNet. Fragment epoxide lactam morpholine oxazole tetrazole NO ether furan guanido halogen piperdine Acc 99.5 99.95 99.45 99.10 98.85 99.60 91.75 99.60 99.55 94.35 95.70 12 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction 0 5 10 15 20 25 30 35 40 45 50 0 25000 50000 75000 100000 125000 150000 175000 200000 Graph Number (a) F ragment per Graph 0 5 10 15 20 25 30 35 40 45 50 0 100000 200000 300000 400000 500000 600000 700000 Graph Number (b) Rings per Graph 0 5 10 15 20 25 30 35 40 45 50 0 50000 100000 150000 200000 250000 300000 Graph Number (c) P aths per Graph 0 5 10 15 20 25 30 35 40 45 50 0 100000 200000 300000 400000 500000 600000 Graph Number (d) Arti per Graph F igure 5. Distribution of structural components in molecular graphs. Each subplot sho ws the distribution of (a) fragment sizes, (b) number of rings, (c) number of paths, and (d) number of articulation points per graph. The x-axis represents the count of each structure, and the y-axis shows the number of graphs with that count. Fragment thiazole thiophene urea allylic oxid amide amidine azo benzene imidazole imide piperzine pyridine Acc 99.05 99.70 99.75 95.45 94.65 99.65 99.80 92.25 98.75 99.4 97.2 97.15 E. Further Related W ork Representation Learning on Molecules. Representation learning on molecules has made use of hand-crafted representation including molecular descriptors, string-based notations, and image ( Zeng et al. , 2022 ). Graph-based representation learning are currently the state-of-the-art methods as it can capture geometric information in molecule structure. In this setting, molecules are typically modeled as 2D graphs, where atoms are represented as nodes and bonds as edges, with associated feature vectors encoding atom and bond types ( Hu et al. , 2020a ). GNN pretraining are commonly used for molecule representation learning using contrastiv e learning ( Y ou et al. , 2020 ; 2021 ; Suresh et al. , 2021 ; Xu et al. , 2021 ), auto- encoding ( Hou et al. , 2022 ; 2023 ), mask ed component modeling ( Xia et al. , 2023 ; Liu et al. , 2024 ), or denoising ( Zaidi et al. , 2022 ; Liu et al. , 2022 ). At the pretraining lev el, methods can be categorized into node-lev el, graph-level, and more recently fragment-lev el ( Guo et al. , 2023 ). Node-level methods capture chemical information at the atomic scale but are limited in representing higher-order molecular semantics, while graph-le vel methods may ov erlook fine-grained structural details. Joint embedding predictive architectur e. Joint-Embedding Predictiv e Architectures ( LeCun , 2022 ; Garrido et al. , 2024 ) are a recently proposed self-supervised learning architecture which combine the idea of both generativ e and contrastiv e learning methods. It is designed to capture the high-level dependencies between the input x and the prediction object y through predicting missing information in an abstract representation space. The JEP A framew ork has been implemented for images ( Garrido et al. , 2024 ), videos ( Bardes et al. , 2023 ) and audio ( Fei et al. , 2023 ) and sho ws a superior performance on multiple do wnstream tasks. It is claimed that JEP A can improve the semantic le vel of self-supervised representations without extra prior kno wledge encoded ( Assran et al. , 2023 ). Ho wev er previous w ork mostly focus on downstream tasks performance without actually provide e vidence to support the semantic le vel comparison. For molecule graph learning, the natural properties of molecules allo w us to ev aluate the semantic information contained in the representation by detecting specific functional groups detection. This allows us to analyze and compare the semantic le vel of learned representation. 13 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction 0 5 10 15 20 25 30 35 40 45 50 0 1 2 3 4 Number 1e6 (a) Rings Graph 0 5 10 15 20 25 30 35 40 45 50 0.0 0.2 0.4 0.6 0.8 1.0 1.2 Number 1e7 (b) P aths size F igure 6. Distribution of (a) Rings and (b) P aths size in ZINC dataset. T able 7. Ablation study on articulation point. T ype Clintox BBBP Sider Bace Mean w/o Arti 70.16 66.84 58.14 77.81 68.24 Full 70.35 67.30 59.61 76.86 68.53 F . LLM Usage Statement W e used large language model (LLM) solely for grammar checking and minor language editing. No part of the research ideation, methodology , analysis, or writing of scientific content relied on LLMs. G. Reproducibility T o ensure reproducibility of our results, we provide the hyperparameter configurations used in both the self-supervised pretraining and downstream fine-tuning stages. T able 9 lists the ke y architectural choices, optimization settings, and training details consistently applied across experiments. The code will be release upon acception. 14 Hierarchical Molecular Representation Learning via Fragment-Based Self-Supervised Embedding Prediction T able 8. T raining time. Model GraphMAE Mole-BER T S2GAE GraphMAE2 SimSGT GraSPNet Pretrain T ime 527 min 2199 min 1763 min 1195 min 645 min 769 min F igure 7. G 1 (top left) and graph G 2 (top right) with their corresponding higher-le vel graph on bottom left and bottom right. Graph G 1 and graph G 2 that are indistinguishable by 1-WL but distinguishable by Fragment-WL with higher -level graph. T able 9. Hyperparameters for Experiments in Self-supervised learning and fine-tuning Setting Self-supervised Learning Fine-tuning Backbone GNN T ype GIN GIN Context Layer 5 5 T arget Layer 1 1 PE type None None Backbone Neurons [300] [300] Fragment layer [2,3] [2,3] Batch size { 32, 64, 128 } { 32 } Fragment GNN T ype GIN GIN Projector Neurons [300, 300] [300, 300] Pooling Layer Global Mean Pool Global Mean Pool Learning Rate { 0.0001 } { 0.0001, 0.005, 0.001 } EMA { 0.996, 1.0 } { None } Masking Ratio { 0.35 } { 0 } T raining Epochs { 100 } { 100 } 15
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment