Enhanced Diffusion Sampling: Efficient Rare Event Sampling and Free Energy Calculation with Diffusion Models
The rare-event sampling problem has long been the central limiting factor in molecular dynamics (MD), especially in biomolecular simulation. Recently, diffusion models such as BioEmu have emerged as powerful equilibrium samplers that generate indepen…
Authors: Yu Xie, Ludwig Winkler, Lixin Sun
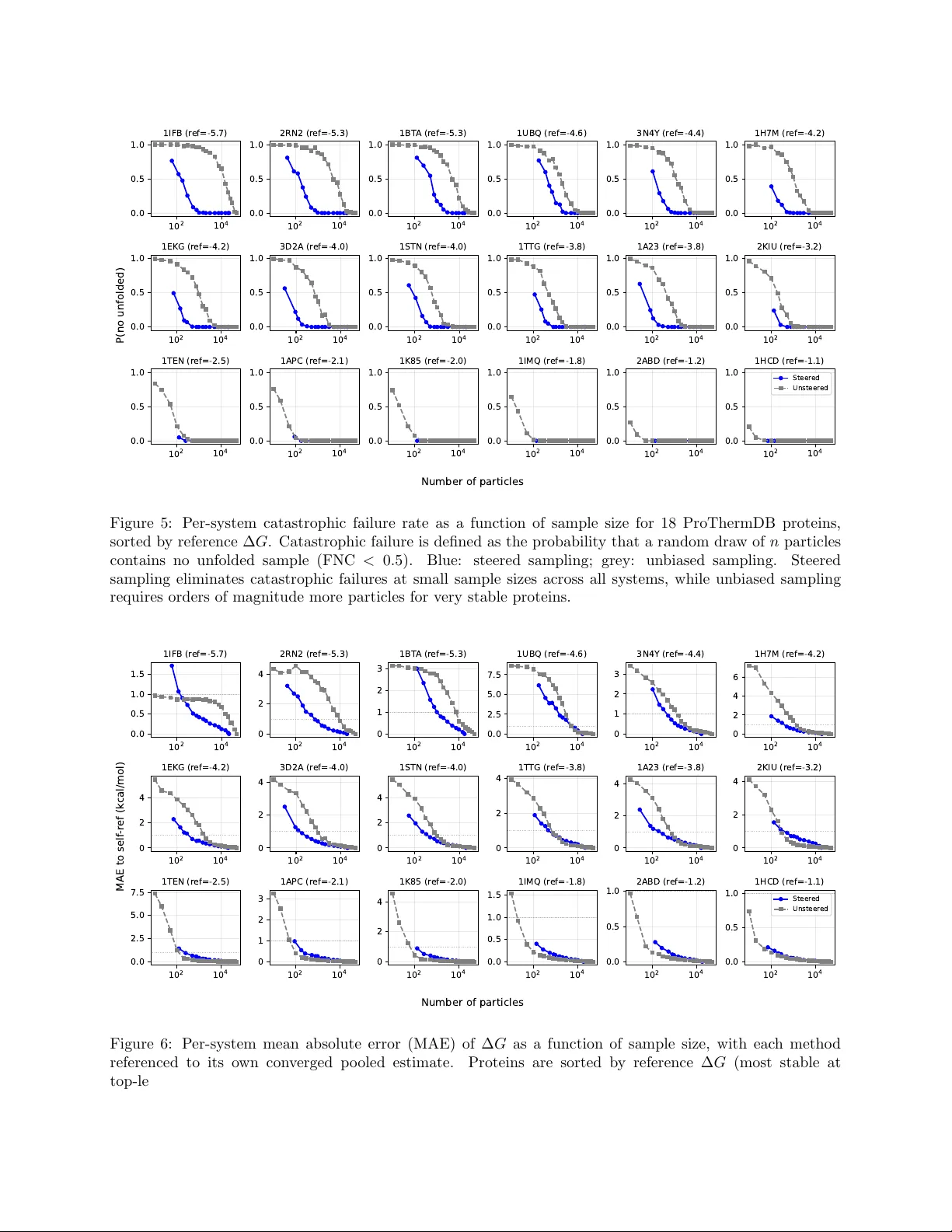
Enhanced Diffusion Sampling: Efficien t Rare Ev en t Sampling and F ree Energy Calculation with Diffusion Mo dels Y u Xie † , Ludwig Winkler † , Lixin Sun † , Sarah Lewis, Adam E. F oster, Jos ´ e Jim ´ enez Luna, Tim Hemp el, Mic hael Gastegger, Y ao yi Chen, Iryna Zap orozhets, Cecilia Clemen ti, Christopher M. Bishop, F rank No ´ e Micr osoft R ese ar ch AI for Scienc e † Equal con tribution. Abstract The rare-even t sampling problem has long been the central limiting factor in molecular dynamics (MD), esp ecially in biomolecular simulation. Recen tly , diffusion mo dels such as BioEmu hav e emerged as p o w erful equilibrium samplers that generate indep enden t samples from complex molecular distributions, eliminating the cost of sampling rare transition ev ents. Ho wev er, a sampling problem remains when computing observ ables that rely on states whic h are rare in equilibrium, for example folding free ener- gies. Here, w e introduce enhanced diffusion sampling, enabling efficien t exploration of rare-ev ent regions while preserving unbiased thermodynamic estimators. The key idea is to p erform quantitativ ely accurate steering proto cols to generate biased ensem bles and subsequently recov er equilibrium statistics via exact rew eighting. W e instantiate our framew ork in three algorithms: Um brellaDiff (umbrella sampling with diffusion mo dels), MetaDiff (a batch wise analogue for metadynamics), and ∆G-Diff (free-energy differ- ences via tilted ensembles). Across to y systems, protein folding landscapes and folding free energies, our metho ds achiev e fast, accurate, and scalable estimation of equilibrium prop erties within GPU-minutes to hours p er system—closing the rare-even t sampling gap that remained after the adven t of diffusion-mo del equilibrium samplers. 1 In tro duction Molecular dynamics (MD) simulation is a widely used computational approach for generating molecular equilibrium ensem bles p ( x ) and predicting exp erimen tal observ ables O = E p ( x ) [ o ( x )], but its effectiveness is limited by the sampling problem, whic h consists of tw o distinct comp onents (T able 1). 1. Slow mixing pr oblem —MD pro duces time-correlated tra jectories x t . Long-liv ed states or phases lead to trapping the sim ulation tra jectory for long times, resulting in slow exploration and slo w conv ergence of exp ectation v alues. 2. R ar e state pr oblem —ev en with indep enden t dra ws from p ( x ), it can b e prohibitive to sample states with small equilibrium probabilities. F or example, the probability ratio of unfolded and folded protein states dep ends exponentially on the folding free energy: p u /p f = exp(∆ G fold /k B T ). A t 300K, ∆ G fold = − 5 k cal/mol implies that ∼ 1 in 4 . 4 × 10 3 equilibrium samples is unfolded. F or a mo derately stable protein (∆ G fold = − 10 k cal/mol) only ∼ 1 in 1 . 9 × 10 7 samples is unfolded. These limitations hav e motiv ated enhanced sampling metho ds ov er the last 70 y ears [1], which address rare states by sampling from a biased distribution and then rew eighting to recov er equilibrium statistics; ho wev er, when implemen ted on top of MD they can remain limited b y slow mixing of the un biased degrees of freedom (T able 1). Recen tly , generative equilibrium samplers based on normalizing flo ws and diffusion mo dels [2, 3] hav e emerged that generate approximately indep enden t equilibrium configurations, removing the slow-mixing b ottlenec k, but rare-state estimation remains when observ ables dep end on lo w-probability 1 Equilibrium sampling Enhanced sampling Molecular Dynamics Slo w mixing problem Rare state problem Slo w mixing problem Rare states sampled Diffusion samplers Indep enden t samples Rare state problem Indep enden t samples Rare states sampled T able 1: The MD sampling problem consists of a slow mixing problem due to rare intercon v ersion b et ween long-liv ed states, and a rare state problem as lo w-probability states are infrequently visited. Diffusion equi- librium samplers tackle the slow mixing problem, enhanced sampling metho ds tac kle the rare state problem. In this pap er we explore the com bination of both: enhanced diffusion samplers. regions of p ( x ) (T able 1). This pap er dev elops a framework for enhanced sampling with diffusion-mo del samplers, addressing b oth b ottlenec ks within a single approac h. T raditional enhanced sampling metho ds include thermo dynamic in tegration [4], free energy p erturbation (FEP) [5], um brella sampling [6, 7], parallel or simulated temp ering [8 – 10] and metadynamics [11]—see [1] for an extensiv e review. All these metho ds sample from a biased distribution, and then later remo ve this bias from the sampled statistics in order to reco ver equilibrium statistics [12–14]. They can accelerate sampling b y orders of magnitude when suitable collectiv e v ariables or thermo dynamic con trols are a v ailable. Represen tative successes include: (i) F r e e ener gy pr ofiles of r e actions and of ion p erme ations thr ough channels [15 – 18], where umbrella sampling can restrain sampling along the w ell-defined reaction co ordinate, while the other degrees of freedom relax quic kly; (ii) smal l-mole cule solvation and pr otein–ligand binding fr e e ener gies [19 – 21], where alc hemical methods relying on free energy p erturbation (FEP) connect nearb y thermo dynamic states with feasible lo cal sampling; (iii) smal l pr otein folding in implicit solvent [22], where replica exc hange remains tractable; and (iv) mutation series in c o arse-gr aine d mo dels [23], where reduced resolution makes otherwise prohibitive transitions amenable to free-energy calculations. F or high-dimensional biomolecular transitions—including protein folding, binding, and conformational c hanges in explicit solv ent—enhanced sampling is often limited by tw o coupled issues. First, suitable low- dimensional bias co ordinates are frequen tly unknown a priori and ma y only b ecome apparent after substantial sampling. This has motiv ated adaptive approac hes that iteratively disco ver reaction coordinates and enhance sampling along them [24 – 32]. Second, these systems typically exhibit a sp e ctrum of slo w relaxation processes rather than a single dominan t timescale, as explored in the MSM literature [33, 34]; when slow mo des are w eakly separated, long sim ulations remain necessary to equilibrate degrees of freedom not directly controlled b y the bias. Consequen tly , successes on explicit-solv ent biomolecular problems ha ve often required sp ecialized com bi- nations of metho ds and/or massiv e compute. F or al l-atom pr otein folding free-energy landscap es, temp era- ture replica exc hange b ecomes increasingly inefficient in explicit solv ent b ecause the n um b er of replicas gro ws with system size, and practical studies hav e relied on hybrids such as REMD+metadynamics [35, 36], bias- exc hange metadynamics [37], or multitemperature MD strategies [38], as w ell as special-purp ose hardware or massiv ely distributed simulations plus MSM analysis to obtain quantitativ e folding landscap es for proteins up to ∼ 100 residues [39 – 42]. F or c omplex c onformational changes , well-c haracterized free-energy landscapes ha ve b een obtained using metadynamics and its v arian ts [43, 44], string-based approac hes com bined with um brella sampling or sw arms of tra jectories [45, 46], and temp erature-accelerated MD in collective v ariables [47]. Large-scale unbiased sim ulation with MSMs has also enabled quan titative conformational landscap es in c hallenging systems [48 – 52], and brute-force sim ulations on sp ecialized hardware hav e resolved long-timescale protein dynamics and GPCR activ ation/binding mec hanisms [53 – 55]. Finally , while alc hemical FEP is w ell established for relative protein–ligand binding or closely related mutan ts, it b ecomes impractical when the alc hemical c hanges induce large-scale conformational rearrangements that are slo w and hard to ov erlap. Recen tly , a complemen tary line of w ork has emerged from generativ e deep learning: Boltzmann gener ators and Boltzmann emulators that aim to generate appro ximately indep enden t configurations from an equilibrium distribution without in tegrating long MD tra jectories. Boltzmann Generators are flo w-based approaches that aim to generate independent samples from a Boltzmann distribution defined via an energy function [2, 56]. Boltzmann em ulators approximate the equilibrium distribution by training on MD simulation data and 2 fine-tuning on exp erimen tal observ ables. This approac h has recently b ecome p opular for sampling protein ensem bles, e.g., BioEm u [3, 57 – 60]. The concept is also emerging in other application areas, suc h as material sciences [59]. By pro ducing effectively iid samples, these mo dels address the slow-mixing b ottlenec k in T able 1. Ho wev er, iid sampling do es not remo ve the r ar e-state b ottlenec k: estimating observ ables con trolled b y lo w- probabilit y regions still requires a num b er of samples that scales exponentially in free-energy differences. F or example, BioEmu-1 estimates protein fold stabilities by directly sampling the equilibrium ensem ble of protein structures and calculating the fraction of unfolded states [3]. F or stabilities of up to ∆ G fold = − 5 kcal/mol this is still feasible within one GPU hour, but this approach quickly b ecomes intractable: sampling ∼ 1 in 1 . 9 × 10 7 unfolded samples for a protein with ∆ G fold = − 10 kcal/mol would tak e on the order of one GPU y ear [3]. Here w e pro vide a solution for the remaining sampling problems of diffusion-based equilibrium samplers b y integrating enhanced sampling mo dels into the diffusion mo del framework. The key comp onen t is a steering algorithm that allo ws to apply the desired bias potentials to a pretrained diffusion model at inference time. This enables the implementation of several classical enhanced sampling methods in the diffusion mo del framew ork, as well as the application of un biasing metho ds suc h as WHAM [13] or MBAR [14]. F urthermore, sev eral tec hnical improv ements are presen ted that pro vide lo w-v ariance estimates of the desired observ ables ev en if only a few thousand diffusion-mo del denoising tra jectories can be afforded. Overall, this framew ork enables conv erged sampling of equilibrium prop erties of complex biomolecular processes with large energy differences within GPU minutes to hours p er calculation, giv en a suitable pretrained diffusion mo del. W e demonstrate our framew ork using the BioEmu model on a biomolecular rare-ev ent problem that is extremely difficult or currently imp ossible with all-atom MD simulations: efficient calculation of folding free energies for a v ariety of proteins ranging betw een 50 and 200 amino acids. Related ideas of combining enhanced sampling metho ds with diffusion mo dels ha ve been presented in sev eral recen t publications: Ref. [61] in tro duces a related approach emplo ying adjoin t sampling to fine-tune a pre-trained diffusion mo del tow ards the sampling of rare states. Ref. [62] introduces a steering approac h for exploring rare ev ents with pretrained diffusion mo dels with an approac h similar to umbrella-sampling that can b e viewed as sp ecial case of the present approach. Ref. [63] hav e presented a steering method whic h c hanges the sampled ensemble to b e consistent with exp erimen tal data or enhance sample diversit y . 2 Biasing and un biasing diffusion mo del ensembles W e assume that we ha ve a pretrained diffusion mo del whose output distribution for a given molecule of in terest is p ( x ) — i.e., the model distribution p ( x ) shall be our unbiase d e quilibrium distribution . W e will equiv alently op erate with probabilities and dimensionless energies, for example: p ( x ) = e − u ( x ) , (1) with dimensionless energy u ( x ). This representation is indep enden t of the thermodynamic ensem ble, e.g., in the canonical ensemble, p ( x ) is the Boltzmann distribution and u ( x ) = U ( x ) /k B T with p oten tial energy U ( x ), Boltzmann constant k B and temp erature T . Subsequently w e will only assume that p ( x ) and u ( x ) exist, but not that they can be explicitly ev aluated. Energy-based diffusion mo dels, for whic h p ( x ) and u ( x ) can b e efficiently ev aluated, hav e b een explored recen tly [64, 65]. Our aim is to compute unbiased exp ectation v alues under the equilibrium distribution of the form O = E p [ o ( x )] . (2) F or example, the probability of b eing in the unfolded state, p u , can b e cast in this form, but accurately estimating it—and th us the folding free energy , relies on sampling rare even ts. As in the traditional enhanced sampling literature, we pro ceed in tw o steps: 1. Generate biased ensembles : given one or m ultiple biasing p otentials b k ( x ), k = 1 , ..., K , draw samples from the biased ensembles with energies u ( x ) + b k ( x ). This is ac hieved b y steering the diffusion mo del at inference time (Section 2.1). 3 2. Recov er unbiased exp ectations : reweigh t the biased samples giv en the kno wn bias potentials in order to recov er the un biased expectation v alues. F or K = 1, rew eighting is trivial, whereas for K > 1 it can b e p erformed with the m ultistate Bennett acceptance ratio (MBAR) metho d (Section 2.2). The choice of biasing p otentials and strategies will b e discussed in Sections 3-5. 2.1 Biased sampling from diffusion mo dels Giv en a pretrained diffusion mo del which samples from p ( x ), w e seek a metho d to draw samples from a single biased ensemble defined via biasing potential b ( x ): q ( x ) := 1 Z p ( x ) e − b ( x ) , Z := Z p ( x ) e − b ( x ) d x, (3) where Z is an unknown normalization constant (partition function) that w e will not need to ev aluate explicitly . Sev eral established families of steering metho ds exist: (i) Sc or e guidanc e , whic h alters the reverse-time score by adding the gradient of a classifier, constraint, or reward to tilt generations to ward desired prop erties [66 – 68]. (ii) Path–inte gr al r eweighting via Sequential Mon te Carlo (SMC) with F eynman–Kac (FK) p oten- tials—k eeping the prop osal dynamics unc hanged and accounting for the bias with incremental imp ortance w eights along the rev erse tra jectory; annealed importance sampling (AIS) app ears as the no-resampling sp e- cial case [69–72]. (iii) Invariant c orr e ctors (e.g., Metrop olis–adjusted Langevin, MALA) that are interlea ved with the reverse dynamics to mix within the curren t biased marginal without changing the FK weigh ts [71, 73]. Score–guidance metho ds are simple and fast but generally do not guarantee un biased sampling of the target density . FK, AIS and SMC giv e explicit imp ortance weigh ts and, with resampling, can target the desired biased marginals exactly . Subsequen tly , w e employ the F eynman-Kac Corrector (FK C) biased sampling metho dology from [74] and [75], describ ed in the follo wing paragraphs. Un biased diffusion mo del. W e assume our diffusion mo del has been trained to reverse the corruption pro cess defined by a sto c hastic differential equation, dx τ = f 1 − τ ( x τ ) dτ + σ 1 − τ dW τ , x τ =0 ∼ p data (4) Here W is a standard Wiener pro cess. The drift f and diffusion σ are c hosen at training time suc h that x ∼ p noise = N (0 , I ) at τ = 1. W e c ho ose to write f and σ with 1 − τ as their argument rather than τ for notational con venience when writing ab out the reverse (denoising) pro cess. W e parameterize rev erse (denoising) time by t := 1 − τ and write p t for the marginal density of x τ at τ = 1 − t . Thus, p 0 = p noise and p 1 = p data . T raining the diffusion mo del consists of learning to compute the score ∇ log p t ( x ) as a function of x and t . Sampling from the diffusion mo del is p erformed by first sampling x from p noise and then sim ulating a reverse pro cess of the form dx t = g t ( x t ) dt + ˜ σ t dW t (5) where g t ( x ) := − f t ( x ) + σ 2 t + ˜ σ 2 t 2 ∇ log p t ( x ) , (6) and ˜ σ t is a hyperparameter w e can c ho ose, with ˜ σ t = σ t b eing the standard choice used in [74]. Biased marginals q t . In order to sample from q ( x ), w e define a family of biased marginal densities q t : q t ( x ) := p t ( x ) e − b t ( x ) Z t , Z t := Z p t ( x ) e − b t ( x ) dx (7) where b 0 ( x ) is constant with resp ect to x , b 1 ( x ) ≡ b ( x ), and b t smo othly in terp olates b et ween the tw o. 4 Generating w eigh ted samples from q t . Ref. [74] provides metho ds to mo dify the dynamics in Eq. 5 so that instead of sampling from p t , w e get weigh ted samples ( x 1 , w 1 ) , ..., ( x N , w N ) from biased marginals q t . The weigh ts w 1 , ..., w N are importance weigh ts, enco ding the discrepancy b et ween prop osal and target biased marginals. In order to sample from the biased marginals in Eq. 7, w e need a slightly more general version of the rew ard-tilted SDE of [74]. W e get this b y using con trol drift ˜ σ 2 t 2 ∇ b t in the framew ork of [75], resulting in the follo wing system of SDEs: x 0 ∼ p noise (8) dx t = − f t + σ 2 t + ˜ σ 2 t 2 ∇ log p t ( x t ) + ˜ σ 2 t 2 ∇ b t ( x t ) dt + ˜ σ t dW t (9) d log w t = F t ( x t ) dt − E x ∼ q t [ F t ( x )] (10) F t ( x ) := − ∂ b t ( x ) ∂ t + ∇ b t ( x ) · f t ( x ) − σ 2 t 2 ∇ log p t ( x ) (11) If we c ho ose σ t ≡ ˜ σ and b t ( x ) ≡ λ t b ( x ) we reco ver the reward-tilted SDE from [74]. Effectiv e sample size W e track the statistical efficiency of a w eighted batc h of n samples using the Kish effectiv e sample size (ESS) [76]: ESS = P i w i 2 P i w 2 i (12) ESS ranges b et w een 1 (all weigh t on one sample) and n (all weigh ts equal). If the weigh ts are normalized as P i w i = 1, it is ESS = 1 / P i w 2 i . Resampling During denoising we p eriodically resample the particles using stratified sampling. Resampling can b e done p eriodically , but should b e done if the ESS falls b elo w a predefined threshold (e.g., n/ 2 [71]). After each resampling step, the w eights are refreshed, i.e., ( x 1 t , w 1 t ) , ..., ( x K t , w K t ) → ( ˜ x 1 t , 1) , ..., ( ˜ x K t , 1) . (13) Optionally , w e can enforce a resampling step after the steering pro cedure at t = 0 p er biased ensem ble. In that case, all steered samples hav e uniform w eights 1 and the subsequen t expressions simplify . 2.2 Un biasing to the equilibrium distribution After pro ducing samples from K biased ensem bles (Sec. 2.1), we ha ve a set of w eighted samples { ( x k,i , w k,i ) } N k i =1 from each biased density q k . Next we com bine all biased samples in to a single sample with w eights W n : { ( x k,i , w k,i ) } N k i =1 → { ( x n , W n ) } N n =1 . (14) The rew eighting co efficients W n com bine (i) the steering importance weigh ts w k,i within eac h biased densit y q k pro duced via (8-11) and (ii) an additional unbiasing factor that maps eac h biased ensemble q k bac k to p . If we hav e c hosen to do a terminal resampling step at the end of steering, w k,i ≡ 1 and W n only contains the un biasing from q k to p . Equilibrium exp ectation v alues The w eights W n m ust be suc h that w e can express equilibrium exp ec- tation v alues of arbitrary observ ables o ( x ) as: b E p [ o ] = P N n =1 W n o ( x n ) P N n =1 W n . (15) in a wa y that is asymptotically un biased in the infinite sample limit, i.e., lim N →∞ b E p [ o ] = E p [ o ]. Expression (15) is sufficiently general that all quantities of the un biased e nsem ble can be expressed with it. F or example, 5 to compute the folding free energy w e define a mem b ership function whic h assigns folded states to 1 and unfolded states to 0: χ ( x ) = ( 0 x unfolded 1 x folded , (16) χ ( x ) can itself be defined in terms of suitable order parameters, such as the fraction of nativ e contacts from the folded structure, and it can optionally tak e in termediate v alues b et w een 0 and 1. In terms of χ , the folding free energy is defined by: ∆ G fold = − k B T ln p folded p unfolded = − k B T ln b E p [ χ ] 1 − b E p [ χ ] (17) using (15). Likewise, a p oten tial of mean force (PMF) along a reaction co ordinate ξ ( x ) can b e formulated b y , e.g., binning the v alue range of ξ and expressing the probabilit y of eac h bin in terms of (15). Practically , w e obtain PMFs b y constructing a w eighted histogram or kernel density estimator from the weigh ted sample { ( x n , W n ) } N n =1 . Single biased ensem ble F or the sp ecial case K = 1, i.e., using a single biased ensem ble with bias potential b ( x ), we hav e generated samples { ( x n , w n ) } N n =1 (dropping the window index). The com bined imp ortance w eights are then trivially giv en b y direct rew eighting: W n = w n e b ( x n ) (18) Un biasing m ultiple biased ensem bles with MBAR A statistically optimal metho d to combine samples from m ultiple biased ensem bles to wards unbiased ensemble estimators is the m ultistate Bennett acceptance ratio (MBAR), also known as binless weigh ted histogram analysis (WHAM) [14, 77, 78]. F or the special case K = 2, MBAR b ecomes traditional BAR [12]. MBAR yields minim um-v ariance, asymptotically unbiased estimates without requiring us to ev aluate log p ( x ). If the bias energy can b e discretized w e can instead work with histograms and binned WHAM [7, 13, 79]. Here we describ e a w eighted MBAR v ariant which takes the weigh ted sample terminals from steering, { ( x k,i , w k,i ) } N k i =1 , and rewrites them in to optimal reweigh ting coefficients for estimating exp ectations under p (Eq. 14). By con ven tion, MBAR op erates on K + 1 ensembles, consisting of the unbiased ensemble b 0 ( x ) ≡ 0 and the biased ensembles b k ( x ) for k = 1 , . . . , K . Note that w e do not dra w samples from ensemble 0, so sums run ov er sampled windows ℓ = 1 , ..., K . T o deal with weigh ted samples, w e define the p er-sample effective mass α k,i ∝ w k,i and normalize so the window mass sums to the effectiv e sample size of ensem ble k , ESS k (Eq. 12): α k,i = ESS k w k,i P N k j =1 w k,j = ⇒ N k X i =1 α k,i = ESS k . (19) If w e hav e chosen to do terminal resampling at the end of steering, the input samples are already unw eighted, w k,i ≡ 1, ESS k = N k , hence α k,i = 1 and we recov er standard MBAR. W e then aggregate all samples as { x n } N n =1 with asso ciated α n (from the bias p oten tial with which they w ere generated). Let M ℓ = P n : orig( x n )= ℓ α n denote the windo w mass, where M ℓ = ESS ℓ under the default ab o v e, and orig( x n ) denotes the index of the window in whic h sample x n w as generated. W e solv e for the reduced free energies of the sampled biased states k = 1 , ..., K via exp( − ˆ f k ) = N X n =1 α n exp − b k ( x n ) P K ℓ =1 M ℓ exp ˆ f ℓ − b ℓ ( x n ) , k = 1 , . . . , K , (20) and forms target-state weigh ts ( a ∈ { 0 , 1 , . . . , K } ), with the unbiased state a = 0 included as the target for rew eighting, as W ( a ) n = α n exp − b a ( x n ) P K ℓ =1 M ℓ exp ˆ f ℓ − b ℓ ( x n ) . (21) 6 W e then compute unbiased expectations in (15) using W n ≡ W (0) n . (22) Using α k,i ∝ w k,i with the normalization ab o v e lets MBAR automatically do wn-weigh t windo ws with highly skew ed particle weigh ts via their ESS k . If desired, one can replace ESS k b y an external effe ctive c ount (e.g. n umber of denoising tra jectories used for window k , or a correlation-corrected N k /g k ); equations (20)– (22) are unchanged. T o compute uncertainties, we can use MBAR’s cov ariance expressions [14], or apply a cluster bo otstrap that resamples at the level of indep enden t denoising tra jectories to accoun t for within-windo w correlations, e.g., from late branc hing. 2.3 Illustration of enhanced diffusion sampling Before moving on to more complex enhanced sampling proto cols, w e first demonstrate that enhanced diffusion sampling is efficient in an idealized setting (Fig. 1). W e define a family of double-well potentials u ( x ) where the energy difference betw een the tw o minima is giv en b y ∆ G ranging from -2 to -14 k B T . The corresponding equilibrium densities are given by p ( x ) ∝ e − u ( x ) , resulting in an equilibrium probabilities betw een 10 − 1 and 10 − 6 for the high-energy state (Fig. 1a). F or each ∆ G v alue, a diffusion mo del with an analytical score function is defined that generates samples from p ( x ) exactly . T o ev aluate the effect of enhanced diffusion sampling on sampling efficiency directly , w e defined a single linear bias p oten tial for each double-well potential, b ∆ G = a (∆ G ) x where a (∆ G ) is chosen suc h that the tw o energy minima are equal in the biased p oten tial (Fig. 1b). F or eac h ∆ G v alue w e then generate N samples with eac h of tw o protocols: (i) direct sampling from the unsteered diffusion mo del with densit y p ( x ), and (ii) steered sampling as describ ed in Sec. 2.1 using b ∆ G and direct reweigh ting of the biased ensemble with (18). W e then estimate ∆ G from the samples and compare with the exact v alue. Both proto cols conv erge to the correct ∆ G v alue in the large N limit (Fig. 1c). Enhanced diffusion sampling conv erges faster than equilibrium sampling for all insp ected ∆ G v alues and the performance gap increases with ∆ G . T o compare sampling efficiencies betw een unbiased and enhanced sampling protocols, w e run fiv e independent rep eats for each N and each proto col and define a sample as b eing conv erged when b oth the absolute difference of the empirical mean of ∆ G from the exact v alue, and the empirical standard deviation of ∆ G are within 1 kcal/mol (at 300K) across rep etitions. As exp ected, the num b er of samples needed to reach conv ergence with un biased sampling increases approximately exp onen tially with ∆ G (Fig. 1d, blue). In contrast, the n umber of samples required by enhanced diffusion sampling only increases mildly with ∆ G and ranges b et ween 10 to 100 samples for all ∆ G v alues insp ected here (Fig. 1d, orange). W e note that this setup describes the b est-case scenario in which only a single biasing p oten tial is needed and the perfect bias is kno wn in adv ance. In a realistic scenario w e will ha ve to try differen t biasing potentials un til all desired states ha ve b een sampled, and then com bine them with MBAR, which will lead to a greater n umber of samples needed to conv ergence and will complicate the relationship betw een free energy differences and the num b er of samples needed. 3 Um brellaDiff — um brella sampling with diffusion mo dels Here we describ e how the classical umbrella sampling method [6] can be adapted to steered diffusion mo dels. The goal of umbrella sampling is to compute the marginal probability density p Ξ ( ξ ) = R p ( x ) δ ( ξ ( x ) − ξ ) dx , or free energy profile—also known as p oten tial of mean force (PMF), u Ξ ( ξ ) = − ln p Ξ ( ξ ) , U Ξ ( ξ ) = k B T u Ξ ( ξ ) , (23) along a user-c hosen reaction co ordinate ξ : X → R , e.g., the end-to-end distance of a protein, or the fraction of nativ e contacts. W e will formulate um brella sampling for the simplest case of one-dimensional co ordinates, but the extension to multidimensional um brella sampling is straightforw ard [7]. 7 a) b) d) c) Figure 1: Enhanced sampling with diffusion mo dels efficiently computes probabilities of rare ev ents a) Un biased diffusion mo del probabilit y densit y p ( x ) and corresp onding energy − ln p ( x ). In this example the free energy difference b et w een the t wo minima is set to ∆ G = − 7 k B T . b) Biased density p ( x )e − b ( x ) using a linear tilt b ( x ) = β x with β chosen to achiev e ∆ G biased = 0 k B T . c) Estimates of ∆ G with an unbiased diffusion mo del and enhanced sampling using a tilting potential that sets ∆ G biased = 0 k B T . Mean (solid line) and 95% confidence interv al (shaded area) are shown. d) Number of samples required to get an estimate within 1 kcal/mol of the exact ∆ G v alue and with a standard deviation of 1 kcal/mol, as a function of ∆ G . Um brella sampling introduces a set of K biased ensembles, each of which restrains sampling near pre- scrib ed cen ters c k in ξ -space. The most common bias potential is harmonic with stiffness κ k > 0: b k ( x ) = 1 2 κ k ∥ ξ ( x ) − c k ∥ 2 , (24) although other c hoices are p ossible, e.g., flat-bottom with harmonic tails. F or eac h bias potential, we generate an ensem ble using the steering procedure of Sec. 2.1 and remo ve bias with w eighted MBAR (Sec. 2.2). MBAR returns a set of equilibrium w eights for eac h sample, ( x n , W n ) whic h can then be used in a histogram or k ernel densit y estimate to obtain p Ξ ( ξ ). Finally we can obtain the PMF via Eq. (23). In order to obtain smo oth estimates of p Ξ ( ξ ) it can b e b eneficial to increase the n umber of samples using late branc hing near t ≈ 0 (Sec. 2.1). Designing bias p oten tials to ensure co verage and ov erlap. W e c ho ose centers { c k } K k =1 to cov er the ξ -range of in terest. Our prior mo del of the biased density is that induced b y the bias p oten tial alone, i.e., a Gaussian with cen ter c k and standard deviation σ k ≈ 1 / √ κ k . As a rule of thum b, space neighboring centers within one or t wo σ to obtain ∼ 10–30% o verlap along ξ . As the effect of p ( x ) will change the resulting distribution of q k ( x ), the bias parameters, ma y b e chosen adaptiv ely , e.g., with a preconditioning suc h as: (i) start with evenly spaced c k and κ k = 1 / ( c k − c k − 1 ) 2 , (ii) run a small initial sample and estimate empirical V ar q k [ ξ ], (iii) update κ k ← 1 / d V ar q k [ ξ ] to equalize widths. Diagnostics W e recommend to monitor standard diagnostics from um brella sampling / free-energy analysis to assess (a) whether each biased ensem ble is w ell sampled and (b) whether the set of windows forms a connected path that p ermits low-v ariance rew eighting [1, 80, 81]. In particular the following diagnostics are suitable: 1. Per-windo w effective sample size (ESS) of the steering terminal w eigh ts w k,i (Eq. 12); when termi- nals may be correlated (e.g. due to late branching or MCMC rejuv enation), we additionally report 8 a correlation-corrected effective count N eff k ≈ N k /g k using the statistical inefficiency g k and use a cluster/b ootstrap analysis for uncertainties [82]. 2. Window ov erlap. Along the um brella coordinate ξ , w e quantify ov erlap b et ween neighboring windo ws using histogram/KDE-based ov erlap co efficients (e.g. O k,k +1 = R p q k ( ξ ) q k +1 ( ξ ) dξ or related normal- ized measures). More generally , w e compute the state overlap matrix O ij from MBAR/WHAM, where O ij estimates the probability that a configuration sampled from state j could b e observ ed in state i . As a practical rule, require neighboring-window ov erlap area in the range 0.1 to 0.3 and a well-connected o verlap matrix (no near-block-diagonal structure) by inserting additional windo ws or revising bias pa- rameters [14, 81, 83] 3. MBAR/WHAM uncertaint y estimates, including the asymptotic cov ariance/standard errors for free energies and target observ ables [14, 79], and s tabilit y of the PMF/observ ables under increasing sample size (and optionally lea ve-one-windo w-out chec ks). F or um brella sampling in particular, w e optionally complemen t MBAR with EMUS error analysis to iden tifying windo ws that dominate v ariance and guide adaptiv e allocation of sampling effort [84]. These diagnostics can b e used to adapt windo w cen ters and force constants, increase sampling in selected windo ws, or insert bridging windo ws to restore ov erlap [80, 81]. Um brellaDiff v ersus traditional umbrella sampling with MD Compared to traditional umbrella sampling with MD, UmbrellaDiff has several adv antages. First, in conv en tional umbrella sampling one m ust prepare initial configurations for eac h window (often via pulling/steered protocols). Then one m ust equilibrate sufficiently so that neighboring windows ov erlap not only in the restrained coordinate ξ but also in orthogonal, slow degrees of freedom [80, 81, 85]. This is not an issue with Um brellaDiff, as each steered sample is indep endently generated from the noisy state using the pro cedure in Sec. 2.1. Secondly , when hidden barriers or m ultiple c hannels exist in directions perp endicular to ξ , traditional um brella sim ulations can remain trapp ed in distinct metastable “orthogonal states”, leading to h ysteresis and very slo w con vergence of the PMF unless additional sampling machinery is introduced (Fig. 2) [86, 87]. This is not an issue for UmbrellaDiff, where the only role of the umbrellas is to ensure that low probability v alues in p Ξ ( ξ ) are sampled, while the iid sampling property of the diffusion mo del av oid problems with kinetic trapping and slo w intra-windo w relaxation. Fig. 2 demonstrates that Um brellaDiff can compute un biased free energy profiles ev en if metastable states that are off of the main path of umbrellas in ξ contribute significan tly to the free energy , whereas traditional umbrella sampling struggles with these cases. 4 MetaDiff — metadynamics with diffusion mo dels Metadynamics [11, 88, 89] is a widely used enhanced sampling metho d with many follow-up v ariants, including w ell-temp ered metadynamics [90, 91] and asso ciated estimators [92]. The original idea of metadynamics is to run an MD sim ulation and to p eriodically deposit repulsiv e hil ls (t ypically Gaussians) in the space of c hosen collectiv e v ariables (CVs) ξ ( x ) at the current CV v alue. The resulting history-dep enden t bias progressively fills free-energy minima and drives exploration of new regions [11, 88]. In standard metadynamics, the bias k eeps gro wing, while in well-tempered metadynamics the hill heights are temp ered so that the bias con verges (up to a known factor) to the negative free energy [90, 91]. Because the bias is explicitly time-dep enden t, metadynamics is generally analyzed via the accum ulated bias and dedicated rew eighting/estimators rather than by treating the tra jectory as equilibrium sampling at a fixed thermo dynamic state; in particular, during the initial “fill-up” p eriod it is a nonequilibrium process, so equilibrium m ulti-state estimators suc h as MBAR are not directly applicable [88, 92]. Here we formulate a metadynamics v ariant for steered diffusion models. In con trast to biased MD tra jec- tories, eac h inv o cation of the steering algorithm (Sec. 2.1) targets the equilibrium distribution of the curr ent bias condition and returns iid (w eighted) samples from that biased ensemble. Th us, eac h bias up date defines a w ell-p osed thermodynamic state, and w e c an apply MBAR online to com bine samples across bias conditions and obtain an unbiased free-energy estimate (and diagnostics) at an y time, without w aiting for a fully “filled” surface. Finally , since diffusion inference is naturally batc hed, MetaDiff up dates the bias from a batc h of samples: instead of a single Gaussian hill, w e add a batch wise kernel (a mixture of Gaussians in ξ - space). 9 Figure 2: Um brellaDiff bypasses kinetic trap problem in traditional umbrella sampling. a) Simple t wo-state 2D potential. Umbrella sampling is p erformed using 8 um brellas that bias ξ = x using regular umbrella sampling with Langevin dynamics (orange) and UmbrellaDiff (blue). Con tours sho w 95% of the sample density of individual umbrellas. b) F ree energy as a function of x using traditional um brella sampling (orange) and UmbrellaDiff (blue). Both metho ds appro ximate the true potential of mean force (grey). c) As a) but with a third high-probability state (top right). T raditional umbrella sampling (orange) do es not sample the third state as it is off-path and long simulation tra jectories in eac h um brella would b e required to sample this state. Um brellaDiff (blue) samples the equilibrium distribution conditioned on the umbrella p otential and therefore samples the third state without issues. d) As b). T raditional um brella sampling estimates a wrong free energy due to failing to sample the third state (orange), UmbrellaDiff estimates the correct free energy (blue). 10 In the limit of batch size 1, the pro cedure reduces to standard metadynamics applied to steered diffusion sampling. W e will again form ulate the metho d on a one-dimensional reaction co ordinate ξ : X → R , the generaliza- tion to multiple dimensions is conceptually straigh tforward. W e represen t the bias at outer iteration k as a sum of Gaussians: b k ( ξ ) = M k X m =1 a m K σ ( ξ ; µ m ) , K σ ( ξ ; µ ) = 1 √ 2 π σ exp − ( ξ − µ ) 2 2 σ 2 , with b 0 ≡ 0. F or eac h outer iteration k , we pro duce a batch of samples with normalized weigh ts using the steering algorithm (Sec. 2.1): { ( x k,i , w k,i ) } N k i =1 with ξ k,i = ξ ( x k,i ). W e then dep osit one Gaussian p er sample sequen tially so that b k remains a sum of Gaussians at all times, in the spirit of classical metadynamics [11] and w ell–temp ered metadynamics [90]. T o control the total amoun t of bias added at iteration k , w e choose a p er-iteration mass h k > 0 and split it across the N k samples using p er-Gaussian masses ˜ h k,i that satisfy P N k i =1 ˜ h k,i = h k . A simple c hoice is ˜ h k,i = h k / N k , for unw eigh ted samples , h k w k,i P j w k,j , for imp ortance–w eighted samples , (25) so eac h iteration dep osits total mass h k regardless of batch size. In practice, standard metadynamics often uses a c onstant base height ( h k ≡ h 0 ) and relies on exploration to distribute hills, whereas well–tempered metadynamics also works robustly with a constant h k b ecause the effectiv e dep osited height deca ys automat- ically via temp ering [90]; optionally , a gen tly decaying schedule h k = h 0 ρ k with ρ ∈ (0 , 1) reduces the noise during late iterations. The final amplitude for eac h Gaussian i is determined by whether we use a standard or w ell-temp ered Metadynamics scheme: (standard) a k,i = ˜ h k,i , (26) (w ell–temp ered, γ > 1) a k,i = ˜ h k,i exp − b ( i − 1) k ( ξ k,i ) γ − 1 . (27) Setting γ = ∞ in (27) recov ers the standard rule (26). Finally , w e up date the bias. Starting from b (0) k ≡ b k w e sequen tially iterate through the batch for i = 1 , . . . , N k : b ( i ) k ( ξ ) = b ( i − 1) k ( ξ ) + a k,i K σ ( ξ ; ξ k,i ) , ξ k,i = ξ ( x k,i ) . (28) After pro cessing all N k terminals, we set b k +1 ≡ b ( N k ) k as the new bias p oten tial. Algorithm (MetaDiff ). Given kernel width σ , temp ering factor γ ≥ 1, and a base-heigh t sc hedule { h k } k ≥ 0 : 1. Initialize b 0 ≡ 0. 2. F or k = 0 , 1 , 2 , . . . : (a) Sample from q k via Steering (Sec. 2.1) and collect w eighted samples { ( x k,i , w k,i ) } N k i =1 . (b) Cho ose ˜ h k,i using Eq. (25). (c) F or i = 1 , . . . , N k : compute a k,i b y (26) (standard) or (27) (w ell–temp ered) using the current b ( i − 1) k , then up date b ( i ) k via (28). (d) Set b k +1 ← b ( N k ) k . Stop when sup ξ | b k +1 ( ξ ) − b k ( ξ ) | < ε b or when an MBAR–based PMF change across selected chec kp oin ts falls b elo w ε F . 11 Mean-field consistency and stationary PMF. As σ → 0 and h k → 0 (small hills), the exp ected increment at ξ under (27)– (28) satisfies E b k +1 ( ξ ) − b k ( ξ ) ∝ q Ξ ,k ( ξ ) e − b k ( ξ ) / ( γ − 1) , whic h is the well–tempered mean-field up date [90]. A t stationarity , u Ξ ( ξ ) = − ( γ / ( γ − 1)) b ∞ ( ξ ) + C (with arbitrary constan t C ); F or γ = ∞ the up date reduces to standard metadynamics; in the idealized fully-filled limit the accumulated bias appro ximates − u Ξ ( ξ ) + C [11]. The typical practice is to keep h k constan t and c ho ose γ to tune smo othness/conv ergence ( γ ≈ 5–20 is common). A modest deca y of h k can further stabilize late iterations. Illustration on a double-well p oten tial Fig. 3 demonstrates MetaDiff on a one-dimensional double-w ell p oten tial whose energy minima ha ve a free energy difference of -13.8 k B T . using 1 . 11 · 10 5 samples, the un biased diffusion sampler only samples the low er energy minimum but fails to sample the second minimum (Fig. 3, first column). MetaDiff is run in batc hes of 1 . 77 · 10 4 samples. The second minimum is explored after the second batc h and an accurate estimate of the free energy difference is immediately achiev ed, and further impro ved in subsequent batc hes (Fig. 3, columns 2-5). Figure 3: MetaDiff illustration on 1-dimensional double-w ell potential. Estimated PMF using iterations of metadynamics at a free energy difference of ∆ G = − 13 . 8 k B T . 5 ∆ G -Diff: F ree energy differences b et w een tw o states Here we aim to compute the free energy difference b et ween t wo configurational states A and B , ∆ G AB . Examples include a folding free energy , binding free energy , the free energy change of a reaction or a confor- mational change. Let ξ ( x ) ∈ [0 , 1] be a smo oth progress co ordinate that indicates how far a configuration x has adv anced from state A to state B : ξ ( x ) = 0 x ∈ A (0 , 1) x / ∈ { A, B } 1 x ∈ B (29) 12 In practice, ξ can b e a sigmoid function of a differentiable fraction-of-native-con tacts (FNC), a normalized ro ot-mean-squared-deviation (RMSD) or a committor function defined b et ween sets of configurations A and B [93 – 96]. T o ensure that b oth states can be sampled, we bias with a cen tered linear tilt in reduced units b a ( x ) = a ξ ( x ) − 1 2 , a ∈ R , so a > 0 fav ors A ( ξ ≈ 0) and a < 0 fav ors B ( ξ ≈ 1). Dominance and ov erlap diagnostics. W e define core sets in the ξ co ordinate as C A = [0 , c ], C B = [1 − c, 1] with, e.g., c = 0 . 4. F rom a weigh ted batch with DM steering (Sec. 2.1) with tilt a we ha ve samples and normalized weigh ts ( ξ ( x i ) , ¯ w i ). W e define the core probabilit y masses as b π A ( a ) = X i ¯ w i 1 { ξ ( x i ) ∈ C A } , b π B ( a ) = X i ¯ w i 1 { ξ ( x i ) ∈ C B } . Sa y a has A -dominanc e if b π A ( a ) ≥ ϑ and B -dominanc e if b π B ( a ) ≥ ϑ with default ϑ = 0 . 5 (can be increased on demand). F or adjacen t tilts a < a ′ , we measure the ov erlap as follo ws b O a → a ′ ∝ X i ∈ a exp − ( a ′ − a ) [ ξ ( x i ) − 1 2 ] , and require b O a → a ′ , b O a ′ → a ≥ ε ov (e.g. 10–30%). Algorithm ( ∆ G-Diff ). Inputs: tilt step s > 0 (default s ≈ 2 in k B T ), initial sample size n (default 100), dominance threshold ϑ (default 0.5), ov erlap threshold ε ov (default 0.25), ESS target (default 100). 1. Initialize: A ← { 0 } . Sample n terminals with a =0. Set a L ← 0 (leftmost), a R ← 0 (rightmost). 2. Increase tilts until both endpoints dominate: (a) If b π A ( a R ) < ϑ : set a R ← a R + s , sam ple n terminals at a R , insert a R in to A . (b) If b π B ( a L ) < ϑ : set a L ← a L − s , s ample n terminals at a L , insert a L in to A . 3. Ensure adjacent ov erlap: Sort A . While any neigh b oring pair ( a, a ′ ) violates b O a → a ′ ≥ ε ov or b O a ′ → a ≥ ε ov , insert the midp oin t ˜ a = ( a + a ′ ) / 2, sample n terminals at ˜ a , insert ˜ a in to A , and up date o verlaps. 4. Refine for accuracy: Add more samples to all a ∈ A to reach ESS target . 5. Unbias and estimate: Solve w eighted MBAR (Sec. 2.2) o ver all tilts using bias p otentials b a ( x ) = a ( ξ ( x ) − 1 2 ) to obtain target ( a =0) weigh ts W (0) n , then compute c ∆ g AB = − log P n W (0) n ξ ( x n ) P n W (0) n [1 − ξ ( x n )] , d ∆ G AB = k B T c ∆ g AB . Uncertain ties follo w from the MBAR cov ariance or a cluster b o otstrap o ver denoising-tra jectory IDs. Illustration and results for protein folding with BioEm u Fig. 4a illustrates how ∆G-Diff works for a t wo-state potential, whic h is representativ e of typical protein folding and protein-ligand binding scenarios. When the free energy difference b etw een the tw o states ∆G is large, almost all equilibrium density is in the more stable state, e.g., the folded or b ound state (Fig. 4a, top left). ∆G-Diff defines a series of tilt p oten tials interpolating b etw een an ensemble in which one state is dominan t (top left) and an ensemble in whic h the other state is dominan t (b ottom right). If a go o d guess of ∆G is av ailable, a single tilted ensemble whic h samples from b oth states is sufficient. The ∆G-Diff ensembles can b e generated with steering and 13 com bined with MBAR to the estimated free energy v alue. Note that in contrast to classical enhanced sampling methods that use MD, there is no need for having ov erlapping distributions in co ordinate space. A single steered diffusion mo del ensemble that is able to sample b oth states (b ottom right) is sufficien t to obtain an unbiased estimate of ∆G. Multiple ensembles are useful to find the optimal tilt, and combining their samples with MBAR improv es statistics. T o connect these trends to practical compute, we implemen ted FKC reward tilting algorithm [74] in com bination with the SDE DPM-Solv er++ [97], a second order denoiser, based on the open source BioEmu [3] co de. W e used BioEmu mo del v ersion 1.1 to generate samples of a set of relatively stable proteins selected from ProThermDB [98] database, with differen t predicted un biased stabilities ranging from 1 to 6 kcal/mol. A t room temp erature, folding free energy of 5 k cal/mol corresp onds to one unfolded sample in 4160 samples, whic h requires on the order of sev eral GPU hours, with the exact timing dep ending on GPU and protein size. W e ha v e c hosen estimated optimal tilts to illustrate the correctness and enhanced sampling efficiency for those proteins. F or each protein, w e generated 25,000 steered samples in total with small batches, using p er-system optimized steering slop es along the collective v ariable defined as the Ro ot Mean Squared Deviation (RMSD) to the nativ e PDB structure. Unbiased statistics were recov ered by direct reweigh ting (18) since it a single bias p oten tial w as used, and folding free energies were computed from the fraction of nativ e contacts (FNC) using a tw o-state mo del. T o quan tify sample efficiency , since different batc hes are indep endently sampled, w e rep eatedly subsampled a giv en n umber of batc hes (and th us a fixed n umber of particles) without replacemen t, and ev aluated the resulting ∆ G estimates as a function of total sample count. W e excluded 8 systems where steering did not yield usable ov erlap, including 5 where the RMSD range is to o large (beyond 2nm), and 3 where the “unbiased” reference estimates were not reliable due to large un biased ∆ G and p otentially m uch larger num b er of un biased samples are needed for con vergence. After filtering, 18 proteins remain for quan titative comparison. In Fig. 4b, we plot the enhanced sampling ∆ G from rew eighted steered samples at approximately 1,000 particles against the con verged unbiased ∆ G , sho wing close agreement across the full stability range. W e next measured the minimum sample count required for con vergence, defining con vergence as a self-reference mean absolute error (MAE) b elo w 1 kcal/mol, as sho wn in Fig. 4c. Ubiquitin (1UBQ) is a notable outlier, lik ely due to the fact that it is not a tw o- state folder. F or un biased sampling, this required sample coun t gro ws exponentially with ∆ G , consisten t with the fact that the probability of observing the thermodynamically rare state decreases exponentially in free energy difference. In contrast, steered sampling sho ws a muc h w eaker scaling o ver the same range, demonstrating a substan tial reduction in sample complexit y for stable proteins. 6 Discussion Diffusion-mo del-based equilibrium samplers remov e one of the tw o fundamental b ottlenec ks of molecular sim ulation: slow mixing due to time-correlated tra jectories. Ho wev er, iid sampling alone do es not resolv e the second b ottlenec k—the exp onen tial sample complexity of estimating observ ables dominated by low- probabilit y regions of the equilibrium distribution, such as in protein folding or protein-ligand binding. In this work, w e show that the classical bias-and-reweigh ting paradigm of enhanced sampling can b e transferred to diffusion samplers by steering pretrained mo dels to generate biased ensem bles and recov ering un biased thermo dynamics with exact reweigh ting (direct reweigh ting, WHAM/MBAR). In this sense, the present framew ork addresses b oth asp ects of the sampling problem: diffusion mo dels address slow mixing, and enhanced sampling addresses rarit y , together enabling conv erged free-energy estimation on GPU timescales that were previously dominated by long MD tra jectories. A k ey consequence of replacing biased MD tra jectories b y indep enden t (possibly w eighted) samples is that sev eral enhanced sampling pro cedures c hange character. In UmbrellaDiff, windows no longer need to b e dynamically connected by slow transitions, and hidden barriers orthogonal to ξ do not cause kinetic trapping within a windo w; ov erlap requiremen ts are purely statistical and can be diagnosed and repaired using standard MBAR/WHAM to ols (ESS, ov erlap matrices, uncertaint y estimates). Likewise, in MetaDiff, history-dep enden t biasing b ecomes an e quilibrium sequence of thermo dynamic states: each bias up date defines a w ell-p osed biased distribution that can b e incorp orated immediately into MBAR, allowing online diagnostics and early stopping without w aiting for a fully “filled” surface. More broadly , once mixing is remo ved from the underlying sampling engine, bias v ariables need not corresp ond to slo w dynamical mo des, suggesting that reaction-co ordinate learning and adaptive windo w placement may become simpler and more 14 1TTG f olded 1TTG un folded a) b) d) c) Figure 4: ∆ G-Diff illustration and protein folding results with BioEmu : a) Processes with large free energy differences (top left, black), e.g., protein folding, sample almost exclusiv ely one state in equilibrium (top left, blue). ∆G-Diff creates a series of tilted biased potentials un til b oth states are dominantly sampled in at least one ensemble eac h (top left, bottom righ t), and these ensembles can b e combined with MBAR to yield an estimate of ∆G. b) Comparison of folding free energies ∆G sampled by unsteered BioEmu and ∆G-Diff with single optimal tilt. c) Sampling efficiency as a function of ∆G: unsteered sampling (grey) requires exp onentially man y samples in ∆G, while the n umber of samples required by ∆G-Diff sho ws m uch w eaker scaling with ∆G. d) Sample folded and unfolded state of a stable protein domain of fibronectin (PDB co de 1ttg). 15 robust than in MD-based enhanced sampling. A t the same time, diffusion-based enhanced sampling inherits—and in some cases amplifies—several familiar limitations. First, the approach relies on ha ving a sufficiently accurate pretrained equilibrium mo del for the molecular system and thermodynamic conditions of interest; an y mo del mismatch propagates in to rew eighted estimates. Second, as in all imp ortance-sampling sc hemes, weigh t degeneracy can arise when biases are to o aggressiv e or when the biased ensembles (and/or the unbiased target) ha ve insufficien t ov erlap, necessitating careful bias design, ov erlap diagnostics, and potentially additional intermediate states. Finally , the present work fo cuses on equilibrium properties; extending these ideas to dynamical observ ables will require additional structure, for example through path rew eighting or consistent generativ e dynamical models. Lo oking forw ard, sev eral directions app ear particularly promising. Metho dologically , the steering frame- w ork can b e com bined with adaptive sc hemes that learn reaction co ordinates or optimal bias p oten tials on the fly , and with automated window/tilt construction guided b y MBAR diagnostics. Arc hitecturally , energy- based or h ybrid diffusion models that allow direct ev aluation of u ( x ) could enable tighter estimator control and new forms of biasing. Bey ond equilibrium ensem ble generators, an activ e line of researc h learns accelerated dynamics or tr ans- p ort op er ators from MD data, including Timewarp [99], implicit transfer op erator learning [100], and tra jec- tory generators such as MDGen [101]. Because these mo dels are trained to repro duce finite-lag transitions rather than the equilibrium distribution, their stationary distribution need not coincide with the desired p ( x ) unless additional constraints or corrections are imp osed. One promising route is to wrap learned transp ort mo dels in to Metrop olis–Hastings or related accept/reject schemes that target a known equilibrium distribu- tion (as done in Timewarp), in which case bias p oten tials can b e incorp orated by changing the target and standard reweigh ting estimators apply . Another route is to extend biasing and reweigh ting to p ath ensem- bles , where one biases tra jectory functionals (e.g., for transition-path sampling) and reweigh ts b y lik eliho od ratios of paths; MDGen already highligh ts transition-path sampling as a target application. Recent w ork on incorp orating equilibrium priors in to transfer-op erator learning (e.g., BoPITO [102]) further suggests opp ortunities to unify learned transport with thermo dynamically consisten t sampling. Finally , while w e ha ve emphasized biomolecular folding and conformational change, the formulation is general and applies to any system where iid equilibrium samplers exist but rare-even t statistics are the remaining b ottlenec k—including materials, soft matter, and condensed-phase chemistry . W e anticipate that in tegrating enhanced sampling principles into diffusion samplers will b ecome a cen tral building blo c k for next-generation molecular simulation workflo ws, enabling routine computation of free energies and rare-state observ ables at scales previously accessible only with sp ecialized hardware or massive distributed MD. 7 Ac kno wledgemen t W e ackno wledge Carles Domingo-Enric h and Akshay Krishnamurth y for helpful discussions. GitHub Copilot has b een used in part of the implemen tation and analysis of this w ork. 8 Co de a v ailabilit y The BioEm u inference code is in https://github.com/microsoft/bioemu . The enhanced sampling feature will b e released so on. 16 References [1] J. H´ enin, T. Leli` evre, M. R. Shirts, O. V alsson, and L. Delemotte. Enhanced sampling metho ds for molecular dynamics simulations [article v1.0]. LiveCoMS , 4:1583, 2022. [2] F. No´ e, S. Olsson, J. K¨ ohler, and H. W u. Boltzmann generators - sampling equilibrium states of man y-b o dy systems with deep learning. Scienc e , 365:eaa w1147, 2019. [3] S. Lewis, t. Hemp el, J. Jim´ enez-Luna, M. Gastegger, Y. Xie, A. Y. K. F o ong, V. Garc ´ ıa Satorras, O. Abdin, B. S. V eeling, I. Zap orozhets, Y. Chen, S. Y ang, A. E. F oster, A. Schneuing, J. Nigam, F. Barb ero, V. Stimp er, A. Campb ell, J. Yim, M. Lienen, Y. Shi, S. Zheng, H. Sch ulz, U. Munir, R. Sordillo, R. T omiok a, C. Clementi, and F. No ´ e. Scalable emulation of protein equilibrium ensembles with generative deep learning. Scienc e , page eadv9817, 2025. doi: 10.1126/science.adv9817. [4] J. G. Kirkw o od. Statistical mec hanics of fluid mixtures. J. Chem. Phys. , 3:300–313, 1935. [5] R. W. Zw anzig. High-temp erature equation of state by a p erturbation metho d. i. nonp olar gases. J. Chem. Phys. , 22:1420–1426, 1954. [6] G. M. T orrie and J. P . V alleau. Nonph ysical Sampling Distributions in Monte Carlo F ree-Energy Estimation: Umbrella Sampling. J. Comp. Phys. , 23:187–199, 1977. [7] M. Souaille and B. Roux. Extension to the w eighted histogram analysis metho d: combining umbrella sampling with free energy calculations. Comput. Phys. Commun. , 135:40–57, 2001. [8] E. Marinari and G. P arisi. Simulated temp ering: A new mon te carlo sc heme. Eur o. Phys. L ett. , 19: 451–458, 1992. [9] U. H. E. Hansmann. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. L et. , 281:140 – 150, 1997. [10] Y. Sugita and Y. Ok amoto. Replica-exchange molecular dynamics metho d for protein folding. Chem. Phys. L ett. , 314:141–151, 1999. [11] A. Laio and M. P arrinello. Escaping free energy minima. Pr o c. Natl. A c ad. Sci. USA , 99:12562–12566, 2002. [12] C. H. Bennett. Efficient estimation of free energy differences from monte carlo data. J. Chem. Phys. , 22:245–268, 1976. [13] A. M. F errenberg and R. H. Sw endsen. Optimized monte carlo data analysis. Phys. R ev. L ett. , 63: 1195–1198, 1989. [14] M. R. Shirts and J. D. Cho dera. Statistically optimal analysis of samples from m ultiple equilibrium states. J. Chem. Phys. , 129:124105, 2008. [15] A. M. T okita, T. Dev ergne, A. M. Saitta, and J. Behler. F ree energy profiles for chemical reactions in solution from high-dimensional neural net work potentials: The case of the streck er synthesis. arXiv pr eprint arXiv:2503.05370 , 2025. [16] S. Sto c ker, H. Jung, G. Cs´ anyi, C. F. Goldsmith, K. Reuter, and J. T. Margraf. Estimating free energy barriers for heterogeneous catalytic reactions with mac hine learning p oten tials and um brella in tegration. J. Chem. The ory Comput. , 19:6796–6804, 2025. [17] J. Hub and B. L. de Gro ot. Mechanism of selectivity in aquap orins and aquaglyceroporins. Pr o c. Natl. A c ad. Sci. USA , 105:1198–1203, 2008. [18] F. T. Heer, D. J. Posson, W. W o jtas-Niziurski, C. M. Nimigean, and S. Bern` eche. Mechanism of activ ation at the selectivity filter of the KcsA K+ channel. eLife , 6:e25844, 2017. 17 [19] Z. Cournia, B. K Allen, and W. Sherman. Relativ e binding free energy calculations in drug disco very: Recen t adv ances and practical considerations. J. Chem. Inf. Mo del. , 57:2911–2937, 2017. [20] J. H. Moore, D. J. Cole, and G. Cs´ anyi. Computing solv ation free energies of small molecules with first principles accuracy . arXiv pr eprint arXiv:2405.18171 , 2025. [21] J. D. Cho dera, D. L. Mobley , M. R. Shirts, R. W. Dixon, K. Branson, and V. S. Pande. Alchemical free energy metho ds for drug disco very: Progress and c hallenges. Curr. Opin. Struct. Biol. , 21:150–160, 2011. [22] J. W. Pitera and W. Sw op e. Understanding folding and design: Replica-exc hange simulations of “trp- cage” miniproteins. Pr o c. Natl. A c ad. Sci. USA , 13:7587—-7592, 2003. [23] N. E. Charron, K. Bonneau, A. S. Pasos-T rejo, A. Guljas, Y. Chen, F. Musil, J. V enturin, D. Gusew, I. Zap orozhets, A. Kr¨ amer, C. T empleton, A. Kelk ar, A. E. P . Durumeric, S. Olsson, A. P ´ erez, M. Ma- jewski, B. E. Husic, A. P atel, G. De F abritiis, F. No ´ e, and C. Clemen ti. Na vigating protein landscap es with a machine-learned transferable coarse-grained model. Natur e Chemistry , 17:1284–1292, 2025. [24] J. M. L. Ribeiro, P . Brav o, Y. W ang, and P . Tiwary . Reweigh ted auto encoded v ariational Ba yes for enhanced sampling (RA VE). J. Chem. Phys. , 149:072301, 2018. [25] Y. W ang, J. M. L. Ribeiro, and P . Tiwary . Past-future information b ottlenec k for sampling molecular reaction co ordinate simultaneously with thermo dynamics and kinetics. Natur e Commun. , 10:3573, 2019. [26] W. Chen, H. Sidky , and A. L. F erguson. Nonlinear disco very of slow molecular mo des using state-free rev ersible v ampnets. J. Chem. Phys. , 150:214114, 2019. [27] A. Mardt, L. Pasquali, H. W u, and F. No´ e. V ampnets: Deep learning of molecular kinetics. Nat. Commun. , 9:5, 2018. [28] L. Bonati, G. Piccini, and M. Parrinello. Deep learning the slow mo des for rare ev ents sampling. Pr o c. Natl. A c ad. Sci. USA , 118:e2113533118, 2021. [29] L. Bonati, V. Rizzi, and M. Parrinello. Data-driv en collective v ariables for enhanced sampling. J. Phys. Chem. L ett. , 11:2998–3004, 2020. [30] D. E. Kleiman and D. Shukla. Activ e learning of the conformational ensemble of proteins using maxi- m um en tropy v ampnets. J. Chem. The ory Comput. , 19:4377–4388, 2023. [31] J. Preto and C. Clemen ti. F ast recov ery of free energy landscap es via diffusion-map-directed molecular dynamics. Phys. Chem. Chem. Phys. , 16:19181–19191, 2014. [32] W. Zheng, M. A. Rohrdanz, and C. Clemen ti. Rapid exploration of configuration space with diffusion- map-directed molecular dynamics. J. Phys. Chem. B , 117:12769–12776, 2013. [33] J.-H. Prinz, H. W u, M. Sarich, B. G. Keller, M. Senne, M. Held, J. D. Cho dera, C. Sc h ¨ utte, and F. No ´ e. Mark ov mo dels of molecular kinetics: Generation and v alidation. J. Chem. Phys. , 134:174105, 2011. [34] F. No´ e, S. Doose, I. Daidone, M. L¨ ollmann, J. D. Cho dera, M. Sauer, and J. C. Smith. Dynamical fingerprin ts for probing individual relaxation pro cesses in biomolecular dynamics with sim ulations and kinetic exp erimen ts. Pr o c. Natl. A c ad. Sci. USA , 108:4822–4827, 2011. [35] J. Juraszek and P . G. Bolhuis. (Un)F olding mechanisms of the FBP28 WW domain in explicit solven t rev ealed b y multiple rare even t simulation metho ds. Biophys. J. , 98:646–656, 2010. [36] S. Ba jpai, C. R. A. Abreu, N. N. Nair, and M. E. T uck erman. Solute temp ered adiabatic free energy dynamics for enhancing conformational space sampling. J. Chem. The ory Comput. , 21:5928–5940, 2025. [37] F. Marinelli, F. Pietrucci, A. Laio, and S. Piana. A kinetic mo del of trp-cage folding from m ultiple biased molecular dynamics sim ulations. PL oS Comput. Biol. , 5:e1000452, 2009. 18 [38] Y. Liu, J. Str ¨ umpfer, L. J. F reddolino, M. Grueb ele, and K. Sc hulten. Structural characterization of λ -repressor folding from all-atom molecular dynamics simulations. J. Phys. Chem. L ett. , 3:1117–1123, 2012. [39] K. Lindorff-Larsen, S. Piana, R. O. Dror, and D. E. Shaw. Ho w fast-folding proteins fold. Scienc e , 334: 517–520, 2011. [40] S. Piana, K. Lindorff-Larsen, and D. E. Shaw. A tomic-level description of ubiquitin folding. Pr o c. Nat. A c ad. Sci. USA , 110:5915–5920, 2013. [41] G. R. Bowman, V. A. V o elz, and V. S. Pande. Atomistic F olding Simulations of the Five-Helix Bundle Protein Lambda 6-85. J. A m. Chem. So c. , 133:664–667, 2011. [42] V. A. V o elz, M. J¨ ager, S. Y ao, Y. Chen, L. Zhu, S. A. W aldauer, G. R. Bowman, M. F riedrichs, O. Bak a jin, L. J. Lapidus, S. W eiss, and V. S. Pande. Slo w unfolded-state structuring in acyl-CoA binding protein folding revealed by sim ulation and exp erimen t. J. Am. Chem. So c. , 134:12565–12577, 2012. [43] D. Prov asi and M. Filizola. T o wards ligand-sp ecific conformations of g-protein coupled receptors: A study of the β 2 -adrenergic receptor. PL oS Comput. Biol. , 7:e1002193, 2011. [44] Y. W ang, E. Papaleo, and K. Lindorff-Larsen. Mapping transiently formed and sparsely populated conformations on a complex energy landscap e. eLife , 5:e17505, 2016. [45] Y. Meng, Y.-L. Lin, and B. Roux. Computational study of the “DF G-flip” conformational transition in c-abl and c-src tyrosine kinases. J. Phys. Chem. B , 119:1443–1456, 2015. [46] O. Fleet woo d, P . Matricon, J. Carlsson, and L. Delemotte. Energy landscap es rev eal agonist control of g protein-coupled receptor activ ation via microswitc hes. Bio chemistry , 59:880–891, 2020. [47] C. F. Abrams and E. V anden-Eijnden. Large-scale conformational sampling of proteins using temp erature-accelerated molecular dynamics. Pr o c. Natl. A c ad. Sci. USA , 107:4961–4966, 2010. [48] K. J. Kohlhoff, D. Shukla, M. La wrenz, G. R. Bo wman, D. E. Konerding, D. Belov, R. B. Altman, and V. S. P ande. Cloud-based simulations on go ogle exacycle reveal ligand mo dulation of gpcr activ ation path wa ys. Nat. Chem. , 6:15–21, 2014. [49] M. M. Sultan, R. A. Denny , R. Un walla, F. Lov ering, and V. S. Pande. Millisecond dynamics of btk rev eal kinome-wide conformational plasticity within the ap o kinase domain. Sci. R ep. , 7:15604, 2017. [50] G. R. Bowman and P . L. Geissler. Equilibrium fluctuations of a single folded protein reveal a m ultitude of p oten tial cryptic allosteric sites. Pr o c. Natl. A c ad. Sci. USA , 109:11681–11686, 2012. [51] D. Shukla, Y. Meng, B. Roux, and V. S. Pande. Activ ation pathw a y of src kinase reveals intermediate states as targets for drug design. Nat. Commun. , 5:3397, 2014. [52] Y. Meng, D. Shukla, V. S. Pande, and B. Roux. T ransition path theory analysis of c-src kinase activ ation. Pr o c. Natl. A c ad. Sci. USA , 113:9193–9198, 2016. [53] D. E. Sha w, P . Maragakis, K. Lindorff-Larsen, S. Piana, R.O. Dror, M.P . East woo d, J.A. Bank, J.M. Jump er, J.K. Salmon, Y. Shan, and W. W riggers. Atomic-Lev el Characterization of the Structural Dynamics of Proteins. Scienc e , 330:341–346, 2010. [54] R. O. Dror, D. H. Arlow, P . Maragakis, T. J. Mildorf, A. C. P an, H. Xu, D. W. Borhani, and D. E. Sha w. Activ ation mechanism of the β 2 -adrenergic receptor. Pr o c. Natl. A c ad. Sci. USA , 108:18684– 18689, 2011. [55] R. O. Dror, A. C. Pan, D. H. Arlow, D. W. Borhani, P . Maragakis, Y. Shan, H. Xu, and D. E. Shaw. P athw ay and mec hanism of drug binding to G-protein-coupled receptors. Pr o c. Natl. A c ad. Sci. USA , 108:13118–13123, 2011. 19 [56] A. J Ha vens, B. K. Miller, B. Y an, C. Domingo-Enrich, A. Sriram, D. S. Levine, B. M W o o d, B. Hu, B. Amos, B. Karrer, X. F u, G.-H. Liu, and R. T. Q. Chen. Adjoint sampling: Highly scalable diffusion samplers via adjoin t matc hing. In A. Singh, M. F azel, D. Hsu, S. Lacoste-Julien, F. Berk enk amp, T. Mahara j, K. W agstaff, and J. Zh u, editors, ICML 42 , volume 267 of Pr o c. Mach. L e arn. R es. , pages 22204–22237. PMLR, 2025. [57] B. Jing, B. Berger, and T. Jaakkola. AlphaFold meets flow matching for generating protein ensem bles. In R. Salakh utdinov, Z. Kolter, K. Heller, A. W eller, N. Oliver, J. Scarlett, and F. Berk enk amp, editors, ICML 41 , volume 235 of Pr o c. Mach. L e arn. R es. , pages 22277–22303. PMLR, 2024. [58] G. Janson, A. Jussup o w, and M. F eig. Deep generativ e mo deling of temp erature-dependent structural ensem bles of proteins. Commun. Chem. , 8(354), 2025. doi: 10.1038/s42004- 025- 01737- 2. [59] S. Zheng, J. He, C. Liu, Y. Shi, Z. Lu, W. F eng, F. Ju, J. W ang, J. Zh u, Y. Min, H. Zhang, S. T ang, H. Hao, P . Jin, C. Chen, F. No´ e , H. Liu, and T.-Y. Liu. Predicting equilibrium distributions for molecular systems with deep learning. Nat. Mach. Intel l. , 6:558–567, 2024. [60] Yik ai Liu, Zongxin Y u, Richard J. Lindsay , Guang Lin, Ming Chen, Abhilash Saho o, and Sony a M. Hanson. Exendiff: An exp erimen t-guided diffusion mo del for protein conformational ensem ble gener- ation. PRX Life , 3(2):023013, 2025. doi: 10.1103/PRXLife.3.023013. URL https://link.aps.org/ doi/10.1103/PRXLife.3.023013 . [61] J. Nam, B. M´ at´ e, A. P . T oshev, M. Kaniselv an, R. G´ omez-Bombarelli, R. T. Q. Chen, B. W o od, G.- H. Liu, and B. Kurt Miller. Enhancing diffusion-based sampling with molecular collective v ariables. arXiv:2510.11923 , 2025. [62] D. D. Richman, J. Karaguesian, C. M. Suomivuori, and R. O. Dror. Unlo c king hidden biomolecular conformational landscap es in diffusion models at inference time. , 2025. [63] H. Y. I. Lam, S. P . Ojeda, M. Brezino v a, J. Hanke, X. E. Ong, Y. Mu, and M. V en- druscolo. Metadiffusion: inference-time meta-energy biasing of biomolecular diffusion mo dels. https://doi.or g/10.64898/2026.02.10.704873 , 2026. [64] M. Arts, V. Garcia Satorras, C.-W. Huang, D. Z ¨ ugner, M. F ederici, C. Clementi, F. No´ e, R. Pinsler, and R. v an den Berg. Two for one: Diffusion mo dels and force fields for coarse-grained molecular dynamics. J. Chem. The ory Comput. , 19:6151–6159, 2023. [65] M. Plainer, H. W u, L. Klein, S. G ¨ unnemann, and F. No´ e. Consisten t sampling and simulation: Molecular dynamics with energy-based diffusion models. In A dv. Neur al Inf. Pr o c. Syst. , 2025. [66] P . Dhariwal and A. Nichol. Diffusion mo dels b eat gans on image synthesis. In A dv. Neur al Inf. Pr o c. Syst. , volume 34, pages 8780–8794, 2021. [67] J. Ho and T. Salimans. Classifier-free diffusion guidance. arXiv pr eprint arXiv:2207.12598 , 2022. [68] Y. Song, J. Sohl-Dickstein, D. P . Kingma, A. Kumar, S. Ermon, and B. P o ole. Score-based generativ e mo deling through sto c hastic differential equations. In H. Daum´ e II I and A. Singh, editors, ICML 37 , v olume 119 of Pr o c. Mach. L e arn. R es. , pages 8960–8970. PMLR, 2020. [69] R. M. Neal. Annealed imp ortance sampling. Statistics and Computing , 11:125–139, 2001. [70] P . Del Moral. F eynman-Kac F ormulae: Gene alo gic al and Inter acting Particle Systems with Applic a- tions . Springer, 2004. [71] A. Doucet and A. M. Johansen. A tutorial on particle filtering and smo othing: Fifteen y ears later. In The Oxfor d Handb o ok of Nonline ar Filtering . 2009. [72] R. Singhal, Z. Horvitz, R. T ee han, M. Ren, Z. Y u, K. McKeown, and R. Ranganath. A general framew ork for inference-time scaling and steering of diffusion models. arXiv pr eprint arXiv:2501.06848 , 2025. 20 [73] G. O. Rob erts and R. L. Tweedie. Exp onen tial conv ergence of Langevin distributions and their discrete appro ximations. Bernoul li , 2:341–363, 1996. [74] M. Skreta, T. Akhound-Sadegh, V. Ohanesian, R. Bondesan, A. Aspuru-Guzik, A. Doucet, R. Brek el- mans, A. T ong, and K. Neklyudov. F eynman-k ac correctors in diffusion: Annealing, guidance, and pro duct of exp erts. arXiv pr eprint arXiv:2503.02819 , 2025. [75] Y. Ren, W. Gao, L. Ying, G. M Rotsk off, and J. Han. Driftlite: Ligh t weigh t drift control for inference- time scaling of diffusion mo dels. arXiv pr eprint arXiv:2509.21655 , 2025. [76] L. Kish. Survey sampling . John Wiley & Sons, 1995. [77] A. Kong, P . McCullagh, X.-L. Meng, D. Nicolae, and Z. T an. A theory of statistical mo dels for monte carlo integration. J. R. Statist. So c. B , 65:585–618, 2003. [78] C. Bartels. Analyzing biased monte carlo and molecular dynamics simulations. Chem. Phys L ett. , 331: 446–454, 2000. [79] S. Kumar, D. Bouzida, R. H. Swendsen, P . A. Kollman, and J. M. Rosenberg. The w eighted histogram analysis method for free-energy calculations on biomolecules .1. the metho d. J. Comput. Chem. , 13: 1011 – 1021, 1992. [80] J. K¨ astner. Umbrella sampling. WIRE Comput. Mol. Sci. , 1:932–942, 2011. [81] P . V. Klimovic h, M. R. Shirts, and D. L. Mobley . Guidelines for the analysis of free energy calculations. J. Comput. A ide d Mol. Des. , 29:397–411, 2015. [82] J. D. Cho dera, P . J. Elms, W. C. Swope, J. H. Prinz, S. Marqusee, C. Bustamante, F. No´ e, and V. S. P ande. A robust approac h to estimating rates from time-correlation functions. http://arxiv.or g/abs/1108.2304 , 2011. [83] M. Shirts, K. Beauchamp, L. Naden, J. Cho dera, J. Rodr ´ ıguez-Guerra, S. Martiniani, C. Stern, M. Henry , J. F ass, R. Go wers, R. T. McGibbon, B. Dice, C. Jones, D. Dotson, and T. Burgin. c ho der- alab/p ymbar: 4.0.0, 2022. URL https://doi.org/10.5281/zenodo.6872022 . [84] E. H. Thiede, B. V an Koten, J. W eare, and A. R. Dinner. Eigenv ector method for um brella sampling enables error analysis. J. Chem. Phys. , 145:084115, 2016. [85] M. Das and R. V enk atramani. AutoSIM: Redesigning and automating umbrella sampling for biomolec- ular conformational transitions. J. Chem. Phys. , 163:014111, 2025. [86] M. Y ang, L. Y ang, Y. Gao, and H. Hu. Combine umbrella sampling with in tegrated tempering metho d for efficient and accurate calculation of free energy changes of complex energy surface. J. Chem. Phys. , 141:044108, 2014. [87] C. F. Sousa, R. A. Beck er, C.-M. Lehr, O. V. Kalinina, and J. S. Hub. Sim ulated temp ering-enhanced um brella sampling improv es con vergence of free energy calculations of drug membrane permeation. J. Chem. The ory Comput. , 19:1898–1907, 2023. [88] A. Laio and F. L. Gerv asio. Metadynamics: a metho d to simulate rare ev ents and reconstruct the free energy in biophysics, chemistry and material science. R ep. Pr o g. Phys. , 71:126601, 2008. [89] A. Barducci, M. Bonomi, and M. Parrinello. Metadynamics. WIREs Comput. Mol. Sci. , 1:826–843, 2011. doi: 10.1002/wcms.31. [90] A. Barducci, G. Bussi, and M. Parrinello. W ell-temp ered metadynamics: A smo othly conv erging and tunable free-energy metho d. Phys. R ev. L ett. , 100:020603, 2008. [91] J. F. Dama, M. Parrinello, and G. A. V oth. W ell-temp ered metadynamics con verges asymptotically . Phys. R ev. L ett. , 112:240602, 2014. 21 [92] P . Tiwary and M. Parrinello. A time-indep endent free energy estimator for metadynamics. J. Phys. Chem. B , 119:736–742, 2015. doi: 10.1021/jp504920s. [93] L. Onsager. Initial recom bination of ions. Phys. R ev. , 54:554+, 1938. [94] P . G. Bolh uis, C. Dellago, and D. Chandler. Reaction co ordinates of biomolecular isomerization. Pr o c. Natl. A c ad. Sci. USA , 97:5877–5882, 2000. [95] W. E and E. V anden-Eijnden. T ow ards a Theory of T ransition Paths. J. Stat. Phys. , 123:503–523, 2006. [96] B. Peters and B. L. T rout. Obtaining reaction coordinates by lik eliho o d maximization. J. Chem. Phys. , 125:054108, 2006. [97] Cheng Lu, Y uhao Zhou, F an Bao, Jianfei Chen, Chongxuan Li, and Jun Zh u. Dpm-solver++: F ast solv er for guided sampling of diffusion probabilistic mo dels. Machine Intel ligenc e R ese ar ch , 22(4): 730–751, 2025. [98] Rahul Nik am, A Kulandaisam y , K Harini, Divy a Sharma, and M Michael Gromiha. ProThermDB: Thermo dynamic database for proteins and m utants revisited after 15 y ears. Nucleic A cids R es. , 49: D420–D424, 2021. [99] L. Klein, A. Y. K. F o ong, T. E. Fjelde, B. Mlo dozeniec, M. Bro c kschmidt, S. Now ozin, F. No´ e, and R. T omiok a. Timew arp: T ransferable acceleration of molecular dynamics b y learning time-coarsened dynamics. arXiv pr eprint arXiv:2302.01170 , 2023. [100] M. Schreiner, O. Winther, and S. Olsson. Implicit transfer operator learning: Multiple time-resolution surrogates for molecular dynamics. arXiv pr eprint arXiv:2305.18046 , 2023. [101] B. Jing, H. St¨ ark, T. Jaakkola, and B. Berger. Generative modeling of molecular dynamics tra jectories. arXiv pr eprint arXiv:2409.17808 , 2024. [102] J. Viguera Diez, M. Schreiner, O. Engkvist, and S. Olsson. Boltzmann priors for implicit transfer op erators. arXiv pr eprint arXiv:2410.10605 , 2025. 22 A BioEm u F olding F ree Energies Details W e selected 26 proteins from the ProThermDB database, ranging from 76 to 372 residues. The steering p oten tial is a clamp ed linear function of the collectiv e v ariable, V = s · clamp(CV , c min , c max ), where s is a p er-system estimated optimal slop e, and c min / max are the clipping bounds to preven t the p otential going un b ounded. Within eac h run, the steered samples are generated in small independent batc hes with batc h size dep enden t of the protein size, allo wing us to po ol and subsample batches across runs to study conv ergence at v arying sample sizes. Specifically , we randomly draw (without replacemen t) subsets of batc hes from the com bined p ool and compute ∆ G for eac h draw, repeating 200 times p er sample size to obtain statistics. W e note the subsampling is performed on the batc h lev el b ecause differen t batches are indep endent, but not within each batc h b ecause samples in the same batc h hav e already b een resampled through the steering algorithm. F olding free energies are estimated from the fraction of nativ e con tacts (FNC) using a t wo-state mo del with inv erse Boltzmann reweigh ting to correct for the steering bias. W e exclude 8 of the 26 systems from the main analysis: 5 proteins (1QLP , 1CEC, 2LZM, 1RHD, 1RIL) whose RMSD ranges go beyond 2nm, leading to difficult steering and extreme rew eighting factors, and 3 proteins (1HK0, 1IOB, 1Y9O) for which the unbiased refere nce ∆ G is unreliable due to uncon verged sampling of the unfolded state. The remaining 18 systems (1A23, 1APC, 1BT A, 1EKG, 1H7M, 1HCD, 1IFB, 1IMQ, 1K85, 1STN, 1TEN, 1TTG, 1UBQ, 2ABD, 2KIU, 2RN2, 3D2A, 3N4Y) span predicted unbiased ∆ G v alues from 1.1 to 5.7 kcal/mol. Fig. 5 sho ws the catastrophic failure rate—defined as the probability that a random dra w of n particles con tains no unfolded sample (FNC < 0 . 5), as a function of sample size for each system. F or steered sam- pling, this rate drops to zero at mo dest sample sizes across nearly all systems, whereas unbiased sampling requires substan tially more particles, particularly for thermo dynamically stable proteins. Fig. 6 shows the corresp onding mean absolute error (MAE) of ∆ G relativ e to each metho d’s o wn con v erged estimate, as a function of sample size. Steered sampling achiev es sub-kcal/mol accuracy at n ≈ 100–1 , 000 dep ending on stabilit y . While unbiased sampling con verges fast on systems with relatively small ∆ G , the required num- b er of samples grow exp onentially with ∆ G . The steered sampling supersedes the unsteered sampling at ∆ G ≈ 3 . 2 k cal/mol, and the adv antage becomes more significant with larger ∆ G . 23 1 0 2 1 0 4 0.0 0.5 1.0 1IFB (r ef=-5.7) 1 0 2 1 0 4 0.0 0.5 1.0 2RN2 (r ef=-5.3) 1 0 2 1 0 4 0.0 0.5 1.0 1BT A (r ef=-5.3) 1 0 2 1 0 4 0.0 0.5 1.0 1UBQ (r ef=-4.6) 1 0 2 1 0 4 0.0 0.5 1.0 3N4Y (r ef=-4.4) 1 0 2 1 0 4 0.0 0.5 1.0 1H7M (r ef=-4.2) 1 0 2 1 0 4 0.0 0.5 1.0 1EKG (r ef=-4.2) 1 0 2 1 0 4 0.0 0.5 1.0 3D2A (r ef=-4.0) 1 0 2 1 0 4 0.0 0.5 1.0 1STN (r ef=-4.0) 1 0 2 1 0 4 0.0 0.5 1.0 1T TG (r ef=-3.8) 1 0 2 1 0 4 0.0 0.5 1.0 1A23 (r ef=-3.8) 1 0 2 1 0 4 0.0 0.5 1.0 2KIU (r ef=-3.2) 1 0 2 1 0 4 0.0 0.5 1.0 1TEN (r ef=-2.5) 1 0 2 1 0 4 0.0 0.5 1.0 1APC (r ef=-2.1) 1 0 2 1 0 4 0.0 0.5 1.0 1K85 (r ef=-2.0) 1 0 2 1 0 4 0.0 0.5 1.0 1IMQ (r ef=-1.8) 1 0 2 1 0 4 0.0 0.5 1.0 2ABD (r ef=-1.2) 1 0 2 1 0 4 0.0 0.5 1.0 1HCD (r ef=-1.1) Steer ed Unsteer ed Number of particles P(no unfolded) Figure 5: P er-system catastrophic failure rate as a function of sample size for 18 ProThermDB proteins, sorted b y reference ∆ G . Catastrophic failure is defined as the probabilit y that a random draw of n particles con tains no unfolded sample (FNC < 0 . 5). Blue: steered sampling; grey: unbiased sampling. Steered sampling eliminates catastrophic failures at small sample sizes across all systems, while unbiased sampling requires orders of magnitude more particles for very stable proteins. 1 0 2 1 0 4 0.0 0.5 1.0 1.5 1IFB (r ef=-5.7) 1 0 2 1 0 4 0 2 4 2RN2 (r ef=-5.3) 1 0 2 1 0 4 0 1 2 3 1BT A (r ef=-5.3) 1 0 2 1 0 4 0.0 2.5 5.0 7.5 1UBQ (r ef=-4.6) 1 0 2 1 0 4 0 1 2 3 3N4Y (r ef=-4.4) 1 0 2 1 0 4 0 2 4 6 1H7M (r ef=-4.2) 1 0 2 1 0 4 0 2 4 1EKG (r ef=-4.2) 1 0 2 1 0 4 0 2 4 3D2A (r ef=-4.0) 1 0 2 1 0 4 0 2 4 1STN (r ef=-4.0) 1 0 2 1 0 4 0 2 4 1T TG (r ef=-3.8) 1 0 2 1 0 4 0 2 4 1A23 (r ef=-3.8) 1 0 2 1 0 4 0 2 4 2KIU (r ef=-3.2) 1 0 2 1 0 4 0.0 2.5 5.0 7.5 1TEN (r ef=-2.5) 1 0 2 1 0 4 0 1 2 3 1APC (r ef=-2.1) 1 0 2 1 0 4 0 2 4 1K85 (r ef=-2.0) 1 0 2 1 0 4 0.0 0.5 1.0 1.5 1IMQ (r ef=-1.8) 1 0 2 1 0 4 0.0 0.5 1.0 2ABD (r ef=-1.2) 1 0 2 1 0 4 0.0 0.5 1.0 1HCD (r ef=-1.1) Steer ed Unsteer ed Number of particles MAE to self -r ef (kcal/mol) Figure 6: Per-system mean absolute error (MAE) of ∆ G as a function of sample size, with each method referenced to its own conv erged p ooled estimate. Proteins are sorted by reference ∆ G (most stable at top-left). Steered sampling (blue) conv erges substan tially faster than un biased sampling (grey), with the adv antage most pronounced for more stable systems. 24
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment