Two-Stage Multiple Test Procedures Controlling False Discovery Rate with auxiliary variable and their Application to Set4Delta Mutant Data
In this paper, we present novel methodologies that incorporate auxiliary variables for multiple hypotheses testing related to the main point of interest while effectively controlling the false discovery rate. When dealing with multiple tests concerni…
Authors: Seohwa Hwang, Mark Louie Ramos, DoHwan Park
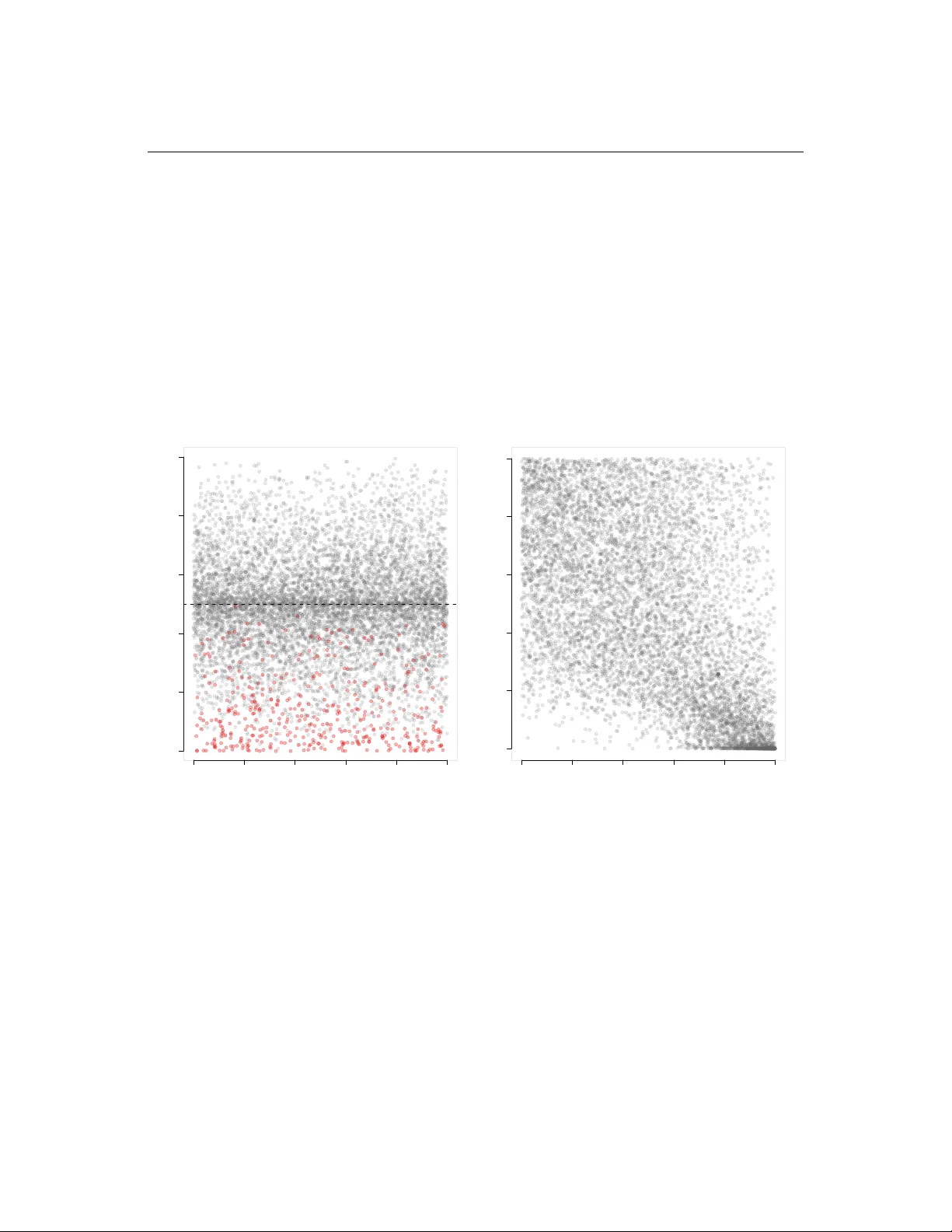
Biometrical Journal 52 (2024) 61, zzz–zzz / DOI: 10.1002/bimj.200100000 T wo-Stage Multiple T est Pr ocedur es Contr olling F alse Discov ery Rate with auxiliary v ariable and their A pplication to Set4 ∆ Mu- tant Data Seohwa Hwang 1 , Mark Louie Ramos 2 , DoHwan Park 3 , Junyong P ark ∗ 1 , Johan Lim 1 , and Erin Green 4 1 Department of Statistics, Seoul National Univ ersity , Seoul, K orea 2 Department of Health Policy and Administration, The Pennsylv ania State Univ ersity , P A, USA 3 Department of Mathematics and Statistics, Univ ersity of Maryland Baltimore County , Baltimore, MD, USA 4 Department of Biological Sciences, Univ ersity of Maryland Baltimore County , Baltimore, MD, USA Receiv ed zzz, revised zzz, accepted zzz In this paper , we present novel methodologies that incorporate auxiliary v ariables for multiple hypotheses testing related to the main point of interest while ef fectiv ely controlling the f alse disco very rate. When deal- ing with multiple tests concerning the primary variable of interest, researchers can use auxiliary variables to set preconditions for the significance of primary variables, thereby enhancing test efficacy . Depending on the auxiliary variable’ s role, we propose two approaches: one terminates testing of the primary variable if it does not meet predefined conditions, and the other adjusts the e valuation criteria based on the auxiliary variable. Employing the copula method, we elucidate the dependence between the auxiliary and primary variables by deriving their joint distribution from individual marginal distributions.Our numerical studies, compared with e xisting methods, demonstrate that the proposed methodologies effecti vely control the FDR and yield greater statistical power than previous approaches solely based on the primary variable. As an illustrativ e example, we apply our methods to the Set4 ∆ mutant dataset. Our findings highlight the distinc- tions between our methodologies and traditional approaches, emphasising the potential advantages of our methods in introducing the auxiliary variable for selecting more genes. K ey wor ds: Copula; False Disco very Rate; Multiple T esting; T wo-Stage Procedure; 1 Introduction In this paper , we consider multiple testing scenarios where additional information beyond p-values is a vail- able. Numerous studies hav e already tackled this subject, predominantly focusing on enhancing rejection rates through the utilisation of covariates. For instance, Lei and Fithian [2018] demonstrated that regard- less of covariate v alues, p -values under the null hypothesis conform to a uniform distribution. In contrast, AdaFDR, proposed by Zhang et al. [2019], relaxes the requirement of a uniform distribution under the null hypothesis but mandates a mirroring property in the distribution of p -v alues given specific co variates, which means the conditional null distribution of p -value giv en the covariate is symmetric around 1 / 2 . Ignatiadis et al. [2016] explore maximising rejections by assigning v arying weights to groups based on cov ariates, while Boca and Leek [2018] estimate the proportion of alternativ e hypotheses given co variates to adjust the rejection region. On the other hand, Cao et al. [2022] proposed a multiple testing procedure when there is auxiliary information such as the order of hypotheses or alternative distribution while there is no specific cov ariate. These methodologies aim to optimise rejection rates while ensuring control over the False Discovery Rate (FDR) across all hypothesis tests. While maximising the number of rejections with controlling an error rate is statistically advantageous, there are instances where such strategies may not ∗ Corresponding author: e-mail: juny ongpark@snu.ac.kr © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 2 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable be desirable when considering scientific knowledge or background. W e incorporate the auxiliary variable more relev antly through modelling the joint distribution of the p -value and the auxiliary v ariable so that we do not require such strong assumptions such as uniform distribution of p -value under the null hypothesis regardless of the co variate or mirroring property of p -v alue under the null hypothesis. One main aim is to incorporate an auxiliary variable to improv e the results of multiple testing compared to the case of using only the primary variable. In addition, scientists may reflect some prior knowledge on the main interest through the auxiliary variable. Some restrictions on the distribution of p value giv en the value of the covariate may not reflect such flexible situations. The reflection of the auxiliary variable in the test of the primary variable essentially signifies that there is a dependency between the two variables, and identifying this dependency is crucial. W e emphasise that only the primary variables are tested and the auxiliary variables provide supplementary information to make better decisions on the primary variables. In this sense, our problem is different from the case of bi variate p -v alues which are declared to be significant when at least one of p -values is significant. See Du and Zhang [2014]. Rather than imposing the restrictions on the distribution of the p value, we use the joint distribution of the auxiliary and primary variables and then propose two types of two-stage procedures controlling FDR. One is called two-stage FDR(H) based on the idea of hard thresholding such as we filter insignificant genes using the auxiliary variable and then only if this stage is passed, the significance of the primary variable is considered. The other one is two-stage FDR(S) where (S) presents the soft threshold which av oids filtering. T wo-stage FDR(S) is testing of the primary variable with adjusted rejection region for the primary variable reflecting the result for the auxiliary variable. As mentioned, our goal is to test only the primary variable while reflecting the information coming from the dependence between the primary and auxiliary variables. The proposed methods are demonstrated through simulation to ef fectiv ely control the FDR while achieving superior testing power compared to existing methods. Existing methods incorporating the auxiliary variable with the restrictions on the distribution of p v alue hav e difficulty in controlling a giv en lev el of FDR and obtaining po wers due to the constraints such as the uniform distribution of p value regardless of the value of covariate and the mirroring property under the null hypothesis. In the context of Set4 ∆ dataset, the two-stage FDR(S) approach successfully identifies a cluster of genes that not only possess statistical significance but are also kno wn as significant from biological experiments. This paper is organised as follows. In section 2, we present the description of Set4 ∆ mutant data, which serves as the motiv ation for this study , along with the necessity of the two-step method proposed. Section 3 introduces the copula that explains distribution estimation and dependence, which are the basic items required for the proposed methods, and explains the joint distribution estimation procedure. Section 4 describes two methodologies based on the tw o-step procedures designed for controlling FDR. Section 5 provides simulation studies to compare the existing methods with the proposed two-stage procedures and section 6 presents an analysis of Set4 ∆ mutant data. Finally , Section 7 contains concluding remarks. 2 Motivating Data set: Set4 ∆ mutant data The moti vating study seeks to identify genes within the genome of the b udding yeast Sacc har omyces cer e- visiae that are differentially expressed when a particular gene expression machinery referred to as Set4 is mutated. The data set is referred to as the Set4 ∆ mutant data set. In order to identify the genes that depend on Set4 during stress, the gene expression profile of the whole genome in cells where Set4 is silenced (knock-out condition or KO) and cells where it is not (wildtype condition or WT) are determined. In ad- dition, cells are treated with hydrogen peroxide to induce oxidative stress. The experiment is replicated thrice. In each replication, the amount of mutation, represented by a weighted count value, was collected for each of about 8,000 RN A species under KO and WT conditions. For each gene, these weighted counts are a veraged to produce a single v alue under each condition. More formally , we hav e the data for M genes and three replicates such as W T ij , K O ij (1) © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 3 under WT and KO conditions, respectively for 1 ≤ i ≤ M and 1 ≤ j ≤ 3 . Let µ W T i and µ K O i be the mean of W ij and K ij , respectiv ely , then we define the vector of logfold changes of M genes as β ′ = ( β 1 , . . . , β M ) ′ = log µ W T 1 µ K O 1 , . . . , log µ W T M µ K O M ′ . (2) From these data matrices, the vector of statistics that is to be used for multiple testing is ˆ β ′ = ( ˆ β 1 , . . . , ˆ β M ) ′ = log ¯ W T 1 . ¯ K O 1 . , · · · , log ¯ W T M . ¯ K O M . ′ (3) where ¯ W T i. = 1 3 P 3 j =1 W ij and ¯ K O i. = 1 3 P 3 j =1 K O ij are the sample means from 3 replicates for the i th gene. The logfold changes statistic is then computed as b β i = log 2 ¯ K O i ¯ W T i (4) where i = 1 , 2 , · · · M and M is the total number of genes. ¯ W T is a mean weighted count measure of mutations under wild type condition and ¯ K O is a mean weighted count measure of mutations under knock- out condition for the i th gene. This statistic ˆ β i represents the doubling effect” of one condition relative to the other condition. A gene is considered interesting” if the weighted count measure is very different between the KO and WT settings, and so correspondingly , if | ˆ β i |≫ 0 , we have supporting evidence for significance of the gene. Ramos et al. [2021] selected genes based on ˆ β i s considering multiple testing procedures controlling FDR. While logfold change is our primary focus, Ramos et al. [2021] presented the results that the genes selected based solely on logfold changes are not biologically significant in most cases. Even if logfold change is considered significant for various reasons, such as e xtremely small values or high v ariance in the data across conditions, it can be influenced by various errors occurring during the experimental process. Therefore, when considering variability in experimental replicates, it is essential to first assess the signifi- cance of changes in logfold changes values. In our analysis, the standard deviation of the logfold changes will serve as an auxiliary variable. The distributions of logfold changes and their standard deviations are shown in Figure 1. [Figure 1 about here.] While the exact distribution of each logfold change remains unknown, it’ s reasonable to assume that different sizes of standard deviation of logfold changes affect the determination of the significance of the logfold change. Based on this idea, we propose two methods for setting rejection criteria based on standard deviation. The first method adjusts rejection regions according to whether the standard de viation exceeds a certain threshold, similar to a diagnostic screening process. The second method v aries the rejection regions with the exact value of the standard de viation, allowing precise adjustments when detailed data on both the primary and auxiliary variables are a vailable. 3 Models and Estimation of Distributions The traditional multiple testing methods with existing cov ariate either assume a uniform distrib ution under the null hypothesis regardless of the covariates or satisfy the mirroring property , where the role of the cov ariate is to maximise the rejection of hypotheses by adjusting rejection regions for different values of the co variate. More specifically , in the existing studies such as Lei and Fithian [2018], Zhang et al. [2019], Ignatiadis et al. [2016] and Boca and Leek [2018], the null density of p value given the auxiliary variable y is either I [0 , 1] ( p ) regardless of y or the mirroring property such as P ( p ≤ t | y ) = P ( p ≥ 1 − t | y ) for t ∈ [0 , 1 / 2] . © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 4 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable [Figure 2 about here.] It’ s worth noting that the mirroring property fails in some cases. This property implies a zero correlation between the p value and the auxiliary v ariable, often indicating a weak relationship between them. Achiev- ing this constraint can be overly restrictive if the auxiliary variable is to be used to increase the power of the test. For e xample, we generated exemplary p -v alues that are symmetric around 0.5, as shown in Figure 2(A). This distribution of p -v alues is suitable for e xisting statistical methods that assume such symmetry in their analyses. Conv ersely , p -values generated from real data typically do not exhibit this symmetry around 0 . 5 , which implies that the underlying assumptions of existing methods are not met. Rather than those constraints on the null distribution of p value given the auxiliary variable, our proposed methods are based on considering the joint distribution of the primary v ariable of interest and an additional auxiliary variable. W e estimate the marginal distribution for each v ariable and derive the joint distribution using the copula method, based on which we propose two methods. In other words, we treat the auxiliary variable as a random variable rather than cov ariate so that we can consider dependent structure between those two variables. Our proposed methods do not request that the marginal distribution of p value is uniformly distributed or has mirroring property . W e use the relationship between the distribution of p value and the auxiliary variable through the joint distribution of them. Therefore, our proposed methods reflect those two variables more flexibly compared to e xisting methods. W e will demonstrate that these proposed methods yield different results compared to the e xisting methods. Suppose we hav e primary and auxiliary variables denoted by β and y respectively . The primary v ariable, β , is modelled as a mixture of two probability density functions, with each being assumed to hav e been generated under null and alternative scenarios, labelled as f 0 and f 1 , respectiv ely . Thus, it is of interest to test M hypotheses of the primary v ariables, H 0 i : β i ∼ f 0 v s. H 1 i : β i ∼ f 1 (5) for 1 ≤ i ≤ M where each β i is modelled as f ( β ) = p 0 f 0 ( β ) + (1 − p 0 ) f 1 ( β ) (6) where p 0 = P ( H 0 i is true ) . This setting has been used in a variety of multiple testing problems. For example, Efron and T ibshirani [2002] used a normal distrib ution for f 0 of which the mean and standard de viation are estimated based on the zero assumption and the mar ginal distrib ution f is estimated using Poisson regression instead of estimating the alternativ e density f 1 . In Ramos et al. [2021], the logfold changes statistics for Set4 ∆ that come from f 0 were modelled as a mixture of two normal distrib utions rather than a single normal distribution to explain two different sources of the null distribution. Once we estimate f 0 , we also obtain the cumulativ e distribution function (cdf), ˆ F 0 ( β ) = R β −∞ ˆ f 0 ( t ) dt . The use of an auxiliary variable ( y ) is also considered. Since the auxiliary variable is used as supple- mentary information, we only need to identify the marginal distrib ution of y such as y ∼ h ( y ) (7) where h ( y ) is the probability density function (pdf) of y . In practice, h ( y ) or the cumulativ e distribution function H ( y ) = R y −∞ h ( t ) dt can be estimated by nonparametric approaches, for e xample, we estimate ˆ H ( y ) = 1 M M X i =1 I ( y i ≤ y ) (8) which is the empirical cdf of y i s. This method helps researchers by allowing them to use extra scientific information that isn’t the main focus of their study , b ut still makes their results stronger and more trustworthy . By estimating ˆ H ( y ) , © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 5 the empirical cumulative distribution function of y , we apply a non-parametric approach without making restrictiv e assumptions about its distribution. Once we hav e the marginal distributions of the primary and auxiliary variables, we use copula models to estimate the dependence structure of y and β of which the joint distribution is G 0 ( y , β ) . The selection of an appropriate copula is a crucial step in modelling the dependence structure in multiv ariate distributions. There are several approaches to choose an appropriate copula. One ad-hoc approach is based on a scatter- plot of the data to gain insight into the dependence structure which can be done by conv erting the data using the data’ s marginal cumulativ e distribution. There are typical patterns of scatterplots for different copulas, so we may choose the closest pattern of the scatterplot. Another approach is comparing Log-Likelihood (LogLik), Akaike Information Criterion (AIC), and Bayesian Information Criterion (BIC). These criteria are widely used for the selection of copulas. For example, see St ¨ ober and Czado [2014] and Fink et al. [2017]. W e present examples of copulas in Figure ?? and numerical studies of the performances of these criteria for selection of copula in T able ?? in supplementary material. In this section, we present our proposed methods depending on how we handle the auxiliary variable. W e first define δ i = I ( H 0 i is rejected ) and θ i = I ( H 1 i is true ) where I ( · ) is an indicator function. Our proposed methods are designed to control the FDR which is F D R = E V R ∨ 1 (9) where V = P M i =1 δ i (1 − θ i ) is the number of false rejections and R = P M i =1 δ i is the number of rejec- tions. W e introduce the marginal p values and then propose two types of methods with some theoretical justification. 3.1 Calculation of Mar ginal p -values W e define p 1 i and p 2 i which are the marginal p -v alues corresponding to the auxiliary variable and the primary variable, respecti vely . For observed y i and ˆ β i , p 1 i = H ( y i ) , (10) p 2 i = 2 min F 0 ( ˆ β i ) , 1 − F 0 ( ˆ β i ) . (11) W e mentioned how f 0 is estimated in the previous section. Based on the estimator of f 0 , we hav e an estimator of F 0 ( β ) = R β −∞ f 0 ( t ) dt . W e also have an estimator of H ( y ) such as the empirical cdf as discussed in the pre vious section. W e assume that p 1 i ∼ U [0 , 1] and p 2 i ∼ U [0 , 1] under the H 0 i : β i ∼ f 0 . Since ( y i , ˆ β i ) are generated from the joint distrib ution G 0 , p 1 i and p 2 i are dependent. Remark 1 Note that p 2 i cov ers the case of two-tailed p -value for non-symmetric distribution. For one-sided cases, we can define F 0 ( ˆ β i ) and 1 − F 0 ( ˆ β i ) for left-tailed and right-tailed cases, respectively . Similarly , p 1 i can also be defined for two-tailed cases. Depending on the marginal p -v alues such as p 1 i and p 2 i , we propose two methods, namely two-stage FDR(H) and two-stage FDR(S) in the follo wing section. 3.2 T wo-stage FDR(H)and T wo-stage FDR(S) As mentioned before, our proposed methods are based on modelling the joint distribution of p 1 i and p 2 i rather than assuming p 1 i is U (0 , 1) regardless of the auxiliary variable p 2 i or the mirroring property such as the density of p 1 i is giv en x under the null hypothesis is symmetric around 1 / 2 . W e present two proposed methods, called T wo-Stage FDR(H) and T wo-Stage FDR(S). T wo-stage FDR(H): For the first type of two-stage procedure two-stage FDR(H), we proceed as fol- lows: Given a value of γ 1 , if p 1 i > γ 1 equiv alently y i > H − 1 ( γ 1 ) , we conclude that the first requirement © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 6 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable via the auxiliary v ariable is not satisfied and hence we do not proceed with testing the significance of the primary variable. On the other hand, if p 1 i ≤ γ 1 equiv alently y i ≤ H − 1 ( γ 1 ) , then we test the significance of the primary variable. Here, γ 1 is a tuning parameter that serves as a cut-off value to determine whether to proceed with testing the primary variable. W e will discuss in section 3.3 how we choose γ 1 to maximise the number of discov eries. Considering this tw o-stage procedure, we define the p -v alue, p i aggregating two p 1 i and p 2 i as follo ws: for a giv en γ 1 ∈ [0 , 1] , p H i ( γ 1 ) = ( C ( γ 1 , p 2 i ) if p 1 i ≤ γ 1 p 1 i if p 1 i > γ 1 (12) where C ( a, b ) = P G 0 ( P 1 i ≤ a, P 2 i ≤ b ) (13) is a 2-dimensional copula distribution ev aluated at ( a, b ) . Here, G 0 stands for the joint distribution of y ′ i s and β ′ i s under null hypothesis. In order to reject the null hypothesis for the primary variable β i , we need two steps such as p 1 i ≤ γ 1 and a small p 2 i , i.e., the event of p 1 i ≤ γ 1 , p 2 i ≤ γ 2 for some γ 2 . In testing the primary variable, it is better not to use γ 2 = α since p 1 i ≤ γ 1 affects the test for the primary variable. More specifically , take γ 2 which should satisfy the uniform distribution of P i such that P G 0 ( P 1 i ≤ γ ) = P G 0 ( P 1 i ≤ γ 1 , P 2 i ≤ γ 2 ) = γ . This means γ 2 depends on γ 1 and γ , say γ 2 ≡ γ 2 ( γ 1 , γ ) . Using the definition of (13), if ˆ β i ≥ D 0 . 5 , then we hav e C ( γ 1 , γ 2 ) = P G 0 ( y ,β ) ( P 1 i ≤ γ 1 , P 2 i ≤ γ 2 ) = Z C 1 − γ 1 0 Z 1 D 1 − γ 2 / 2 G 0 ( y , β ) dβ dy = γ (14) where C α and D α are α quantiles of f 0 and h 0 , respectiv ely .; if ˆ β i < D 0 . 5 C ( γ 1 , γ 2 ) = P G 0 ( y ,β ) ( P 1 i ≤ γ 1 , P 2 i ≤ γ 2 ) = Z C 1 − γ 1 0 Z D γ 2 / 2 0 G 0 ( y , β ) dβ dy = γ (15) equiv alently , we can simply define γ 2 as the value which satisfies C ( γ 1 , γ 2 ) = γ (16) This method determines whether to proceed to the next step based on the value of p 1 i in the first step, and if p 1 i > γ 1 , the corresponding primary variable y i as a whole is not considered significant although the second p-value, p 2 i , is very small. In other words, such a thresholding in the first step plays a role of screening genes which do not satisfy the condition based on the auxiliary v ariable y i . T o highlight this characteristic, we will refer to it as “two-stage FDR(H)”, denoting the implementation of two-stage FDR with hard thresholding. From two-stage FDR(H), it is crucial to check whether p i has a desirable property such that p H i has the uniform distribution under G 0 . Before we state Theorem 3.1 showing the uniform distribution of p i in both (12) and (18) under G 0 , we present the following lemma which is used in the proof of Theorem 3.1. Lemma 1 From the definition of p H i ( γ 1 ) in (12), we ha ve P ( P 1 i ≤ γ 1 , P 2 i ≤ γ 2 ) = P ( P H i ( γ 1 ) ≤ γ , P 1 i ≤ γ 1 ) (17) for γ = C ( γ 1 , γ 2 ) in (16) where γ 1 and γ 2 are in [0 , 1] . P r o o f. See the supplementary material. Using Lemma 1, we present the following theorem showing that p H i ( γ 1 ) is uniformly distributed re- gardless of the choice of γ 1 . © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 7 Theorem 3.1 If ( ˆ β i , y i ) ∼ G 0 ( β , y ) , p H i ( γ 1 ) ’ s defined in (12) follow the uniform distribution in (0 , 1) . P r o o f. See the supplementary material. T wo-stage FDR(S): Instead of this stringent thresholding procedure in two-stage FDR(H), we also provide the follo wing method, two-stage FDR(S). The basic idea is to adjust the distrib ution of the second p 2 i based on the given value of the first p 1 i , by using the estimated copula. In fact, this will be implemented by computing the conditional distribution of p 2 i giv en p 1 i . More specifically , p S i aggregating p 1 i and p 2 i is defined as follows: p S i = C ( p 2 i | p 1 i ) ≡ P G 0 ( P 2 i ≤ p 2 i | P 1 i = p 1 i ) (18) which is the conditional cumulativ e distribution of p 2 i giv en p 1 i . As in two-stage FDR(H), we can show that p S i defined in (18) also follo ws uniform distribution under G 0 . Theorem 3.2 If ( ˆ β i , y i ) ∼ G 0 ( β , y ) , p S i ’ s defined in (18) follow the uniform distribution in (0 , 1) . P r o o f. See the supplementary material. Comparision of T wo Methods: T wo-stage FDR(S) differs from two-stage FDR(H) by introducing multiple thresholds. Notably , two-stage FDR(S) does not require a tuning parameter such as γ 1 in two- stage FDR(H). Instead of relying on one threshold γ 1 in two-stage FDR(H), two-stage FDR(S) uses p 1 i to adjust the decision rule for p 2 i through the conditional probability . W e call this process as soft thresholds in the sense that we adjust rejection region for the primary v ariable in a smooth way which is demonstrated in Figure 3. T o emphasise the contrast with “two-stage FDR(H)”, we refer to this procedure as “two-stage FDR(S)”, representing the concept of a soft margin. T o clarify the differences between two-stage FDR(H) and two-stage FDR(S), we present Figure 3 in- cluding (A), (B) and (C) for two methods, respecti vely . In each figure, we consider three distinct points, A , B and C which are corresponding to ( p 1 i , p 2 i ) = (0 . 4 , 0 . 1) , (0 . 8 , 0 . 1) and (0 . 1 , 0 . 143) . In Figure 3, (A) and (B) are the cases of rejection regions for dif ferent γ 1 values, γ 1 = 0 . 9 and 0 . 7 respectiv ely . W e see that the rejection regions have rectangular forms which means that the cut-of f value for p 2 i is identical when- ev er p 1 i < γ 1 . From (A) and (B), the rejections for B and C are different for different γ 1 values. When γ 1 = 0 . 9 , all A , B and C are proceeded to test the primary v ariable p 2 i , howe ver C is not rejected since p 2 i is larger than the cut-off value. On the other hand, when γ 1 = 0 . 7 , B does not mov e to the testing of the primary variable, ho we ver C is rejected to be significant. This shows that there is trade-off relationship between γ 1 and the cut-off value for p 2 i . For larger value of γ 1 (equiv alently , weak requirement on the auxiliary variable), the cut-off value for p 2 i is small, i.e., more stringent on testing the primary variable. Con versely , for small value of γ 1 (stringent on the auxiliary variable), testing for the primary variable is less stringent, i.e., the cut-of f value is larger for p 2 i . Regarding two-stage FDR(S), we observe similar patterns, howe ver the rejection region is not a rectangular form which means the rejection for p 2 i depends on p 1 i coming from the conditional distribution. T o summarise differences between two methods, both methods ha ve a trade-off relationship between the first and second tests, but the major difference lies in the shape of the rejection regions. As mentioned earlier , in the case of two-stage FDR(H), if a v ariable passes the first stage through p 1 i ≤ γ 1 , the same cutoff value is applied to p 2 i in the second stage. In this case, if it was eliminated in the first stage ( p 1 i > γ 1 ), there is no possibility of rejecting the primary variable regardless of the value of p 2 i . On the other hand, in the case of two-stage FDR(S), the rejection region of p 2 i gradually becomes larger depending on the significance strength shown by the auxiliary variable (small p 1 i ). Unlike two-stage FDR(H), two- stage FDR(S) has a possibility that the primary variable can be rejected in all range of p 1 i . [Figure 3 about here.] © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 8 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable 3.3 F alse discovery rate contr olling procedur es In the pre vious section, Theorem 3.1 and 3.2 sho w that suggested p -values for both methods are uniformly distributed when ( ˆ β i , y i ) ∼ G 0 ( β , y ) . Based on this property of p i s, we apply a method to control the FDR using p H i s and p S i s in (12) and (18). Our goal is to estimate the FDR and ensure that its value does not exceed a giv en FDR le vel, denoted as α . W e test M hypotheses in (5), for example, in the Set4 ∆ data introduced in section 2, we test the significance of logfold changes . For a gi ven γ , the estimated FDR is expressed as: \ F D R ( γ ) = c π 0 γ M max( R M ( γ ) , 1) (19) where • M represents the number of hypotheses tested, • c π 0 is an estimate of the proportion of true negati ves among M , • R M ( γ ) = # { i : p i ≤ γ , 1 ≤ i ≤ M } is the number of rejections, where p i is either p H i ( γ 1 ) or p S i . The denominator in (19) takes the maximum value between the number of rejected hypotheses and 1. Here, c π 0 is estimated by using c π 0 = # { p i > λ } (1 − λ ) M (20) and we set λ = 0 . 5 for this study . Selection of γ 1 in two-stage FDR(H) Since p H i ( γ 1 ) is uniformly distributed with any γ 1 ∈ [0 , 1] , we select γ 1 which rejects most to maximise the true positiv e rate. i.e. b γ = arg max γ \ F D R ( γ ) ≤ α , (21) b γ 1 = arg max γ 1 # { i : p H i ( γ 1 ) ≤ b γ , 1 ≤ i ≤ M } , (22) p H i = p H i ( ˆ γ 1 ) . (23) W e summarise our proposed methods controlling FDR in Algorithm 1 and Algorithm 2 . Algorithm 1 T wo-stage FDR(H) 1: Estimate null distribution of β 2: Compute p 2 i under the null distribution of β , and p 1 i using the empirical distribution of Y 3: Select the copula between p 1 i and p 2 i 4: for γ 1 ∈ { r 1 , r 2 , . . . , r K } ⊂ [0 , 1] do 5: Compute p H i ( r k ) using p 1 i , p 2 i 6: Compute c π 0 7: For fix ed values of α and λ , check the number of rejections ( m k ) 8: end for 9: Compare the number of rejections for each r k and choose b γ 1 = r k as the one that maximises m k 10: Reject if P H i ( b γ 1 ) ≤ b γ , where γ is defined in (21) with b γ 1 © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 9 Algorithm 2 T wo-stage FDR(S) 1: Estimate null distribution of β 2: Compute p 2 i under the null distribution of β , and p 1 i using the empirical distribution of Y 3: Select the copula between p 1 i and p 2 i 4: Compute p S i using p 1 i , p 2 i with estimated copula 5: Compute c π 0 6: Estimate b γ satisfying (21) 7: Reject if p S i ≤ b γ 4 Simulation Studies In our study , we generate a dataset of M = 8 , 000 pairs of variables, ( β i , y i ) , for 1 ≤ i ≤ 8 , 000 . This dataset is designed to simulate the characteristics of our real data example, the Set4 ∆ data, but in a simplified form. The auxiliary variable y i y i ∼ Gamma (3 , 4) (24) with the mean value 3 / 4 and the primary variable β i β i ∼ f ( β ) = p 0 f 0 ( β ) + (1 − p 0 ) f 1 ( β ) = 0 . 95 ϕ ( β ) + 0 . 05(0 . 5 ϕ ( β − µ ) + 0 . 5 ϕ ( β + µ )) for some µ > 0 . T o closely replicate the joint distribution observed in the Set4 ∆ data, we model the pairs ( β i , y i ) to be jointly distrib uted following a Clayton copula. In our first scenario we used the Clayton copula to calculate p values. W e v aried the parameters µ and K endall’ s τ and assessed the F D R in (9). Our Monte Carlo simulations provide an approximate FDR which is F D R ≈ 1 K P K k =1 V k R k ∨ 1 where R k and V k are the number of rejections and that of false rejections, respectiv ely at k th simulation among K simulations. Let θ i = I ( H 1 i is true ) . As statistical powers, we use the true positiv e rate (TPR) which is T P R = E P M i =1 θ i δ i P M i =1 θ i ∨ 1 ! (25) where a ∨ 1 = max( a, 1) is the maximum of a and 1, P M i =1 θ i is the number of H 1 i and P M i =1 θ i δ i is the number of the true positives. Similarly , an approximate TPR is calculated 1 K P K k =1 S k M k where S k and M k are the number of true rejections and that of H 1 i at k th simulation among K simulations. W e use K = 1000 in our simulation studies. W e consider the v arious methods as follows : locfdr in Efron [2004], Storey in Storey [2002], IHW in Ignatiadis et al. [2016], Boca and Leek in Boca and Leek [2018] and our proposed two-stage FDR methods such as T ype H and T ype S. The results shown in T ables 1 show the ef fects of dif ferent degrees of dependence ( τ ) with a constant distance between the null and alternativ e distributions ( µ = 3 . 0 ), and µ changes while τ remains fixed at -0.4. Notably , all methods indicate an increase in TPR as µ increases. The one-stage FDR methods, which depend only on ˆ β i values, sho w consistent FDR and TPR, highlighting their limitations in adapting to dif- ferent lev els of dependence. Conv ersely , both covariate-assisted and two-stage FDR methods sho w higher TPR with increasing dependence. Ho we ver , their effecti veness in controlling FDR v aries. Co variate- assisted methods, despite extending the rejection region to reject more hypotheses, fail to maintain FDR control when dependencies are present. This suggests that cov ariate-assisted methods extend the rejection region where null hypotheses accumulate. Meanwhile, two-stage FDR methods effecti vely maintain FDR control ev en as rejection rates increase with increasing dependency , thereby achieving a better TPR. This result suggests that the inclusion of auxiliary variables can improv e the discovery of significant results, © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 10 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable but the results should be interpreted with caution. In Section 5, we demonstrate the potential for increased discov ery using auxiliary variables. Still, the results of covariate-assisted methods should be treated with particular caution. [T able 1 about here.] Our simulation results indicate that our proposed methods can control the False Discovery Rate (FDR) when the dependency structure is known. Ho wev er , in real data, the copula is often unknown. T o address the question, ”What happens if we mis-specify the copula in the models?” we explore the consequences of choosing an incorrect copula for calculating p i in our proposed methods. While the correct copula selection is essential for controlling the FDR, it is equally crucial to understand the consequences of using an incorrect copula. T o e valuate the robustness of our methodology to the selection of copula, we choose a copula that does not align with the true underlying dependencies. Specifically , we chose the BB7, BB6 and Joe copulas for comparison. These were chosen because of their similarity to the Clayton copula, which allowed us to e xplore the ef fects of potential misspecification when replacing the Clayton copula with these alternativ es. T able 2 displays the estimated FDR and true positive rate (TPR) for each of the four copulas when µ = 3 and τ = − 0 . 4 . [T able 2 about here.] From T able 2, we see that all four methods control a given lev el of FDR for all settings while locfdr and two-stage FDR (S) obtain lower FDR v alues compared to the nominal level α = 0 . 05 . Howe ver , two- stage FDR (S) and two-stage FDR (H) obtain highest and the second highest TPRs, respecti vely across all settings. These results show that the supplementary information from the auxiliary v ariable may af fect the power of testing procedures when it is appropriately reflected in testing procedure. 5 Real Data Analysis 5.1 Estimation of the Marginal p -values and Optimal Copula auxiliary variable ( p 1 i ) : The standard deviation of the logfold changes of each gene is used as an aux- iliary variable. Since we have small number of samples, we estimated the standard deviation by bootstrap method for all possible combinations, resulting in 729 (i.e. 3 3 × 3 3 ) possible combinations, and deriv ed the p 1 i values from their empirical distrib ution. The detailed proceudure is described in Algorithm 3. Algorithm 3 Estimation of Standard Deviation using Bootstrap for Set4 ∆ data 1: f or i ← 1 to 8220 do ▷ Iterate through all genes 2: for b ← 1 to 729 do ▷ Iterate through all bootstrap samples 3: Sample three knockouts ( K O b i (1) , K O b i (2) , K O b i (3) ) from the original knockouts ( K O i 1 , K O i 2 , K O i 3 ) with replacement. 4: Sample three wildtypes ( W T b i (1) , W T b i (2) , W T b i (3) ) from the original wildtypes ( W T i 1 , W T i 2 , W T i 3 ) with replacement. 5: Calculate logfold changes: ˜ β b i = log ¯ K O b ¯ W T b 6: end for 7: Compute the sample standard deviation of ˜ β b i values 8: Determine the p 1 i value based on the empirical distrib ution of these standard deviations 9: end f or primary variable ( p 2 i ) : For the primary variable, we adopt the approach proposed by Ramos et al. [2021] to evaluate the log change values of each gene. This method estimates the null distribution as a mixture of two normal distrib utions.i.e. f 0 ( β ) = 0 . 615 ϕ β 0 . 063 + 0 . 385 ϕ β + 0 . 002 0 . 205 © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 11 where ϕ is a probability distrib ution function of a normal distribution. Based on this null, p 2 i is calculated as described in (11). Optimal Copula : we ev aluated the validity of various copulas - including Gaussian, Frank, Clayton, Gumbel and Joe - for modelling the dependencies between p 1 i and p 2 i . This ev aluation, based on Log- Likelihood (LogLik), Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC), is illustrated in T able 3. The Clayton copula was selected o wing to its outstanding performance in all criterion, indicating an optimal balance between model complexity and fit. [T able 3 about here.] 5.2 Application of FDR Controlling Pr ocedure Based on the estimation of the distribution G 0 in the previous section, we apply the Algorithm 1 and Algorithm 2 with α = 0 . 10 . For the distributions of p H i and p S i , see Figure ?? in the supplementary material. In Algorithm 1 , the process of selecting γ 1 in volv ed analysing the relationship between the number of hypothesis rejections and various γ 1 values. Using the equation (22), we determined the optimal value of ˆ γ 1 as the value that maximises the number of rejections. Figure 4 illustrates the relationship between γ 1 values and the number of rejections. It shows that the number of rejections increases as γ 1 increases, peaking at γ 1 = 0 . 987 , after which it starts to decrease. W ith the choice of ˆ γ 1 = 0 . 987 , this resulted in the rejection of 492 null hypotheses out of a total of 8 , 220 . [Figure 4 about here.] In order to assess the performance of four different FDR controlling methods, we compare the number of rejections for both one-stage and two-stage approaches. Firstly , employing the one-stage FDR(locfdr), we identified 249 rejected genes. A slightly higher number of genes, 424 , were rejected when applying the one-stage FDR(Storey). Howe ver , when utilizing two-stage FDR methods, we observed a significant increase in gene rejections. Specifically , the two-stage FDR(H) resulted in 485 rejected genes, while the two-stage FDR(S) led to a 582 rejections. These findings support the simulation results presented in T able 1, demonstrating the greater power of two-stage FDR methods in detecting deferentially expressed genes (DEG). Figure ?? in online supplementary matierial offers an in-depth comparativ e analysis of the proposed two-stage FDR methods introduced in this study versus traditional one-stage FDR methods (local FDR and Storey’ s method), applied to the set4 ∆ mutant dataset. Figure 5 shows four different rejection regions corresponding to the two one-stage FDR methods and the two two-stage FDR methods. These different rejection re gions provide further insight into the comparison between one-stage and two-stage FDR methods. As sho wn in the figures, the one-stage FDR methods determine the rejection region based on only the logfold changes in the y -axis, while in the case of the two-stage FDR(H), it exhibits a rectangular -shaped rejection re gion determined independently by cutoff values for y and β . On the other hand, in the case of two-stage FDR(S), it shows a curved rejection region, indicating that the dependence between the two variables has been taken into account. The further result compared to DESeq2 (Love et al. [2014]), a well-known DEG analysis, is also represented in the supplementary material. HXT gene family : W e conducted an in vestigation into the functional role of these ne wly rejected gene sets using GO and MIPs databases of the two rejected gene lists in Robinson et al. [2002]. In both cases, we observed an enrichment of genes associated with stress response and cell wall maintenance among the differentially e xpressed genes, consistent with our previous findings in Jethmalani et al. [2021]. Addition- ally , the new analysis re vealed an interesting enrichment of genes in volv ed in hexose transport, particularly from the HXT gene family . Figure 5 include the locations of the HXT gene family represented by crosses and show that the two-stage FDR(S) reject all of them while the others reject part of them. These rejected genes mainly belong to the minor cate gory of he xose transporters, kno wn for primarily transporting manni- tol and sorbitol (Diderich et al. [1999]). Remarkably , under stress conditions like hypoxia or anaerobia, the © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 12 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable transcription of these transporters is altered, likely leading to increased transport of the preferred carbon source, glucose, into cells (Rintala et al. [2008]). This finding identifies another class of genes regulated by Set4 during hypoxia that play a crucial role in the cell’ s adaptation to low oxygen. Moreover , a number of these genes are located within approximately 40 kB of the chromosome end, including HXT15, HXT13, and HXT8, consistent with previous findings that Set4 regulates genes within the subtelomeric regions of chromosomes (Jethmalani et al. [2021]). In conclusion, this differential gene expression analysis method has successfully identified a new class of genes in volved in stress responses, regulated by the Set4 protein in yeast grown under h ypoxic conditions. [Figure 5 about here.] [T able 4 about here.] 6 Concluding Remarks In this paper, we dev elop the two stage procedures controlling FDR based on incorporating an auxiliary variable to improv e the power of testing the primary variable. Furthermore, we also increase the credibility of the results via incorporating prior kno wledge through the auxiliary variable. Rather than assuming the structure of standard de viation of logfold changes, we simply used the copula to model the dependence between the logfold changes and their standard deviations. Our methods allo w for a more flexible and accurate representation of the joint distribution of these two variables. T wo proposed methods select the significant genes in different ways. One takes the form of screening, while the other manifests as a progres- siv e form by showing different rejection regions based on the values of auxiliary variable. Both methods are superior to existing methods without using an y auxiliary variable. By taking into account the variability of the experiments and controlling for false discovery rate, the proposed methods can help to increase the confidence and reproducibility of gene expression analysis. W e present numerical studies supporting our proposed methods and the real data example of Set4 ∆ mutant data. Our proposed methods can control a given level of FDR and are more powerful in detecting the true alternati ve hypotheses than one-stage procedures and some existing methods with cov ariate. Especially in the real data, tw o-stage FDR (S) is more efficient to select all the genes in the HXT gene f amily compared to all the other methods. Overall, the proposed methods incorporating additional information into the problem of testing the primary v ariable are a v aluable contribution to the field of gene e xpression analysis, and ha ve the potential to improv e the quality and reliability of experimental results. Additionally , the proposed methods can be widely applied beyond the gene-related data we introduced, to multiple testing problems where there e xist both the primary and auxiliary variables. 7 Data and Code A v ailability The dataset(Set4 ∆ ) and source code de veloped for this research can be found at the following GitHub repository: https://github .com/twostageFDR/.github. This repository includes all scripts and instructions necessary to replicate the computational procedures and analyses presented in this paper . Conflict of Interest The authors hav e declared no conflict of interest. Supporting Inf ormation Additional Supporting Information may be found in the online version of this article. © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 13 Acknowledgements This work was supported by the National Research Foundation of Korea (NRF) grant funded by the K orea gov ernment (MSIT) (No. 2020R1A2C1A01100526). References S. M. Boca and J. T . Leek. A direct approach to estimating false discovery rates conditional on covariates. P eerJ , 6: e6035, 2018. H. Cao, J. Chen, and X. Zhang. Optimal false discovery rate control for large scale multiple testing with auxiliary information. The Annals of Statistics , 50(2):807–857, 2022. J. A. Diderich, M. Schepper, P . van Hoek, M. A. Luttik, J. P . van Dijken, J. T . Pronk, P . Klaassen, H. F . Boelens, M. J. T . de Mattos, K. v an Dam, et al. Glucose uptake kinetics and transcription of hxtgenes in chemostat cultures of saccharomyces cerevisiae. Journal of Biological Chemistry , 274(22):15350–15359, 1999. L. Du and C. Zhang. Single-index modulated multiple testing. The Annals of Statistics , pages 1262–1311, 2014. B. Efron. Large-scale simultaneous hypothesis testing: the choice of a null hypothesis. Journal of the American Statistical Association , 99(465):96–104, 2004. B. Efron and R. T ibshirani. Empirical bayes methods and false discovery rates for microarrays. Genetic epidemiology , 23(1):70–86, 2002. H. Fink, Y . Klimova, C. Czado, and J. St ¨ ober . Regime switching vine copula models for global equity and v olatility indices. Econometrics , 5(1):3, 2017. N. Ignatiadis, B. Klaus, J. B. Zaugg, and W . Huber . Data-driv en hypothesis weighting increases detection power in genome-scale multiple testing. Natur e methods , 13(7):577–580, 2016. Y . Jethmalani, K. Tran, M. Y . Negesse, W . Sun, M. Ramos, D. Jaiswal, M. Jezek, S. Amos, E. J. Garcia, D. Park, et al. Set4 regulates stress response genes and coordinates histone deacetylases within yeast subtelomeres. Life Science Alliance , 4(12), 2021. L. Lei and W . Fithian. Adapt: An interacti ve procedure for multiple testing with side information. Journal of the Royal Statistical Society Series B: Statistical Methodology , 80(4):649–679, 2018. M. I. Love, W . Huber , and S. Anders. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome biology , 15(12):1–21, 2014. M. L. Ramos, D. Park, J. Lim, J. Park, K. Tran, E. J. Garcia, and E. Green. Adaptiv e local false discov ery rate proce- dures for highly spiky data and their application rna sequencing data of yeast set4 deletion mutants. Biometrical Journal , 63(8):1729–1744, 2021. E. Rintala, M. G. W iebe, A. T amminen, L. Ruohonen, and M. Penttil ¨ a. T ranscription of he xose transporters of saccha- romyces cerevisiaeis af fected by change in oxygen provision. BMC microbiolo gy , 8(1):1–12, 2008. M. D. Robinson, J. Grigull, N. Mohammad, and T . R. Hughes. Funspec: a web-based cluster interpreter for yeast. BMC bioinformatics , 3(1):1–5, 2002. J. St ¨ ober and C. Czado. Regime switches in the dependence structure of multidimensional financial data. Computa- tional Statistics & Data Analysis , 76:672–686, 2014. J. D. Storey . A direct approach to false discovery rates. Journal of the Royal Statistical Society: Series B (Statistical Methodology) , 64(3):479–498, 2002. M. J. Zhang, F . Xia, and J. Zou. Fast and cov ariate-adaptive method amplifies detection power in large-scale multiple hypothesis testing. Natur e Communication , 10(1):3433, 2019. doi: 10.1038/s41467- 019- 11247- 0. © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 14 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable List of Figures 1 The histograms show that the distributions of (A) the logfold changes and (b) their esti- mated standard deviations. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15 2 Comparison of p -value distributions: (A) p -values generated symmetrically around 0.5, and (B) p -values generated from Set4 ∆ data . . . . . . . . . . . . . . . . . . . . . . . . 16 3 p i ’ s for two-stage procedures (A),(B) of two-stage FDR(H) and (C) of two-stage FDR(S). In (A), p H i with the threshold γ 1 = 0 . 9 and (B) with the threshold γ 1 = 0 . 7 . . . . . . . . 17 4 Selection of γ in two-stage FDR(H). The x-axis displays γ and the y-axis represents the corresponding number of rejection with two-stage FDR(H). . . . . . . . . . . . . . . . . . 18 5 Comparison of Rejection Regions: One-Stage vs. T wo-Stage FDR Methods. The x-axis represents the standard deviation of logfold changes on a logarithmic scale as an auxiliary variable, while the y-axis sho ws the logfold changes as the primary variable. The sky- blue points corresponds to the rejection region at α < 0 . 05 , and the darker blue points represents the rejection region at α < 0 . 10 . In the legend, ✕ and ✴ signify the HXT gene family rejection with α = 0 . 05 and α = 0 . 10 , respectively , while ▲ indicates non-rejected genes in the HXT gene family . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19 © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 15 −4 −2 0 2 4 0 1000 2000 3000 4000 (A) Logfold changes −6 −5 −4 −3 −2 −1 0 1 0 500 1000 1500 2000 (B) Standard deviation of logf old changes(log−scale) Figure 1 The histograms show that the distributions of (A) the logfold changes and (b) their estimated standard deviations. © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 16 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable 0.0 0.2 0.4 0.6 0.8 1.0 0.0 0.2 0.4 0.6 0.8 1.0 Auxiliary p−values Primary p−values (A) p−values Generated Symmetrically ar ound 0.5 0.0 0.2 0.4 0.6 0.8 1.0 0.0 0.2 0.4 0.6 0.8 1.0 Auxiliary p−values (standard de viation of logfold changes) Primary p−values (logfold changes) (B) p−values Generated b y Real Data Figure 2 Comparison of p -value distributions: (A) p -v alues generated symmetrically around 0.5, and (B) p -values generated from Set4 ∆ data © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 17 0.0 0.2 0.4 0.6 0.8 1.0 0.0 0.2 0.4 0.6 0.8 1.0 (A) A B C 0.0 0.2 0.4 0.6 0.8 1.0 0.0 0.2 0.4 0.6 0.8 1.0 (B) A B C 0.0 0.2 0.4 0.6 0.8 1.0 0.0 0.2 0.4 0.6 0.8 1.0 (C) A B C Figure 3 p i ’ s for two-stage procedures (A),(B) of two-stage FDR(H) and (C) of two-stage FDR(S). In (A), p H i with the threshold γ 1 = 0 . 9 and (B) with the threshold γ 1 = 0 . 7 © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 18 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable 0.5 0.6 0.7 0.8 0.9 1.0 0 100 200 300 400 0.989 0.989 Figure 4 Selection of γ in two-stage FDR(H). The x-axis displays γ and the y-axis represents the corre- sponding number of rejection with two-stage FDR(H). © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 19 −6 −5 −4 −3 −2 −1 0 1 −4 −2 0 2 4 (A) One−stage (locfdr) −6 −5 −4 −3 −2 −1 0 1 −4 −2 0 2 4 (B) One−stage (Storey) −6 −5 −4 −3 −2 −1 0 1 −4 −2 0 2 4 (C) T wo−stage (H) −6 −5 −4 −3 −2 −1 0 1 −4 −2 0 2 4 (D) T wo−stage (S) Figure 5 Comparison of Rejection Regions: One-Stage vs. T wo-Stage FDR Methods. The x-axis rep- resents the standard deviation of logfold changes on a logarithmic scale as an auxiliary variable, while the y-axis sho ws the logfold changes as the primary v ariable. The sky-blue points corresponds to the rejection region at α < 0 . 05 , and the darker blue points represents the rejection re gion at α < 0 . 10 . In the le gend, ✕ and ✴ signify the HXT gene family rejection with α = 0 . 05 and α = 0 . 10 , respecti vely , while ▲ indicates non-rejected genes in the HXT gene family . © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 20 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable List of T ables 1 Comparison of False Discov ery Rate (FDR) and T rue Positiv e Rate (TPR) across dif ferent settings for the mean parameter , µ , and Kendall’ s τ coefficient. . . . . . . . . . . . . . . . 21 2 Comparison of False Discov ery Rate (FDR) and True Positi ve Rate (TPR) in the Presence of Misspecified Copulas. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22 3 The Log-likelihood(LogLik), AIC, BIC of Gaussian, Frank, Clayton, Gumbel and Joe cop- ulas with set4 ∆ dataset . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 4 Comparison of total and HXT family gene rejections using one-stage (locfdr , Storey), cov arate-assisted (IHW , Boca and Leek, AdaFDR) and two-stage (types H and S) FDR methods at α = 0 . 05 and α = 0 . 10 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24 © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 21 T able 1 Comparison of False Discovery Rate (FDR) and T rue Positiv e Rate (TPR) across dif ferent set- tings for the mean parameter , µ , and Kendall’ s τ coefficient. τ One-stage FDR Cov ariate-assisted FDR T wo-stage FDR locfdr Storey IHW Boca and Leek AdaFDR type H type S \ F DR 0.0 0.013 (0.013) 0.046 (0.020) 0.043 (0.020) 0.000 (0.006) 0.048 (0.024) 0.046 (0.021) 0.045 (0.020) -0.2 0.063 (0.032) 0.000 (0.006) 0.160 (0.061) 0.044 (0.019) 0.038 (0.016) -0.4 0.070 (0.039) 0.000 (0.007) 0.480 (0.085) 0.038 (0.016) 0.028 (0.012) -0.6 0.074 (0.041) 0.014 (0.020) 0.837 (0.019) 0.032 (0.018) 0.017 (0.009) -0.8 0.063 (0.045) 0.945 (0.002) 0.873 (0.007) 0.029 (0.028) 0.005 (0.006) [ T P R 0.0 0.213 (0.038) 0.378 (0.051) 0.367 (0.052) 0.002 (0.006) 0.375 (0.059) 0.379 (0.051) 0.376 (0.051) -0.2 0.364 (0.050) 0.004 (0.008) 0.380 (0.073) 0.490 (0.042) 0.577 (0.042) -0.4 0.364 (0.047) 0.014 (0.020) 0.500 (0.062) 0.643 (0.033) 0.804 (0.026) -0.6 0.392 (0.039) 0.115 (0.052) 0.656 (0.037) 0.739 (0.025) 0.928 (0.014) -0.8 0.466 (0.039) 0.582 (0.028) 0.672 (0.051) 0.790 (0.022) 0.980 (0.007) µ One-stage FDR Cov ariate-assisted FDR T wo-stage FDR locfdr Storey IHW Boca and Leek AdaFDR type H type S \ F D R 2.0 0.022 (0.079) 0.026 (0.065) 0.174 (0.084) 0.000 (0.000) 0.643 (0.247) 0.030 (0.022) 0.024 (0.016) 2.5 0.016 (0.026) 0.038 (0.030) 0.121 (0.057) 0.000 (0.011) 0.691 (0.101) 0.033 (0.018) 0.026 (0.012) 3.0 0.013 (0.013) 0.046 (0.020) 0.070 (0.039) 0.000 (0.007) 0.580 (0.085) 0.038 (0.016) 0.028 (0.012) 3.5 0.010 (0.008) 0.049 (0.017) 0.081 (0.047) 0.003 (0.007) 0.442 (0.088) 0.045 (0.018) 0.030 (0.012) 4.0 0.008 (0.006) 0.051 (0.016) 0.130 (0.053) 0.008 (0.008) 0.404 (0.080) 0.050 (0.018) 0.031 (0.012) [ T P R 2.0 0.010 (0.007) 0.015 (0.013) 0.073 (0.026) 0.000 (0.000) 0.183 (0.090) 0.190 (0.035) 0.308 (0.045) 2.5 0.065 (0.022) 0.127 (0.041) 0.168 (0.034) 0.001 (0.002) 0.348 (0.070) 0.420 (0.039) 0.595 (0.038) 3.0 0.213 (0.038) 0.378 (0.051) 0.364 (0.047) 0.014 (0.020) 0.500 (0.062) 0.643 (0.033) 0.804 (0.026) 3.5 0.428 (0.044) 0.642 (0.041) 0.614 (0.037) 0.175 (0.060) 0.681 (0.055) 0.803 (0.024) 0.920 (0.016) 4.0 0.643 (0.038) 0.828 (0.027) 0.781 (0.026) 0.454 (0.063) 0.838 (0.035) 0.902 (0.017) 0.973 (0.009) © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 22 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable T able 2 Comparison of False Discovery Rate (FDR) and T rue Positive Rate (TPR) in the Presence of Misspecified Copulas. Copula One-stage FDR T wo-stage FDR locfdr Storey type H type S \ F D R Clayton 0.013 (0.013) 0.046 (0.020) 0.038 (0.016) 0.028 (0.012) BB7 0.006 (0.012) 0.001 (0.0.02) BB6 0.008 (0.009) 0.002 (0.003) Joe 0.049 (0.019) 0.032 (0.014) [ T P R Clayton 0.213 (0.038) 0.378 (0.051) 0.643 (0.033) 0.804 (0.026) BB7 0.466 (0.035) 0.588 (0.030) BB6 0.518 (0.037) 0.630 (0.032) Joe 0.660 (0.032) 0.805 (0.026) Copula One-stage FDR T wo-stage FDR locfdr Storey type H type S \ F D R Clayton 0.013 (0.013) 0.046 (0.020) 0.038 (0.016) 0.028 (0.012) BB7 0.038 (0.017) 0.029 (0.012) BB6 0.036 (0.016) 0.034 (0.013) Joe 0.031 (0.014) 0.028 (0.011) [ T P R Clayton 0.213 (0.038) 0.378 (0.051) 0.643 (0.033) 0.804 (0.026) BB7 0.643 (0.033) 0.805 (0.026) BB6 0.641 (0.033) 0.800 (0.026) Joe 0.643 (0.033) 0.804 (0.026) © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com Biometrical Journal 52 (2024) 61 23 T able 3 The Log-likelihood(LogLik), AIC, BIC of Gaussian, Frank, Clayton, Gumbel and Joe copulas with set4 ∆ dataset Family LogLik AIC BIC Gaussian 306.45 -610.91 -604.80 Frank 332.84 -663.68 -657.57 Clayton 393.08 -784.16 -778.05 Gumbel 201.79 -401.57 -395.46 Joe 96.29 -190.57 -184.46 © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com 24 Hwang et al. , Ramos, Park, P ark, Lim, and Green: T wo-Stage Multiple T esting with auxiliary variable T able 4 Comparison of total and HXT family gene rejections using one-stage (locfdr, Storey), cov arate- assisted (IHW , Boca and Leek, AdaFDR) and two-stage (types H and S) FDR methods at α = 0 . 05 and α = 0 . 10 . One-stage FDR Cov ariate-assisted FDR T wo-stage FDR α locfdr Storey IHW Boca and Leek AdaFDR type H type S # rejected genes 0.05 230 361 486 110 844 407 503 0.10 249 424 633 116 1329 485 582 # of rejected HXT family 0.05 3 4 5 3 6 3 4 0.10 3 4 5 3 6 4 6 © 2024 WILEY -VCH V erlag GmbH & Co. KGaA, W einheim www .biometrical-journal.com
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment