AgriVariant: Variant Effect Prediction using DeepChem-Variant for Precision Breeding in Rice
Predicting functional consequences of genetic variants in crop genes remains a critical bottleneck for precision breeding programs. We present AgriVariant, an end-to-end pipeline for variant-effect prediction in rice (Oryza sativa) that addresses the…
Authors: Ankita Vaishnobi Bisoi, Bharath Ramsundar
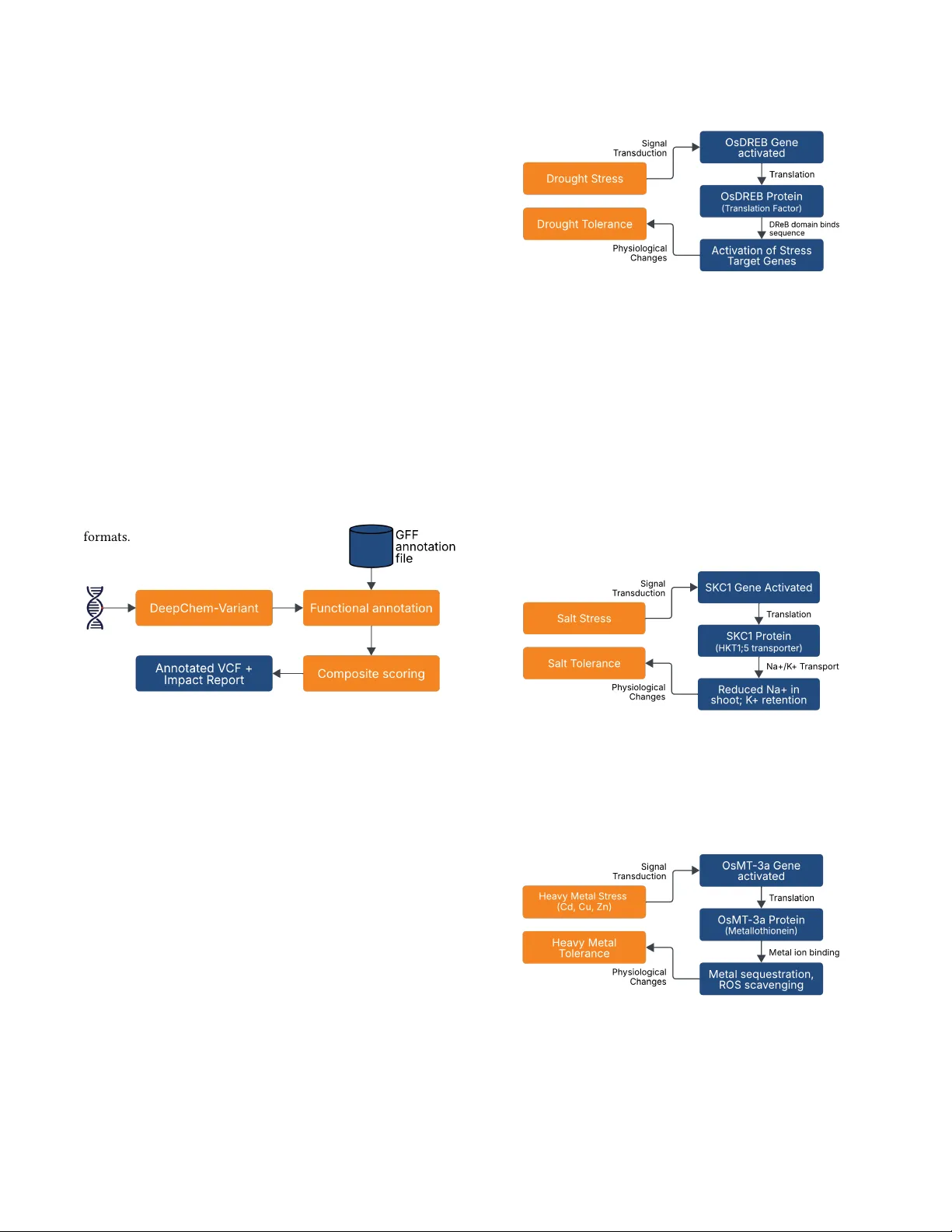
AgriV ariant: V ariant Ee ct Prediction using DeepChem- V ariant for Precision Breeding in Rice Ankita V aishnobi Bisoi f20212306@goa.bits- pilani.ac.in BI TS Pilani Goa Campus, India Goa, India Bharath Ramsundar bharath@deepforestsci.com Deep Forest Sciences California, USA Abstract Predicting functional consequences of genetic variants in crop genes remains a critical bottleneck for pr ecision br eeding programs. W e present AgriV ariant, an end-to-end pipeline for variant-eect prediction in rice (Oryza sativa) that addresses the lack of crop- specic variant-interpretation tools and can be extended to any crop spe cies with available r eference genomes and gene annota- tions. Our approach integrates deep learning-based variant calling (DeepChem- V ariant [ 2 ]) with custom plant genomics annotation using RAP-DB[ 23 ][ 11 ] gene models and database-independent dele- teriousness scoring that combines the Grantham[ 9 ] distance and the BLOSUM62 [ 10 ] substitution matrix. W e validate the pipeline through targeted mutations in str ess-response genes (OsDREB2a, OsDREB1F, SKC1), demonstrating correct classication of stop- gained, missense, and synonymous variants with appropriate HIGH / MODERA TE / LO W impact assignments. An e xhaustive mutagen- esis study of OsMT -3a analyzed all 1,509 possible single-nucleotide variants in 10 days, identifying 353 high-impact, 447 medium- impact, and 709 low-impact variants —an analysis that would have required 2-4 years[ 5 ] using traditional wet-lab approaches. This computational framework enables breeders to prioritize variants for experimental validation across div erse crop species, reducing screening costs and accelerating dev elopment of climate-resilient crop varieties. CCS Concepts • Applied computing → Bioinformatics ; Computational ge- nomics ; Precision Breeding ; • Computing methodologies → Supervise d learning by classication ; Neural networks . Ke ywords variant eect prediction, deep learning, crop genomics, functional annotation, rice breeding, precision agriculture A CM Reference Format: Ankita V aishnobi Bisoi and Bharath Ramsundar. 2026. AgriV ariant: V ariant Eect Prediction using DeepChem- V ariant for Precision Breeding in Rice. In . ACM, Ne w Y ork, N Y , USA, 9 pages. https://doi.org/10.1145/nnnnnnn. nnnnnnn Permission to make digital or hard copies of all or part of this work for personal or classroom use is granted without fee provided that copies are not made or distributed for prot or commercial advantage and that copies bear this notice and the full citation on the rst page. Copyrights for components of this work owned by others than the author(s) must be honor ed. Abstracting with credit is permitted. T o copy otherwise, or republish, to post on servers or to redistribute to lists, r equires prior spe cic permission and /or a fee. Request permissions from permissions@acm.org. Conference’17, Washington, DC, USA © 2026 Copyright held by the owner/author(s). Publication rights licensed to A CM. ACM ISBN 978-x-xxxx-xxxx-x/Y YY Y/MM https://doi.org/10.1145/nnnnnnn.nnnnnnn 1 Introduction Climate change threatens global fo od se curity through increased fre- quency and severity of environmental stresses. Rice (Oryza sativa), a staple crop feeding over half the world’s population, faces partic- ular vulnerability to drought and salinity stress. Genetic variants in stress-response genes dictate crop durability , yet identifying which variants functionally impact pr otein activity remains a critical chal- lenge despite vast available genomic data. W e address this challenge by developing AgriV ariant, a vari- ant eect prediction pipeline that integrates deep learning-based variant calling with plant-specic functional annotation and quan- titative deleteriousness scoring. Building upon our previous w ork on De epChem- V ariant [ 2 ], a modular implementation of Deep- V ariant [ 19 ][ 20 ] within the DeepChem [ 21 ] framework, we extend variant calling capabilities to downstream functional interpreta- tion spe cically for crop genomes. DeepChem- V ariant leverages convolutional neural networks [ 13 ] to reframe variant calling as image classication, generating multi-channel pileup images from sequencing reads and processing them through Inception V3 [ 26 ] or MobileNetV2 [ 24 ] architectures, achieving 95.3% accuracy on rice genomic variants. DeepChem, the underlying framework, is an open-source Python library designed for scientic machine learning that has estab- lished itself as a versatile platform for applications ranging from the Mole culeNet b enchmark suite [ 29 ] to protein-ligand interac- tion modeling [ 8 ] and generative modeling of molecules [ 7 ]. This ecosystem enables AgriV ariant to leverage mature molecular ma- chine learning infrastructure while pro viding modular , extensible components specically tailored for crop genomics applications. The primary bottlene ck in crop br eeding lies not in variant de- tection, but in variant interpretation. W e target stress-response genes including the DREB transcription factor family (OsDREB2a, OsDREB1F) [ 6 ] [ 28 ], ion homeostasis genes (SK C1) [ 22 ], and met- allothionein genes (OsMT -3a) [ 18 ], which collectively regulate drought tolerance, salinity str ess, and heavy metal detoxication in rice. Howe ver , existing variant eect prediction tools (SIFT[ 25 ], PolyPhen-2[ 1 ], CADD[ 12 ]) rely heavily on human-specic databases and evolutionary conser vation patterns, leaving plant genomics without equivalent prediction frameworks. Existing plant annota- tion tools like SnpE[ 3 ] provide basic functional classication but oer limited species coverage and lack crop-specic databases for many reference genomes. W e overcome this limitation by developing a custom functional annotation pipeline that maps variants to coding sequences using Rice Annotation Project Database (RAP-DB)[ 23 ][ 11 ] gene models and classies functional eects (stop-gained, missense, synony- mous) based on aected codons and amino acid translations. W e Conference’17, July 2017, W ashington, DC, USA Bisoi et al. implement a database-indep endent deleteriousness scoring st rategy that merges Grantham[ 9 ] distance matrices (which quantify amino acid divergence based on composition, polarity , and volume) with BLOSUM62 [ 10 ] substitution matrices (reect ev olutionary toler- ance to amino acid changes). This combined scoring framework enables quantitative variant severity assessment without requiring organism-specic training databases, proving particularly valuable for understudied crops where extensive variant annotations ar e unavailable. 2 Methods The complete pipeline for A griV ariant (Figure 1) combines three complementary methods for variant interpretation in rice genomes. Sequencing data undergoes variant calling via DeepChem- V ariant, followed by functional annotation using custom scripts that map mutations to coding sequences and classify eects based on RAP-DB gene models. Detecte d variants are then scored for deleteriousness using a composite metric derived from Grantham distance and BLOSUM62 substitution matrices. The output consists of annotate d V ariant Call Format (VCF) les containing variant positions, func- tional classications, and quantitative severity scores. This modular architecture enables independent updating of individual compo- nents while maintaining compatibility with standard genomic le formats. Figure 1: AgriV ariant pipeline integrating de ep learning- based variant calling, plant-sp ecic functional annotation, and quantitative deleteriousness scoring. 2.1 T arget Gene Selection and Biological Context W e selected target genes based on documente d roles in abiotic stress tolerance and ion homeostasis. The DREB (Dehydration-Responsive Element-Binding) gene family belongs to the AP2/ERF superfamily of plant transcription factors. These genes regulate responses to abiotic stresses(dr ought, cold, and salt) by binding to DRE/CRT cis- elements in DNA, thereby activating downstream str ess-responsive genes and improving plant tolerance. T wo DREB family members, OsDREB2a [ 6 ] and OsDREB1F [ 28 ], were selected as primary targets for loss-of-function variant anal- ysis. When a plant experiences stress, expression of OsDREB2a and OsDREB1F is induced (Figure 2). The resulting proteins act as transcriptional activators, switching on expression of target genes. This activation leads to accumulation of osmolytes such as soluble sugars and free proline, which help the plant adjust osmotic poten- tial, maintain cell structure, and protect cellular components under stress. Figure 2: OsDREB-me diated drought stress response path- way includes str ess-induced gene activation, transcription factor production, downstream target gene regulation, and resulting drought tolerance. Beyond transcriptional r egulation, ion homeostasis represents another critical mechanism for stress tolerance. SK C1 (Shoot K+ Concentration 1), also known as OsHKT1;5, is a high-anity K + transporter that regulates sodium-potassium balance in rice tissues during salt stress [ 22 ]. Genetic variants in SKC1 inuence salt toler- ance (Figure 3) across rice cultivars, making it an ideal candidate for neutral variant analysis. Additionally , we examined OsMT -3a [ 18 ], a metallothionein gene involved in heavy metal detoxication and oxidative stress responses (Figure 4), to demonstrate AgriV ariant’s applicability across diverse stress-r esponse mechanisms. Figure 3: SKC1-mediated salt stress response pathway . Salt stress activates SKC1 gene expression, producing HKT1;5 transporter protein that regulates Na+/K+ homeostasis by reducing sodium accumulation in sho ots while maintaining potassium levels, resulting in salt tolerance . Figure 4: OsMT -3a-me diated heav y metal stress response pathway . Heav y metal exposure (Cd, Cu, Zn) induces OsMT - 3a expression, producing metallothionein protein that binds and sequesters toxic metal ions while scavenging reactiv e oxygen sp ecies, conferring heavy metal tolerance . AgriV ariant: V ariant Eect Prediction using DeepChem- V ariant for Precision Breeding in Rice Conference’17, July 2017, W ashington, DC, USA 2.2 Computational Mutation Framework Rationale for in silico Approach: T raditional functional vali- dation of genetic variants r equires laboratory mutagenesis using CRISPR-Cas9 gene editing, followed by phenotypic screening across multiple growing seasons—a process r equiring 2-4 years and sig- nicant resources. For breeding programs evaluating thousands of candidate variants across multiple genes, this approach becomes expensive and time-consuming. AgriV ariant’s computational muta- tion framework enables rapid in silico scr eening of variant eects, allowing breeders to prioritize the most promising candidates for subsequent wet-lab validation. W e implemented a synthetic variant generation pipeline that mimics the CRISPR mutagenesis workow computationally ( Algo- rithm 1). This approach cr eates articial genomes containing tar- geted mutations, generates synthetic sequencing reads from these modied genomes, and pr ocesses them through DeepChem- V ariant to validate detection accuracy and annotation correctness. Algorithm 1 Synthetic V ariant Generation and V alidation Frame- work Require: Reference genome (F ASTA ), target gene coordinates, de- sired mutation Ensure: V alidate d variant in synthetic dataset 1: // Phase 1: Mutation Design 2: Extract CDS coordinates from GFF annotation for target gene 3: Identify target codon position and genomic coordinate 4: Design mutation: ( 𝑐ℎ𝑟 𝑜𝑚, 𝑝 𝑜 𝑠 , 𝑟 𝑒 𝑓 _ 𝑎𝑙 𝑙 𝑒 𝑙 𝑒 , 𝑎 𝑙 𝑡 _ 𝑎𝑙 𝑙 𝑒𝑙 𝑒 ) 5: V erify mutation type: stop-gained, missense, or synonymous 6: 7: // Phase 2: Synthetic Genome Generation 8: Create VCF le with mutation: { 𝑐ℎ𝑟 𝑜𝑚, 𝑝 𝑜 𝑠 , 𝑟 𝑒 𝑓 , 𝑎𝑙 𝑡 , 𝐺𝑇 = 1 / 1 } 9: Compress and index V CF: bgzip , tabix 10: Generate mutant genome: bcftools consensus 𝑟 𝑒 𝑓 . 𝑓 𝑎 𝑚𝑢𝑡 𝑎𝑡 𝑖𝑜 𝑛 .𝑣𝑐 𝑓 .𝑔 𝑧 11: Index mutant genome: samtools faidx 12: 13: // Phase 3: Synthetic Read Generation 14: Extract target region: [ 𝑔 𝑒 𝑛𝑒 _ 𝑠𝑡 𝑎𝑟 𝑡 − 5 𝑘 𝑏, 𝑔𝑒𝑛𝑒 _ 𝑒𝑛𝑑 + 5 𝑘𝑏 ] 15: Generate paired-end reads: wgsim -N 1000 -1 150 -2 150 16: 17: // Phase 4: V ariant Calling V alidation 18: Align synthetic reads to original reference: bwa mem 19: Sort and index BAM le: samtools sort , samtools index 20: V erify mutation presence: samtools mpileup at target p osition 21: Run DeepChem- V ariant pipeline (Algorithm 2) 22: 23: // Phase 5: V alidation 24: if called variant matches designed mutation then 25: Success: Mutation correctly detected 26: else 27: Failure: Investigate detection/annotation errors 28: end if 29: return Annotate d variant with functional classication AgriV ariant ser ves as a proof-of-concept for computational vari- ant prioritization. In production breeding worko ws, researchers would use these predictions to rank thousands of naturally occur- ring or CRISPR-induced variants, selecting only the highest-impact candidates for costly eld trials. 2.3 V ariant Calling with Deep-Chem- V ariant DeepChem- V ariant performe d alignment-based variant calling be- tween reference and simulated genomes. Unlike traditional statis- tical variant callers (GA TK[ 17 ], SAMtools[ 15 ]) that rely on hand- crafted probabilistic models, DeepChem- V ariant leverages convolu- tional neural networks to learn complex inter-read dependencies directly from data (Figure 5). DeepChem- V ariant converts aligned sequencing reads into multi- channel pileup images (6 channels encoding base identity , base quality , mapping quality , strand orientation, variant support, and reference match indicators acr oss 221-bp windows with 100-read depth). These images are processed through a MobileNetV2 or In- ceptionV3 CNN that outputs probabilistic genotype classications: homozygous r eference, heterozygous, or homozygous alternate ( Al- gorithm 2). This image-based representation enables the model to capture spatial patterns in read alignments that traditional methods cannot detect. Figure 5: DeepChem-V ariant workow: Reference genome and aligne d reads are processe d through candidate detection, pileup image generation (6-channel tensors), CNN classica- tion (MobileNetV2 or InceptionV3), and VCF conversion to produce annotated variant calls. AgriV ariant is built within the DeepChem framework and is fully open source, with the entire codebase written in Python. This design enables spe cies specic optimization that is not possible with closed source or human centric variant callers. W e trained the model on Indica rice genomic data, and the same architecture allows researchers to retrain it on their own crop varieties, adapt it to new sequencing platforms, and integrate variant calling dir ectly with downstream molecular machine learning workows. These capabilities are especially important for resource limited breeding programs working with understudied crops. Conference’17, July 2017, W ashington, DC, USA Bisoi et al. Algorithm 2 DeepChem- V ariant Require: Aligned reads (BAM 𝐵 ), Reference genome (F AST A 𝐹 ) Ensure: V ariants with genotyp e probabilities (V CF) 1: C ← CandidateFeaturizer ( 𝐵, 𝐹 ) ⊲ Realign, detect candidates 2: I ← PileupFeaturizer ( C ) ⊲ Generate 6 × 100 × 221 tensors 3: P ← CNN MobileNetV2 ( I ) ⊲ [ 𝑃 ( 0/0 ) , 𝑃 ( 0/1 ) , 𝑃 ( 1/1 ) ] 4: Filter variants where arg max ( P ) ≠ hom-ref 5: Compute quality: 𝑄 = − 10 log 10 ( 1 − max ( P ) ) 6: return VCF with genotypes and quality scores V ariant calling generated V ariant Call Format (V CF) les contain- ing chromosome positions, reference and alternate alleles, quality scores, and lter status for each detected variant. W e validated detection accuracy by conrming that simulated variants appeared in output V CFs with correct genomic coordinates and allelic states. 2.4 Functional Annotation Absence of crop-specic databases in standard annotation tools (SnpE ) ne cessitated development of custom annotation infras- tructure. W e obtained gene structure information from RAP-DB in General Feature Format (GFF) les containing exon coordinates, coding sequence (CDS) boundaries, and transcript identiers for all annotated rice genes. Our annotation pipeline (Figure 6) parsed GFF les to extract CDS coordinates for target genes and mapped variant positions from V CF les to corresponding exonic regions. For each detected variant, the algorithm identied ov erlapping CDS features based on chromosome and genomic coordinates, calculated relative posi- tion within the CDS while accounting for strand orientation, and determined the aecte d codon. The reference codon sequence was retrieved from the genome F AST A le, the variant nucleotide sub- stitution was applied to generate the alternate codon, and both codons were translated using the standard genetic code. V ariants were classied as missense, nonsense , or silent based on resulting amino acid changes. Figure 6: Functional annotation workow mapping variants from genomic position through codon extraction and trans- lation to amino acid change The codon extraction process accounted for multi-exon genes by retrieving nucleotides from adjacent exons to assemble com- plete codons for variants near exon b oundaries. Strand orientation determined whether codons were r ead 5’ to 3’ or re verse comple- mented before translation. Functional eect classication followed standard nomenclature: stop-gained variants introduced prema- ture stop codons, missense variants changed amino acid identity , and silent (synonymous) variants maintained the same amino acid despite nucleotide changes. Impact severity was assigned using established guidelines: HIGH for stop-gained and frameshift vari- ants, MODERA TE for missense variants, and LOW for synonymous variants. 2.5 Database-Independent Deleteriousness Scoring Most widely used deleteriousness prediction tools (SIFT , PolyP hen- 2, CADD) depend heavily on human-specic reference databases and evolutionary conservation patterns derived from animal genomes, limiting their applicability to plant genomes. T o enable quantitative assessment of variant severity in rice genes, we implemented a database-independent scoring strategy based on amino acid physic- ochemical properties and evolutionary substitution likelihoods, making this an approach that generalizes to any organism without requiring pre-trained species-specic mo dels. W e combined two complementary scoring metrics (Figure 7). The Grantham distance matrix quanties amino acid dissimilarity based on composition (atomic weight ratios of non-carb on elements), polarity (hydrophobicity measurements), and molecular volume. Grantham scores range from 5 ( conservative substitutions) to 215 (radical substitutions). Reference and alternate amino acids from annotation output were use d to quer y the Grantham matrix and retrieve distance scor es. BLOSUM62 (BLOcks SUbstitution Matrix 62) derives from ob- served substitution frequencies in protein alignments sharing at least 62% sequence identity . Positive scores indicate substitutions occurring more frequently than expected by chance ( conservative), whereas negative scores correspond to substitutions less favored in nature (radical). Stop-gained mutations ar e scored 1 by A griV ari- ant as they introduce premature stop codons with HIGH impace severity . This metric estimates evolutionary tolerance of spe cic amino acid changes. Figure 7: Composite Scoring combining Grantham Scor e and BLOSUM62 Score T o enable integration of both metrics, Grantham scores wer e normalized to 0–1 range by dividing by maximum observed value (215), while BLOSUM62 scores were normalized by shifting by minimum matrix value and dividing by overall score range . The AgriV ariant: V ariant Eect Prediction using DeepChem- V ariant for Precision Breeding in Rice Conference’17, July 2017, W ashington, DC, USA resulting composite score provides a quantitative measure of variant severity independent of categorical impact classications: Composite Score = 1 2 Grantham 215 + BLOSUM62 min − BLOSUM62 BLOSUM62 range By using this framework, rather than training organism-sp ecic machine learning models (which require extensive labeled data), we leverage universal biochemical principles encoded in substitution matrices. This approach provides immediate generalizability to understudied crops without requiring training data. 3 Case Studies T o validate AgriV ariant’s performance, w e obtained BAM les for three rice mutant lines the MiRiQ Database [ 14 ] and analyzed them. These variants span the functional impact sp ectrum—MODERA TE impact missense substitutions (OsDREB2a, SK C1) and a LOW im- pact synonymous control (OsDREB1F), thus demonstrating the pipeline’s ability to correctly classify variant eects across diverse stress-response genes and severity le vels. 3.1 OsDREB2a Missense Mutation The variant Os01t0165000-01:c.794G > A (p.G265E) introduced a guanine-to-adenine substitution at nucleotide position 794, con- verting a glycine codon to glutamate at amino acid position 265 (T able 1). AgriV ariant successfully detected this substitution, r e- porting it in the VCF at position chr01:3358191 with reference allele G and alternate allele A. The substitution replaces glycine, a small non-polar amino acid that confers conformational exibility , with glutamate, a larger negatively charged residue. This physicochemical change scored moderately severe on b oth substitution matrices: Grantham dis- tance of 98 ( on a 0-215 scale where higher values indicate mor e radical changes) and BLOSUM62 score of -2 (negativ e values indi- cate substitutions disfavored by ev olution). The combined delete- riousness score of 0.137 reected the intermediate severity of this substitution. AgriV ariant correctly classied this variant as missense with MODERA TE impact severity . The glycine-to-glutamate substitution at position 265 introduces b oth steric bulk and electrostatic charge in a region that may aect protein stability or regulatory interac- tions. The missense variant likely r educes but does not eliminate OsDREB2a activity , potentially resulting in intermediate drought tolerance phenotypes. Plants carrying this variant would likely exhibit reduce d drought tolerance compared to wild-type but retain partial stress response capacity , making it a candidate for functional validation studies. 3.2 OsDREB1F Synonymous Mutation The variant Os01g0968800-01:c.165G > A (p.G55G) replaces guanine with adenine at nucleotide position 165, maintaining glycine at amino acid position 55 (Table 2). A griV ariant successfully detected this substitution, reporting it in the VCF at position chr01:42727659 with reference allele G and alternate allele A. Annotation analysis identied the variant within the OsDREB1F coding se quence. The substitution altered the third position of the glycine codon, changing GGG to GGA. Both of these encode T able 1: AgriV ariant validation of a missense OsDREB2a vari- ant. Simulated gene context (top) and automated functional annotation with MODERA TE-impact classication (b ottom). Green highlights denote correct predictions. Background Context (Known from Simulation) Gene ID Os01g0165000 Gene Name DREB2A Chromosome:Position chr01:3358191 Ref → Alt Nucleotide G → A AgriV ariant Predictions (V alidated Outputs) Ref → Alt Codon G G G → G A G Amino Acid Change G → E (Glycine → Glutamate) Grantham Score 98 BLOSUM62 Score -2 Combined Score 0.137 Functional Eect Missense Impact Severity MODERA TE T able 2: AgriV ariant validation of a synonymous OsDREB1F variant. Simulated gene context (top) and automated func- tional annotation with LO W -impact classication (bottom). Green highlights denote correct predictions. Background Context (Known from Simulation) Gene ID Os01g0968800 Gene Name DREB1F Chromosome:Position chr01:42727659 Ref → Alt Nucleotide G → A AgriV ariant Predictions (V alidated Outputs) Ref → Alt Codon GG G → GG A Amino Acid Change G → G (Glycine → Glycine) Grantham Score 0 BLOSUM62 Score 7 Combined Score -0.50 Functional Eect Synonymous Impact Severity LO W the same amino acid due to the genetic code’s degeneracy . This represents a synonymous or silent mutation where the nucleotide sequence changes but the protein sequence remains identical. AgriV ariant correctly classied this variant as synonymous with LO W impact severity . The Grantham score of 0 reects no amino acid change (glycine to glycine), while the BLOSUM62 scor e of 7 (the maximum value, indicating identical amino acids) conrms the neutral nature of this substitution. This contr ol variant validates the pipeline ’s ability to distinguish functionally neutral changes from deleterious mutations. Plants carrying this variant would exhibit normal OsDREB1F function, as the protein sequence remains unchanged despite the genomic alteration. Conference’17, July 2017, W ashington, DC, USA Bisoi et al. T able 3: AgriV ariant validation of a missense SKC1 variant. Simulated gene context (top) and automate d functional an- notation with MODERA TE-impact classication (bottom). Green highlights denote correct predictions. Background Context (Known from Simulation) Gene ID Os01g0307500 Gene Name SKC1 Chromosome:Position chr01:11462201 Ref → Alt Nucleotide T → A AgriV ariant Predictions (V alidated Outputs) Ref → Alt Codon T GT → A GT Amino Acid Change C → S (Cysteine → Serine) Grantham Score 112 BLOSUM62 Score -1 Combined Score 0.124 Functional Eect Missense Impact Severity MODERA TE 3.3 SK C1 Missense Mutation The SKC1 variant Os01t0307500-01:c.1075T > A (p.C359S) r eplaced thymine with adenine at nucleotide position 1075, converting cys- teine at position 359 to serine (T able 3). AgriV ariant successfully de- tected this substitution, reporting it in the VCF at position chr01:11462201 with reference allele T and alternate allele A. Annotation analysis identied the variant within the SKC1 cod- ing sequence at amino acid p osition 359. The substitution replaces cysteine, a sulfur-containing amino acid capable of forming disul- de bonds, with serine, a polar hydroxyl-containing residue . While both are small polar amino acids, the loss of cysteine’s sulfur group eliminates potential disulde bond formation, which could aect protein structure if position 359 participates in structural stabi- lization. The Grantham score of 112 indicates moderate physico- chemical divergence, while the BLOSUM62 score of -1 suggests this substitution is slightly disfavored in evolution. The combined deleteriousness score of 0.124 reected intermediate severity . AgriV ariant correctly classied this variant as missense with MODERA TE impact severity . The cysteine-to-serine substitution at position 359 represents a moderately conser vative change (both residues are small and polar ) but the functional consequences de- pend on whether this cysteine participates in disulde bonding or active site chemistry . In the context of the HKT1;5 sodium trans- porter , this position may inuence ion selectivity or transport ki- netics, potentially aecting salt tolerance phenotypes. 4 Exhaustive Mutation Analysis of OsMT -3a 4.1 Rationale and Gene Selection OsMT -3a encodes a type 3 metallothionein involv ed in heavy metal detoxication and oxidative stress r esponse in rice [ 18 ]. Metalloth- ioneins are cysteine-rich proteins that bind and se quester toxic metal ions (cadmium, copper , zinc), protecting cells from oxidativ e damage. The gene ’s 183-bp coding sequence produces a compact T able 4: Deleteriousness distribution across mutation types for OsMT -3a variants. V ariant T ype High Impact Medium Impact Low Impact T otal Substitutions (SNV s) 112 171 284 567 Insertions 157 238 361 756 Deletions 84 38 64 186 T otal 353 447 709 1,509 61-amino acid protein, making it an ideal candidate for exhaustive computational mutagenesis. Traditional breeding approaches to assess all possible single- nucleotide variants in OsMT -3a would require generating 1,509 individual mutant lines (accounting for all substitutions, insertions, and deletions), followed by 8-10 growing seasons for phenotypic evaluation. The selection and screening would span 2-4 years and require e xtensive greenhouse facilities. Our computational pipeline completed this analysis in appr oximately 10 days on standard hard- ware, demonstrating its utility for rapid variant prioritization in breeding programs. W e systematically generated all possible single-nucleotide sub- stitutions, insertions, and deletions across the 183-bp OsMT -3a coding sequence. For each mutation, a synthetic variant was in- troduced into the reference genome using bcftools consensus , followed by generation of synthetic sequencing reads using wgsim with 1000 × coverage. V ariants were detected using DeepChem- V ariant (Algorithm 2), annotated for functional eects using the custom plant genomics pipeline, and scored for deleteriousness via the Grantham-BLOSUM62 composite metric. V ariants were classied into three deleteriousness categories based on composite scores: High (scor e > 0.3), Medium (0 < score <= 0.3), and Low (scor e <= 0). Functional eects were categorized as missense ( amino acid substitution), synonymous (silent muta- tion), nonsense (pr emature stop codon), or stop loss (stop codon mutation). 4.2 Results The exhaustive analysis revealed 1,509 total possible variants across the OsMT -3a coding sequence, with deleteriousness distribution showing 353 high-impact, 447 medium-impact, and 709 low-impact variants (T able 4). Substitutions exhibited the highest proportion of low-impact variants (284 of 567 or 50.1%), while deletions show ed elevated high-impact frequency (84 of 186 or 45.2%). Insertions demonstrated intermediate distribution across impact categories. This exhaustive analysis demonstrates the pipeline’s capacity to comprehensively map mutational landscapes of target genes, enabling design of crop varieties with precisely engineered traits. The 10-day computational analysis r eplaced what would have r e- quired 2-4 y ears of sequential CRISPR generation and molecular characterization of 1,509 mutant lines. For a breeding program de- veloping cadmium-tolerant rice for contaminated soils, these results enable negative selection by identifying 353 high-impact variants to avoid during screening, while simultaneously prioritizing 709 low-impact variants as candidate tolerance alleles that preser ve protein function under metal stress. AgriV ariant: V ariant Eect Prediction using DeepChem- V ariant for Precision Breeding in Rice Conference’17, July 2017, W ashington, DC, USA T able 5: Model performance across training and test datasets. The training subspecies was Indica, while the dierent sub- species tested was T emperate Japonica. Precision, Recall, and F1-Scores are weighted averages. Dataset Accuracy Precision Recall F1-Score Training/V alidation 0.9537 0.9529 0.9537 0.9532 T est Set (Same Subspecies) 0.9530 0.9539 0.9530 0.9532 T est Set (Dierent Subspecies) 0.9594 0.9606 0.9594 0.9598 Beyond immediate breeding applications, this approach enables forward engineering of crop genomes with designer properties. While our study employed computational simulation for proof-of- concept validation, these analytical steps apply equally to variants generated through established mutagenesis methods including X - ray or gamma irradiation, ethyl methanesulfonate (EMS) treatment, or CRISPR-induced mutations, thus enabling bree ders to prioritize among thousands of laboratory-generate d variants before pheno- typic screening. Rather than relying on random mutagenesis or conventional crossing to identify favorable alleles, breeders can computationally predict which specic nucle otide changes will produce desired phenotypes, then introduce only those variants via CRISPR gene editing. For complex traits requiring modica- tion of multiple genes simultaneously , exhaustive analysis of each target gene generates a searchable database of predicted variant eects, allowing optimization algorithms to identify optimal allele combinations before any seeds are planted. While wet-lab valida- tion remains essential for high-priority candidates, this in silico pre-screening reduces experimental scope, focusing resour ces on variants most likely to yield relevant phenotypes. 5 Discussion W e developed a complete variant eect prediction pipeline for crop genomics by e xtending De epChem- V ariant with plant-specic func- tional annotation and database-independent deleteriousness scor- ing. The pipeline successfully predicted functional consequences of variants in rice stress-response genes, demonstrating its utility for computational variant prioritization in breeding pr ograms. Our exhaustive mutagenesis study of OsMT -3a analyzed 1,509 variants in 10 days, replacing what would require 2-4 years of wet-lab char- acterization and enabling breeders to focus experimental resources on high-priority candidates. 5.1 Model Performance and Generalization DeepChem- V ariant achieved 95.37% accuracy on training and vali- dation sets, with consistent performance on same-subspecies test data (95.30%) and notably str ong cross-subspecies generalization to T emperate Japonica (95.94%, Table 5). The model was trained on ten Indica rice genes using Google Colab’s L4 GPU infrastructure [ 4 ]. This capability proves particularly valuable for crop genomics, where generating large training datasets for each cultivar remains impractical. The model was trained exclusively on Indica rice but maintained high precision (0.9606) and recall (0.9594) on a phy- logenetically distinct subspecies ( Japonica), demonstrating that CNN-based variant calling learns generalizable features of sequenc- ing read patterns rather than subspecies-specic artifacts. All genes used during training and testing were do wnloaded from the 3000 Rice Genomes Project [27] [16]. The comparable F1-scores across all datasets (0.9532-0.9598) indi- cate robust performance without overtting to training data. How- ever , these metrics reect variant detection accuracy rather than functional annotation correctness. 5.2 Accuracy Estimation Acr oss Diverse Gene Functions T o assess AgriV ariant’s performance beyond stress-r esponse genes, we conducted a small-scale accuracy study across 40 variants se- lected from published functional genomics studies, using real se- quencing data (BAM les) from the MiRIiQ Database [ 14 ]. The variants spanned diverse functional categories: salt tolerance , cy- tokinin signaling, blast fungus sensitivity , bacterial blight sensitiv- ity , fragrance biosynthesis, panicle architecture , grain morphology , and heavy metal uptake. The variant set included single-nucleotide substitutions, insertions, and deletions to evaluate detection across mutation types. AgriV ariant successfully detecte d 36 of 40 variants (90.0% detec- tion rate). All 36 detected variants were correctly annotated for func- tional eect (missense, nonsense, synonymous) and assigned ap- propriate impact severity classications (HIGH/MODERA TE/LO W) based on amino acid changes. Deleteriousness scores correlated with expected functional impacts, with stop-gained variants scoring highest and synonymous variants scoring lowest. This estimate pr ovides preliminary evidence of AgriV ariant’s generalizability beyond the primary case studies but should b e interpreted cautiously given the small sample size. The 90.0% detec- tion rate demonstrates robust performance on r eal sequencing data containing authentic coverage patterns and sequencing artifacts. Current scoring methods also cannot distinguish functionally criti- cal positions—a variant’s impact depends not only on the amino acid change but also on whether it occurs in catalytic sites, binding domains, or structurally important regions. Comprehensive bench- marking against large-scale functional genomics datasets will be necessary to establish r obust performance estimates for production breeding applications. 5.3 Database-Independent Scoring and Limitations The Grantham-BLOSUM62 composite scoring framework enabled quantitative variant assessment without requiring organism-specic training databases, addressing a critical gap for understudied crops. This approach correctly identied high-impact variants and neu- tral variants, validating the scoring calibration. However , these matrices capture only amino acid-level eects and cannot account for structural context, protein-pr otein interactions, or regulatory mechanisms. The exhaustive OsMT -3a analysis rev ealed an unbalance d delete- riousness distribution (23.4% high, 29.6% medium, 47.0% low), with nearly half of all variants classied as lo w-impact. This distribution suggests that simple physicochemical metrics may lack discrimi- natory power for functionally critical residues. For instance, a con- servative substitution in a catalytic site would score as low-impact Conference’17, July 2017, W ashington, DC, USA Bisoi et al. despite potentially ab olishing protein function, while a radical sub- stitution in a non-conserved loop might scor e high-impact with minimal phenotypic consequence. This limitation underscores the need for structure-aware and context-dependent scoring methods. 5.4 Implications for Precision Breeding AgriV ariant addresses a fundamental bottlene ck in molecular breed- ing: identifying which naturally occurring or CRISPR-induced vari- ants merit experimental validation. Traditional appr oaches screen variants sequentially through multi-season eld trials, limiting breeding programs to evaluating dozens of candidates annually . Our computational framework enables parallel in silico screening of thousands of variants, with the OsMT -3a study demonstrating 150-fold acceleration (10 days versus 2-4 years[ 5 ] for equivalent coverage). This acceleration proves particularly valuable for multi-gene trait engineering, where bree ders must optimize allele combinations across multiple loci. For dr ought tolerance involving OsDREB2a, OsDREB1F, and additional regulatory genes, exhaustive analysis of each gene generates a sear chable database of predicted eects, enabling combinatorial optimization algorithms to identify syner- gistic allele combinations before any experimental validation. The reduction in experimental scope translates directly to cost savings and faster cultivar development timelines, critical for responding to rapidly changing climate conditions. Beyond variant prioritization, AgriV ariant can enable reverse ge- netics approaches where breeders specify desired phenotypes and the system identies which spe cic nucleotide changes would pro- duce those traits. This transitions br eeding from selection (choosing among existing variation) to design (engineering targeted impr ove- ments), though wet-lab validation remains essential. 5.5 Open Source Implementation The complete pipeline is freely available as open-source software under the MI T License at https://github.com/KitVB/AgriV ariant and https://github.com/deepchem/deepchem. All components for vari- ant calling, functional annotation, deleteriousness scoring, and syn- thetic mutagenesis are implemented within the DeepChem frame- work in Python. This enables resear chers to adapt the pipeline to their crop spe cies, retrain models on proprietar y data, and integrate with existing breeding workows. Documentation and installation instructions are provided in the repository . 5.6 Future W ork While Grantham-BLOSUM62 scoring successfully distinguished extreme cases in our validation studies, integrating protein struc- ture prediction would enable me chanistic explanations (quanti- fying binding anity changes or protein destabilization) rather than categorical classications. Machine learning models trained on experimentally validated variants could predict quantitativ e phe- notypic impacts (percent yield r eduction, stress tolerance scores) that directly inform breeding decisions. Systematic ecacy studies comparing predictions against eld trial data would establish condence thresholds and r eveal whether scoring adjustments are needed for specic protein families or ge- nomic contexts where physicochemical metrics prov e insucient. The pipeline currently handles single-nucleotide variants, as demonstrated in our case studies. Extending annotation to large indels, structural variants, copy number variations and variations in non coding regions would address frameshift mutations and gene fusions that represent important phenotypic diversity sources. A s long-read sequencing impro ves detection of such variants, interpre- tation methods must evolve to maintain comprehensiv e coverage. 6 Conclusion Engineering climate-resilient crops is paramount for global food se- curity in the 21st century . W e present AgriV ariant, preliminary sys- tem integrating deep learning base d variant calling via DeepChem- V ariant with plant-specic annotation and database-independent scoring, enabling rapid in silico estimation of variant eects in rice genes. AgriV ariant achieved 95.3% variant detection accuracy and correctly classied functional impacts across diverse stress- response mechanisms, with the exhaustive OsMT -3a analysis being completed in 10 days instead of 2-4 years by traditional wet lab approaches. Implemented entirely in Python as modular open-source com- ponents within the DeepChem framework, AgriV ariant enables researchers to adapt variant calling, annotation, and scoring inde- pendently . This establishes a foundation for computational crop design where breeders specify desired traits and algorithms identify which genetic changes will produce them. Rather than screening random mutations across growing seasons, quick computational estimates enable targeted introduction of benecial variants. Fu- ture ecacy studies validating in silico predictions against eld phenotypes will determine how reliably these estimates translate to agronomic performance, advancing toward pr ecision engineering of crops that survive environmental stresses more eectively . 7 Limitations and Ethical Considerations The pipeline requires high-quality reference genomes and annota- tions, currently handles only single-nucleotide variants and small indels, and evaluates amino acid changes without structural context. Model training on rice requires validation for phylogenetically dis- tant crops. All predictions must b e experimentally validated b efore breeding deployment. This work used publicly available data; no proprietary germplasm was employed. The open-source implementation enables e quitable access. Breeding pr ograms using proprietary data should ensure compliance with data protection policies. Computational predictions should complement rather than r e- place comprehensive br eeding strategies that maintain genetic di- versity , incorporate farmer preferences, and ensur e biosafety evalu- ation before cultivar release. 8 GenAI Disclosure Generative AI and Large Language Models were used to improve the language, grammar and clarity of the manuscript. All scientic contributions and analyses are entirely the work of the authors. References [1] Ivan A. Adzhubei, Steen Schmidt, Leonid Peshkin, V asily E. Ramensky , Alisa Gerasimova, Peer Bork, Alexey S. K ondrashov , and Shamil R. Sunyae v . 2010. A AgriV ariant: V ariant Eect Prediction using DeepChem- V ariant for Precision Breeding in Rice Conference’17, July 2017, W ashington, DC, USA method and server for predicting damaging missense mutations. Nature Methods 7, 4 (2010), 248–249. doi:10.1038/nmeth0410- 248 [2] Ankita V aishnobi Bisoi, Shreyas V , Jose Siguenza, and Bharath Ramsundar . 2024. A Modular Open Source Framework for Genomic V ariant Calling. arXiv preprint arXiv:1909.13334 (2024). [3] P. Cingolani, A. Platts, M. Coon, T . Nguyen, L. W ang, S.J. Land, X. Lu, and D .M. Ruden. 2012. A program for annotating and predicting the eects of single nu- cleotide polymorphisms, SnpE: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 2 (2012), 80–92. [4] Google Colab. [n. d.]. Google Colaboratory. https://colab.research.google.com/. [5] Bertrand C. Y . Collard, Joseph C. Beredo, Bert Lenaerts, Rhulyx Mendoza, Ronald Santelices, Vitaliano Lopena, Holden V erdeprado, Chitra Raghavan, Glenn B. Gregorio, Leigh Vial, Matty Demont, Partha S. Biswas, Khandakar M. Iftekharud- daula, Mohammad Akhlasur Rahman, Joshua N. Cobb, and Mohammad Raqul Islam. 2017. Revisiting rice br eeding methods - evaluating the use of rapid gen- eration advance (RGA) for routine rice breeding. Plant Production Science 20, 4 (2017), 337–352. doi:10.1080/1343943X.2017.1391705 [6] Min Cui, W ei Zhang, Qiang Zhang, Zhen Xu, Zhiguo Zhu, Feng Duan, and Rongling Wu. 2011. Induced ov er-expression of the transcription factor Os- DREB2A improves drought tolerance in rice . Plant Physiology and Biochemistry 49, 12 (2011), 1384–1391. doi:10.1016/j.plaphy .2011.09.012 [7] Nathan C. Frey , Vijay Gadepally , and Bharath Ramsundar . 2022. FastF lows: Flow- Based Models for Mole cular Graph Generation. arXiv:2201.12419 [physics.chem- ph] [8] Joseph Gomes, Bharath Ramsundar , Evan N. Feinberg, and Vijay S. Pande. 2017. Atomic Convolutional Networks for Predicting Protein-Ligand Binding Anity . arXiv:1703.10603 [cs.LG] [9] Richard Grantham. 1974. Amino acid dierence formula to help explain pr otein evolution. Science 185, 4154 (1974), 862–864. doi:10.1126/science.185.4154.862 [10] Steven Heniko and Jorja G. Heniko. 1992. Amino acid substitution matrices from pr otein blocks. Procee dings of the National Academy of Sciences of the United States of America 89, 22 (1992), 10915–10919. doi:10.1073/pnas.89.22.10915 [11] Y oshihiro Kawahara, Marion de la Bastide , John P. Hamilton, Hiroyuki Kanamori, W . Richard McCombie, Shu Ouyang, David C. Schwartz, T akuji T anaka, Jianzhong Wu, Shengqiang Zhou, Kevin L. Childs, Robert M. Davidson, Hsin- Yi Lin, Lina M. Quesada-Ocampo, Bruno V aillancourt, Hiroaki Sakai, Sung Soo Lee, Jae-Y oung Kim, Hisataka Numa, T akeshi Itoh, and T akeshi Matsumoto. 2013. Impro vement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 1 (2013), 4. doi:10.1186/1939- 8433- 6- 4 [12] Martin Kircher , Daniela M. Witten, Preti Jain, Brian J. O’Roak, Gregor y M. Cooper , and Jay Shendure. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics 46 (2014), 310–315. doi:10.1038/ng.2892 [13] Alex Krizhevsky , Ilya Sutskever , and Georey E. Hinton. 2012. ImageNet Classication with Deep Convolutional Neural Networks. In A dvances in Neu- ral Information Processing Systems , V ol. 25. Curran Associates, Inc., 1097– 1105. https://papers.nips.cc/pap er/4824- imagenet- classication- with- de ep- convolutional- neural- networks.pdf [14] T akahiko Kubo, Y oshiyuki Y amagata, Hir oaki Matsusaka, Atsushi T oy oda, Y utaka Sato, and T oshihiro Kumamaru. 2024. MiRiQ Database: A Platform for In Silico Rice Mutant Screening. P lant and Cell P hysiology 65, 1 (2024), 169–174. doi:10. 1093/pcp/pcad134 [15] Heng Li, Bob Handsaker , Alec W ysoker , Tim Fennell, Jue Ruan, Nils Homer , Gabor Marth, Goncalo Abecasis, Richard Durbin, and 1000 Genome Project Data Pr ocessing Subgroup. 2009. The sequence alignment/map format and SAMtools. bioinformatics 25, 16 (2009), 2078–2079. [16] Jinyin Li, Jun W ang, and Robert S. Zeigler . 2014. The 3,000 rice genomes project: new opportunities and challenges for future rice research. GigaScience 3 (2014), 8. doi:10.1186/2047- 217X- 3- 8 [17] Aaron McKenna, Matthew Hanna, Eric Banks, Andrey Sivachenko , Kristian Cibulskis, Andrew Kernytsky , Kiran Garimella, David Altshuler , Stacey Gabriel, Mark Daly , et al . 2010. The Genome Analysis T oolkit: a MapRe duce framework for analyzing next-generation DNA sequencing data. Genome research 20, 9 (2010), 1297–1303. [18] Ahmad Mohammad M. Mekawy, Dekoum V . M. A ssaha, Riko Munehiro, Eri Kohnishi, T oshinori Nagaoka, Akihiro Ueda, and Hirofumi Saneoka. 2018. Char- acterization of type 3 metallothionein-like gene (OsMT -3a) from rice revealed its ability to confer tolerance to salinity and heav y metal stresses. Environmental and Experimental Botany 147 (2018), 157–166. doi:10.1016/j.enve xpbot.2017.12.002 [19] De Pristo Poplin. 2017. Deep V ariant: Highly Accurate Genomes With Deep Neural Networks. https://research.google/blog/deepvariant- highly- accurate- genomes- with- deep- neural- networks/ Go ogle Research Blog. [20] Ryan Poplin, Pi-Chuan Chang, David Alexander , Scott Schwartz, Thomas Colthurst, Alexander Ku, Dan Newburger , Jojo Dijamco, Nam Nguyen, Pegah T Afshar , et al . 2018. A universal SNP and small-indel variant caller using deep neural networks. Nature biotechnology 36, 10 (2018), 983–987. [21] Bharath Ramsundar , Peter Eastman, Patrick W alters, Vijay Pande, Karl Leswing, and Zhenqin Wu. 2019. Deep Learning for the Life Sciences . O’Reilly Media. https: //www.amazon.com/Deep- Learning- Life- Sciences- Microscopy/dp/1492039837. [22] Zonghua Ren, Jianping Gao, Liguo Li, Xiaoli Cai, W ei Huang, Deyong Chao, Meizhong Zhu, Zhiyong W ang, Sheng Luan, and Hongxuan Lin. 2005. A rice quantitative trait locus for salt tolerance encodes a sodium transporter . Nature Genetics 37, 10 (2005), 1141–1146. doi:10.1038/ng1643 [23] Hiroaki Sakai, Sung Soo Lee, T akuji Tanaka, Hisataka Numa, Jae- Y oung Kim, Y oshihiro Kawahara, Hironori W akimoto, Chi-Cheng Y ang, Masahiro Iwamoto, T akehiko Abe, Y uko Y amada, Akiko Muto, Hiroshi Inokuchi, T oshimichi Ikemura, T akeshi Matsumoto, Takashi Sasaki, and T akeshi Itoh. 2013. Rice Annotation Project Database (RAP-DB): an integrative and interactive database for rice genomics. Plant & Cell P hysiology 54, 2 (2013), e6. doi:10.1093/pcp/pcs183 [24] Mark Sandler , Andrew Howar d, Menglong Zhu, Andrey Zhmoginov , and Liang- Chieh Chen. 2018. MobileNetV2: Inverted Residuals and Linear Bottlenecks. The IEEE Conference on Computer Vision and Pattern Re cognition (CVPR) (2018), 4510–4520. [25] Ngak-Leng Sim, Prateek Kumar, Jing Hu, Steven Heniko, Georg Schneider , and Pauline C. Ng. 2012. SIFT web server: predicting eects of amino acid substitutions on proteins. Nucleic Acids Research 40, W1 (2012), W452–W457. doi:10.1093/nar/gks539 [26] Christian Szegedy , Vincent V anhoucke, Serge y Ioe, Jonathon Shlens, and Zbig- niew W ojna. 2015. Rethinking the Inception Architecture for Computer Vision. arXiv:1512.00567 [cs.CV] https://arxiv .org/abs/1512.00567 [27] The 3,000 Rice Genomes Project. 2014. The 3,000 rice genomes project. Giga- Science 3, 1 (2014), 7. doi:10.1186/2047- 217X- 3- 7 [28] Qing W ang, Y ujie Guan, Y anhua Wu, Hong Chen, Feng Chen, and Chengcai Chu. 2008. Overe xpression of a rice OsDREB1F gene increases salt, drought, and low temperature tolerance in both Arabidopsis and rice. Plant Molecular Biology 67, 6 (2008), 589–602. doi:10.1007/s11103- 008- 9340- 6 [29] Zhenqin Wu, Bharath Ramsundar , Evan N. Feinberg, Joseph Gomes, Caleb Ge- niesse, Aneesh S. Pappu, Karl Leswing, and Vijay Pande. 2018. MoleculeNet: A Benchmark for Molecular Machine Learning. arXiv:1703.00564 [ cs.LG]
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment