Clapeyron Neural Networks for Single-Species Vapor-Liquid Equilibria
Machine learning (ML) approaches have shown promising results for predicting molecular properties relevant for chemical process design. However, they are often limited by scarce experimental property data and lack thermodynamic consistency. As such, …
Authors: Jan Pavšek, Alex, er Mitsos
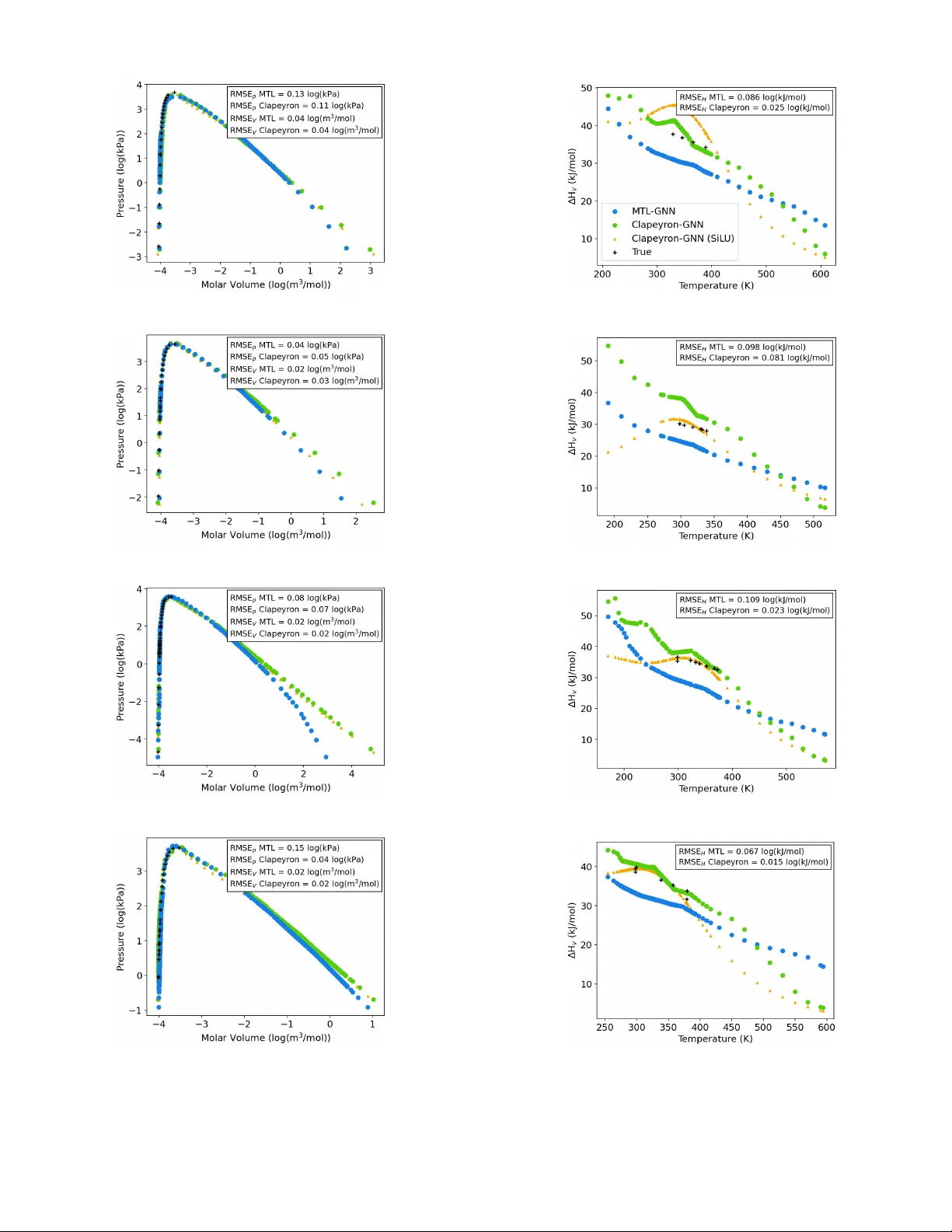
C L A P E Y R O N N E U R A L N E T W O R K S F O R S I N G L E - S P E C I E S V A P O R - L I Q U I D E Q U I L I B R I A Jan Pa všek 1 , Alexander Mitsos 1,2,3 , Elvis J. Sim 1 , Jan G. Rittig 1* 1 Process Systems Engineering (A VT .SVT), R WTH Aachen Uni versity 2 J ARA Center for Simulation and Data Science (CSD) 3 Institute of Climate and Energy Systems ICE-1: Energy Systems Engineering, F orschungszentrum Jülich GmbH * Corresponding author , jan.rittig@rwth-aachen.de A B S T R AC T Machine learning (ML) approaches ha ve sho wn promising results for predicting molecular properties rele vant for chemical process design. Howe ver , they are often limited by scarce experimental property data and lack thermodynamic consistenc y . As such, thermodynamics-informed ML, i.e., incorporating thermodynamic relations into the loss function as re gularization term for training, has been proposed. W e herein transfer the concept of thermodynamics-informed graph neural networks (GNNs) from the Gibbs-Duhem to the Clapeyron equation, predicting sev eral pure component properties in a multi-task manner , namely: vapor pressure, liquid molar volume, vapor molar v olume and enthalpy of vaporization. W e find improv ed prediction accuracy of the Clapeyron-GNN compared to the single- task learning setting, and improved approximation of the Clapeyron equation compared to the purely data-driv en multi-task learning setting. In fact, we observe the largest improvement in prediction accuracy for the properties with the lowest a vailability of data, making our model promising for practical application in data scarce scenarios of chemical engineering practice. 1 Introduction Process design requires information on multiple thermodynamic properties for the species included in the process, such as density , enthalpy and vapor pressure. Here, molecular ML has sho wn to provide accurate predictions for a variety of such properties, including both pure component properties, e.g., density [ 1 ] and phase transition enthalpy [ 2 ], and mixtures, especially for activity coef ficients [3, 4, 5, 6, 7]. Recently , research has particularly focused on advancing molecular ML by incorporating thermodynamic relations into the model architecture and training, in the form of hybrid and physics-informed ML, see overvie w in [ 8 ]. Thermodynamics-informed approaches are particularly promising, as the y do not rely on semi-empirical thermo- dynamic models, which introduce corresponding modeling limitations and assumptions; rather , the y use dif ferential equations to thermodynamic potentials. These thermodynamics-enriched ML approaches reduce the data required for training and enhance - in some cases, ev en guarantee - thermodynamic consistency of the predictions [ 9 , 4 , 5 ], making them highly promising for chemical process design applications. Howe ver , most thermodynamics-informed ML models focus on predicting a single property of interest. W e transfer the concept of thermodynamics-informed ML to single-species vapor -liquid equilibria prediction and e xtend it with multi-task training. That is, we train a GNN to predict v apor and liquid molar volume, vapor pressure and enthalpy of v aporization of a single-species system at v apor-liquid equilibrium. W e jointly train on all four properties in a multi-task learning setting. W e use the Clapeyron equation as physics regularization [ 10 ] in the training loss function to enhance physical consistency of the network predictions, i.e., follo wing a thermodynamics-informed approach. Notably in preliminary studies, we also tested a thermodynamics-consistent approach by directly embedding the Clapeyron equation into the GNN architecture. This, ho wev er , resulted in significantly lower prediction accuracy , which we attrib ute to the imbalanced dataset of the four independently measured properties making model training challenging, also cf. [ 11 ]. The Clape yron-informed approach is more flexible here — albeit at the expense of guaranteed consistency P avšek et al. A preprint — as the Clapeyron equation acts as a soft constraint in the training loss and can be adapted using a weighting factor . W e refer to our architecture as Clapeyron-GNN. A comparable approach has recently been published by Park et al. [ 12 ], who directly predict single-species vapor-liquid equilibria with a GNN. Howe ver , the y not only provide the graph structure of the molecules as input but also additional graph le vel features such as the acentric factor , which might not necessarily be av ailable when making predictions for new molecules. Additionally , Kochi et al. [ 13 ] employ the Clausius-Clape yron equation for physics-informed v apor pressure prediction, using c hempr op as graph encoder [ 14 ], but only predict vapor pressure and dynamic viscosity . W ith Clapeyron-GNN, we train an architecture that predicts all four properties, v apor pressure, liquid and vapor molar volumes and enthalp y of vaporization, in a multi-task learning setting and relies purely on the molecular structure and temperature, and thereby enables easier prediction for new molecules. W e benchmark the predicti ve performance of Clapeyron-GNN against a purely data-driv en multi-task GNN and against four individual GNNs predicting all four properties in a single-task learning setting. 2 Clapeyron Graph Neural Network Figure 1: Schematic illustration of Clapeyron-GNN The Clapeyron-GNN architecture is based on our pre viously employed GNNs [ 15 ]. That is, we map molecular graphs via graph con v olutional layers and a multi-layer perceptron to molecular properties. The prediction tar gets are the temperature-dependent vapor and liquid molar v olume, the vapor pressure and the enthalpy of v aporization (see Figure 1). T emperature is considered as additional input by concatenation with the molecular fingerprint. W e incorporate the Clapeyron equation in the GNN model training. The Clapeyron equation is an exact equation deri ved from the equality of Gibbs free energy in the v apor and liquid phase; it provides a relationship between the four target properties and the temperature: dp sat dT = ∆ H V T ( V V − V L ) (1) where p sat is the v apor pressure, ∆ H V is the enthalpy of vaporization, V V is the v apor molar v olume and V L is the liquid molar volume. All four properties depend on the temperature T . W e reformulate the Clapeyron equation to a Clapeyron error , which measures relativ e deviation of the network predictions from the nearest point fulfilling the Clapeyron equation: L Clapeyron = ( d ˆ p sat dT T ( ˆ V V − ˆ V L ) ˆ ∆ H V − 1) 2 . (2) Similarly to our previous thermodynamics-informed approach [ 16 ], we e xtend the training loss by adding the Clape yron error as additional re gularization to the prediction error using a weighting factor λ , see Figure 1. The gradient of the vapor pressure with respect to the temperature is obtained during training from backpropagation, preserving end-to-end 2 P avšek et al. A preprint (a) p sat (T) (b) V V (T) (c) V L (T) (d) ∆ H V (T) Figure 2: Histograms of data distributions for four target properties and 40 bins for numeric values across all properties learning. In contrast to the prediction loss, which can only be e v aluated for each tar get property if data e xists for the particular property , the Clapeyron regularization provides a loss signal for all four properties, ev en for temperature points for which only data on a subset of the target properties is a v ailable. This is also a major practical difference between employing a thermodynamics-informed and a thermodynamics-consistent approach. As thermodynamics-consistent models embed the thermodynamics equations directly in the output head of the model, they rely on the same number of loss signals as purely data-driven models. W e performed first in vestigations of employing a thermodynamics-consistent approach, ie., directly embedding the exact Clapeyron equation into the output head, which resulted in non-con ver ging training and lower prediction performance than purely data-dri v en models. Hence, in the present work we employ the thermodynamics- informed Clape yron-GNN. 3 Case Study 3.1 Dataset W e extract e xperimental data for the v apor and liquid molar volume, the v apor pressure and the enthalpy of v aporization from the NIST ThermoData Engine [ 17 ]. Notably , we manually remo ved 10 outliers, whose numeric values de viated by at least an order of magnitude from the rest of the dataset. Our dataset contains 879 molecules at varying temperatures, ranging from 56.75 K to 1021 K, with 102,121 data-points in total. The molecules are organic compounds, including amines, esters, alcohols, carboxylic acids, ketones, phenols, amines, nitro compounds and amides, and range in 3 P avšek et al. A preprint molecular weight from 27 to 493 g mol . The distribution of data points across dif ferent molecules and the four dif ferent properties is highly une ven (see Figure 2). Specifically , the amount of data points varies from a single point to more than a thousand for dif ferent molecules. Considering the properties, our data sets contains 78,840 data points for the vapor pressure, 43,056 for liquid molar volume, 2,206 for v apor molar volume and 1,057 for enthalp y of vaporization. In fact, there is abundant training data across the temperature domain for the vapor pressure and the liquid molar volume, whereas for the vapor molar volume and the enthalpy of vaporization, most molecules have either a single or no data point at all. Therefore, learning the temperature dependency for these two properties is inherently more dif ficult. Not only does the number of av ailable datapoints v ary largely between properties, but also within one property , the distribution of numeric values is ske wed (see Figure 2). T o account for this, all four properties are predicted on a logarithmic scale; temperature v alues are linearly normalized to range 0-1. All error metrics are therefore reported on the logarithmic scale. 3.2 Prediction Scenario & Benchmark W e ev aluate the prediction performance for extrapolating to new molecules. Hence, we randomly select all 80% of the molecules with corresponding data points for the training set and keep the remaining 20% of molecules and associated data points for model testing. T o e valuate the contrib ution of the multi-task learning setting and the physics regularization, we compare Clapeyron-GNN against tw o base cases: (i) a purely data driven multi-task learning setting, which we refer to as MTL-GNN, and (ii) a purely data driven single-task learning setting, where we train separate models for each property , which we refer to as STL-GNN. W e train and ev aluate each model 10 times with different seeds, and report av erage and standard deviation across these runs in all metrics. 3.3 Implementation & Hyperparameters The model is implemented using PyT orch and PyT orch Geometric [ 18 ] within our GMoLprop framew ork, which is av ailable as open source. Hyperparameters are optimized with grid search, where the optimized parameters are the batch size ∈ {64, 128}, the activ ation function ∈ {LeakyReLU, SiLU}, the fingerprint dimension ∈ {64, 128} and for Clapeyron-GNN, the weighting factor of the Clapeyron loss ∈ {0.1, 0.5, 1}. This results in a batch size of 64, a fingerprint dimension of 64 and LeakyReLU as acti v ation function for the STL-GNN, a batch size of 64, a fingerprint dimension of 64 and LeakyReLU as activ ation function for the MTL-GNN, and a batch size of 64, a fingerprint dimension of 128, LeakyReLU as activ ation function and a weighting factor of 0.1 for the Clapeyron-GNN. This value of the weighting factor yields the best performance as it does not ne gativ ely impact data approximation or destabilize training, which is observed at larger values for the weighting factor , while still achie ving good approximation of the Clapeyron equation. For the activ ation function, Leak yReLU results in better predicti ve performance than SiLU. While SiLU yields smooth functions with respect to temperature and lower Clape yron errors, it significantly decreases predictiv e performance. Hence, we choose LeakyReLU as acti vation function. 4 Results & Discussion W e first compare the Claperyon-GNN to the other models, and then sho w ex emplary predictions for individual molecules. 4.1 Model comparison The ov erall prediction accuracy for all four properties across the three dif ferent models, STL-GNN, MTL-GNN and Clapeyron-GNN o ver 10 runs is sho wn in T able 1. Comparing the performance of the MTL-GNN to the STL-GNNs, a clear increase in prediction performance can be observed for the v apor molar v olume and the enthalpy of v aporization, for which data is rather scarce, see Section 3.1. For the liquid molar v olume and the v apor pressure with relativ ely high data a vailability , the performance is comparable across the MTL-GNN and the STL-GNN. The relative improvement in the prediction performance of vapor molar volume and enthalp y of vaporization by the introduction of multi-task learning is on par, i.e. the RMSE reduces from 0.31 to 0.17 and from 0.15 to 0.11, respecti vely . As the amount of data on vapor molar v olume is twice as large as on the enthalpy of v aporization, a notable dif ference in the performance improv ement through introduction of multi-task learning might have been expected. Howe ver , as this is not observed, small variations in dataset size appear not to hav e a significant influence on the value of multi-task learning. Performance improvements become observable at dataset size differences across orders of magnitude in our case. Hence in our case, multi-task learning introduces a clear adv antage for properties where data are scarce, while for properties where data are abundant in the temperature domain, the prediction performance is not altered significantly . This aligns with the findings by bin Jav aid et al. [ 19 ], 4 P avšek et al. A preprint T able 1: Performance metrics, root mean squared error (RMSE), mean absolute error (MAE), and coefficient of determination ( R 2 ) ev aluated on the logarithmic scale on the test set for the three different models and for all four properties, and Clapeyron error (see Equation 2) ev aluated on the test set. Lar ge font values are av erages and small fonts standard deviations. Property STL-GNN MTL-GNN Clapeyron-GNN RMSE MAE R 2 RMSE MAE R 2 RMSE MAE R 2 p sat (T) 0.27 ± 0.019 0.14 ± 0.0074 0.97 ± 0.0040 0.26 ± 0.013 0.14 ± 0.0076 0.97 ± 0.0025 0.26 ± 0.019 0.14 ± 0.0065 0.97 ± 0.0040 (#78,840) V V (T) 0.31 ± 0.028 0.18 ± 0.022 0.84 ± 0.028 0.17 ± 0.022 0.13 ± 0.021 0.95 ± 0.013 0.18 ± 0.019 0.14 ± 0.016 0.95 ± 0.011 (#2,206) V L (T) 0.048 ± 0.0057 0.020 ± 0.0012 0.95 ± 0.013 0.048 ± 0.0029 0.030 ± 0.0022 0.95 ± 0.0064 0.048 ± 0.0050 0.029 ± 0.0046 0.95 ± 0.012 (#43,056) ∆ H V (T) 0.15 ± 0.024 0.099 ± 0.017 0.71 ± 0.096 0.11 ± 0.016 0.083 ± 0.017 0.85 ± 0.046 0.10 ± 0.023 0.075 ± 0.018 0.85 ± 0.063 (#1,057) L Clapeyron 0.45 ± 0.12 0.14 ± 0.050 0.0069 ± 0.0051 who find that MTL impro ves prediction accurac y only for tasks that ha ve strong relationships among each other and limited data av ailability . Standley et al. [ 20 ] report that in data scarce scenarios the change in predictive performance of MTL compared to STL depends largely on the combination of tasks that are trained jointly , significantly improving the performance for some and even w orsening it for others, which can depend on the relationships between tasks. As in the present work, all four properties are strongly related - the existence of the Clapeyron equation serves as proof for that - our results align with literature reports with respect to the effect of MTL on predicti v e performance. The prediction performance of the Clapeyron-GNN is on par with the high prediction performance of the MTL-GNN for all four prediction tar gets. Howev er , in the Clape yron error (see T able 1) a significant dif ference can be observed between the Clapeyron-GNN and the MTL-GNN. The Clape yron error is with 0.007 compared to 0.138, respecti vely , two orders of magnitude smaller when using the thermodynamics-informed approach. Thus, for the single-species VLE prediction, Clapeyron regularization can significantly enhance the approximation of fundamental thermodynamics relations, without negati v ely impacting the performance in data approximation. The reason why it does not impro ve the prediction performance lies in the fact that the Clapeyron regularization provides additional information on the consistency of indi vidual molecules. Ho we ver , it does not give additional information on ne w molecules. As we test on unseen molecules, the prediction performance of the Clapeyron-GNN remains on the same lev el but rather shows improved consistenc y of predictions. Notably , also the introduction of MTL alone enhances adherence to the Clapeyron equation; for the STL-GNN, the Clape yron error is significantly higher than for the MTL-GNN, with 0.45 compared to 0.14, respectiv ely (see T able 1). So interestingly , the introduction of multi-task learning also improves the approximation of the Clapeyron equation, albeit significantly less than the introduction of Clape yron regularization. Overall, the Clape yron-GNN achie ves high prediction accurac y on the same le vel as MTL, while significantly better approximating fundamental thermodynamic relations. W e thus further in vestigate the prediction capabilities of the Clapeyron-GNN for the best models and indi vidual molecules in the follo wing. W e show the parity plots of the best model out of the 10 different seeds, for all four properties for the MTL-GNN and the Clapeyron-GNN, see Figure 3. W e do not show the STL-GNN due to lo w accuracy . Overall, the best model of the Clapeyron-GNN slightly outperforms that of the MTL-GNN in two of the four properties ( V V (T) , ∆ H V (T) ). In all four properties, parallel lines to the diagonal are visible both for the predictions by the Clapeyron-GNN and by the MTL-GNN. This indicates that for indi vidual molecules, the predictions follo w the trend in the temperature dependenc y but contain an of f-set. For the vapor molar volume and for the enthalpy of vaporization, see Figure 3(c)-(d) & (g)-(h), the predictions of the Clapeyron-GNN align more closely aligned with the diagonal than for the MTL-GNN. Ho wev er , both models ov erestimate the vapor molar volume in lo w molar v olume regions (numeric v alues under -3.5). For most molecules, this region is close to the critical point, where predictions are inherently more dif ficult. Overall – also for the best models of the MTL-GNN and the Clapeyron-GNN – the predictions are on a high lev el and capture the temperature dependency of the properties, with minor systematic deviations in both models on the v erge of the respectiv e data domains. 4.2 Predictions for individual molecules In Figure 4, we sho w the p(V)-plot and ∆ H v (T)-plot for predictions of the MTL-GNN (in blue) the Clapeyron-GNN (in green), and the Clapeyron-GNN with SiLU as activ ation function (in orange) for four molecules from the test set, 5 P avšek et al. A preprint (a) V apor pressure MTL-GNN (b) V apor pressure Clapeyron-GNN (c) V apor molar volume MTL-GNN (d) V apor molar volume Clapeyron-GNN (e) Liquid molar volume MTL-GNN (f) Liquid molar volume Clape yron-GNN (g) Enthalpy of v aporization MTL-GNN (h) Enthalpy of v aporization Clapeyron-GNN Figure 3: Parity plots of test set MTL-GNN and Clapeyron-GNN 6 P avšek et al. A preprint (a) 3-Methylthiophene (b) 3-Methylthiophene (c) 2-Bromopropane (d) 2-Bromopropane (e) 1-Bromobutane (f) 1-Bromobutane (g) Piperidine (h) Piperidine Figure 4: p(V)-plots and ∆ H v (T)-plots for four exemplary molecules of the test set: experimental data in black crosses, multi-task learning in blue dots, Clapeyron-informed learning with LeakyReLU in green dots, and Clapeyron-informed learning with SiLU in orange triangles. 7 P avšek et al. A preprint namely 1-Bromobutane, 2-Bromopropane, 3-Methylthiophene and Piperidine. The four representative molecules are randomly selected from the subset of molecules in the test set that contain more than one datapoint for the enthalpy , to allow for comparison with e xperimental data. In the p(V)-plots, good alignment of both the Clapeyron-GNN and MTL-GNN predictions with the e xperimental data can be observed across orders of magnitude in the pressure and the molar v olumes. In the lower pressure region, the vapor molar v olume predictions deviate significantly between the MTL-GNN and the Clapeyron-GNN, especially for 1-Bromobutane 4 (e). As in this area, there is no experimental data, it is not clear which prediction describes the true behavior more closely . For 1-Bromob utane and 2-Bromopropane, both the Clape yron-GNN and the MTL-GNN approximate the critical point well. For 3-Methylthiophene both models underestimate the numeric values around the critical point, with the MTL-GNN underestimating more significantly , while for Piperidine the MTL-GNN o verestimates the numeric v alues. Accurate predictions close to the critical point are inherently dif ficult, underlining the quality of the Clapeyron-GNN and MTL-GNN predictions, particularly as the models hav e not been trained on data from these specific molecules. For the enthalp y , a significant difference in the prediction accuracy is observ able. The Clapeyron-GNN approximates the data significantly better but exhibits a corner , while the predictions by the MTL-GNN have a constant of f-set to the experimental data. This off-set is also visible in the parity plot for enthalp y of v aporization of the MTL-GNN, where most prediction lie below the diagonal (see Section 4.1). Apart from the approximation of the data, there is also a large difference in the trend of the MTL-GNN and the Clapeyron-GNN at higher temperatures. While the MTL-GNN predicts a linear trend with constant gradient with respect to temperature which seems close to the gradient in the experimental data, the Clapeyron-GNN predicts a steeper curve at higher temperatures, predicting significantly lower enthalpies at high temperatures than the MTL-GNN. These predictions are more consistent as the enthalpy of v aporization approaches zero, as the temperature approaches the critical temperature. Given the fact, that there is little data on the enthalpy across the temperature domain for most molecules, learning this trend from data alone is challenging. This is where the strength of the Clapeyron-re gularization can be observed, as it improves the consistenc y of predictions particularly in these areas close to the critical point where data are scarce, and the purely data-driv en MTL-GNN fails to make consistent predictions. Howe v er , flaws can also be observed in the Clapeyron-GNN predictions. At medium to lower temperatures corner points are visible in the Clapeyron-GNN predictions. This behavior is non-physical as the enthalpy of vaporization decreases in a smooth curve from its maximum at the triple point until it approaches zero at the critical point. Notably , when employing SiLU as activ ation function, the Clapeyron-GNN yields smooth output functions, but has a local maximum in the enthalpy for all four example molecules. While there are corners in the prediction of the enthalp y when using LeakyReLU as activ ation function for the Clapeyron-GNN, it does follow the overall trend of the data better, ev en beyond the temperature range in the training data. Hence, LeakyReLU is the better choice for balancing accuracy and consistency (see Section 3.3). As data are scarce for the enthalpy , it is likely that the loss signal during training is dominated by the Clapeyron re gularization, which might introduce corner points when there is inconsistencies in the experimental data between the four properties. This underlines that thermodynamics-informed models only promote but do not guarantee consistency . If data were suf ficiently dense across the temperature domain for all four properties, the minimization of the data approximation error would make such non-physical predictions unlikely . Howe ver , especially when data are scarce, non-physical predictions are possible. Nonetheless, Clapeyron-GNN shows highly promising performance, enabling good approximation of Clapeyron- consistent VLE calculation for single-species systems, for which experimental data is lacking for conv entional VLE calculations, e.g. via Peng Robinson [21]. 5 Conclusion W e incorporate the Clapeyron equation into the training of GNNs for predicting single-species v apor-liquid equilibria from molecular graph structure and temperature in a multi-task learning setting. W e find Clapeyron regularization of model training to greatly increase the approximation of the exact Clape yron equation while maintaining the same lev el of prediction accuracy . Additionally , multi-task learning of vapor molar volume, liquid molar volume, vapor pressure and enthalpy of vaporization significantly increases prediction accuracy compared to single-task learning. Clapeyron-GNN is therefore a highly promising architecture for single-species VLE predictions for molecular and process design applications. Future work should inv estigate the possibility of using the Clapeyron equation as a hard constraint in the model architecture, following our recently introduced thermodynamics-consistent GNN approach [ 4 ], ie., embedding the Clapeyron equation directly into the output head of the model. This would be particularly interesting, as the present 8 P avšek et al. A preprint results, in particular for the enthalpy , show that employing physics-based regularization does not guarantee that all predictions are physically consistent in all scenarios. Notably , our first inv estigations of a thermodynamics-consistent GNN with embedded Clape yron equation did result in lo wer prediction accurac y , requiring further tests to be performed in future work, targeting a wider range of properties and molecules. For extending data sets and model testing in practical scenarios, collaboration with industry thus remains critical. CRediT authorship contribution statement Jan Pa všek: Conceptualization, Methodology , Software, F ormal analysis, In v estigation, Writing - original draft, V isualization. Alexander Mitsos: Conceptualization, Writing - revie w & editing, Supervision, Funding Acquisition. Elvis J . Sim: Methodology , Software, Inv estigation, Writing - revie w & editing, V isualization. Jan G. Rittig: Conceptualization, Software, Writing - original draft, Supervision, Funding Acquisition. Data and Software A vailability The data used for training of the presented models are confidential and can be accessed through the NIST Thermodata engine. The code is av ailable in our GitLab repository GMoLprop . Acknowledgments This project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 466417970 – within the Priority Programme “SPP 2331: Machine Learning in Chemical Engineering”. Model training was performed with computing resources granted by R WTH Aachen University . The authors thank René Görgen for his softw are engineering support. References [1] B. W inter , P . Rehner , T . Esper , J. Schilling, A. Bardow , Understanding the language of molecules: predicting pure component parameters for the pc-saft equation of state from smiles, Digital Discov ery 4 (5) (2025) 1142–1157. [2] R. Leenhouts, S. Jankele vitch, R. Raik e, S. Müller , F . V ermeire, Thermodynamics-informed graph neural networks for phase transition enthalpies, Systems and Control T ransactions (2025) 1662–1669. [3] S. Qin, S. Jiang, J. Li, P . Balaprakash, R. C. V an Lehn, V . M. Za v ala, Capturing molecular interactions in graph neural networks: a case study in multi-component phase equilibrium, Digital Discovery 2 (1) (2023) 138–151. [4] J. G. Rittig, A. Mitsos, Thermodynamics-consistent graph neural networks, Chemical Science 15 (44) (2024) 18504–18512. [5] T . Specht, M. Nagda, S. Fellenz, S. Mandt, H. Hasse, F . Jirasek, Hanna: hard-constraint neural network for consistent activity coef ficient prediction, Chemical Science 15 (47) (2024) 19777–19786. [6] E. I. S. Medina, K. Sundmacher , Graph neural networks embedded into mar gules model for v apor –liquid equilibria prediction, Fluid Phase Equilibria 599 (2026) 114520. [7] A. W ahyudi, N. Sue viriyapan, T . Rirksomboon, U. Suriyapraphadilok, et al., Deepthermomix: a local composition graph neural networks model for multicomponent acti vity coefficients (2026). [8] J. G. Rittig, M. Dahmen, M. Grohe, P . Schwaller , A. Mitsos, Molecular machine learning in chemical process design, arXiv preprint arXi v:2508.20527 (2025). [9] D. Rosenberger , K. Barros, T . C. Germann, N. Lubbers, Machine learning of consistent thermodynamic models using automatic differentiation, Ph ysical Revie w E 105 (4) (2022) 045301. [10] M. Raissi, P . Perdikaris, G. E. Karniadakis, Ph ysics-informed neural networks: A deep learning framew ork for solving forward and in verse problems in v olving nonlinear partial differential equations, Journal of Computational physics 378 (2019) 686–707. [11] M. Z. Alam, A. Alqallaf, C. McGill, Equinet: Modeling vapor -liquid equilibrium using simultaneous neural- network predictions for acti vity coefficient and v apor pressure (2026). 9 P avšek et al. A preprint [12] J. Park, R. M. Muthoka, S. Jang, Y . Lee, A multi-stage graph neural network–physics-informed neural network (gnn–pinn) framework for thermodynamic property prediction, Industrial & Engineering Chemistry Research 64 (40) (2025) 19722–19734. [13] M. R. K ochi, H. Rezaei, S. T . Khan, B. T . Mamillapalli, M. Ebrahimiazar , H. Y e, R. Moosavian, M. Zar gartalebi, D. Sinton, S. M. Moosa vi, Thermodynamics-informed machine learning for predicting temperature-dependent chemical properties (2025). [14] E. Heid, K. P . Greenman, Y . Chung, S.-C. Li, D. E. Graff, F . H. V ermeire, H. W u, W . H. Green, C. J. McGill, Chemprop: a machine learning package for chemical property prediction, Journal of Chemical Information and Modeling 64 (1) (2023) 9–17. [15] A. M. Schweidtmann, J. G. Rittig, A. Konig, M. Grohe, A. Mitsos, M. Dahmen, Graph neural networks for prediction of fuel ignition quality , Energy & fuels 34 (9) (2020) 11395–11407. [16] J. G. Rittig, K. C. Felton, A. A. Lapkin, A. Mitsos, Gibbs–duhem-informed neural networks for binary acti vity coefficient prediction, Digital Disco very 2 (6) (2023) 1752–1767. [17] National Institute of Standards and T echnology (NIST), Thermodata engine, nist standard reference database 103b, version 10.4.5, accessed June 20, 2025; critically e v aluated thermodynamic and transport property data (2025). URL https://www.nist.gov/mml/acmd/trc/thermodata- engine/srd- nist- tde- 103b [18] M. Fey , J. E. Lenssen, Fast graph representation learning with PyT orch Geometric, in: ICLR W orkshop on Representation Learning on Graphs and Manifolds, 2019. [19] M. bin Jav aid, T . Gervens, A. Mitsos, M. Grohe, J. G. Rittig, Exploring data augmentation: Multi-task methods for molecular property prediction, Computers & Chemical Engineering (2025) 109253. [20] T . Standley , A. Zamir , D. Chen, L. Guibas, J. Malik, S. Sav arese, Which tasks should be learned together in multi-task learning?, in: International conference on machine learning, PMLR, 2020, pp. 9120–9132. [21] D. B. Robinson, D.-Y . Peng, S. Y . Chung, The de velopment of the peng-robinson equation and its application to phase equilibrium in a system containing methanol, Fluid Phase Equilibria 24 (1-2) (1985) 25–41. 10
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment