Toward computational cumulative biology by combining models of biological datasets
A main challenge of data-driven sciences is how to make maximal use of the progressively expanding databases of experimental datasets in order to keep research cumulative. We introduce the idea of a modeling-based dataset retrieval engine designed fo…
Authors: Ali Faisal, Jaakko Peltonen, Elisabeth Georgii
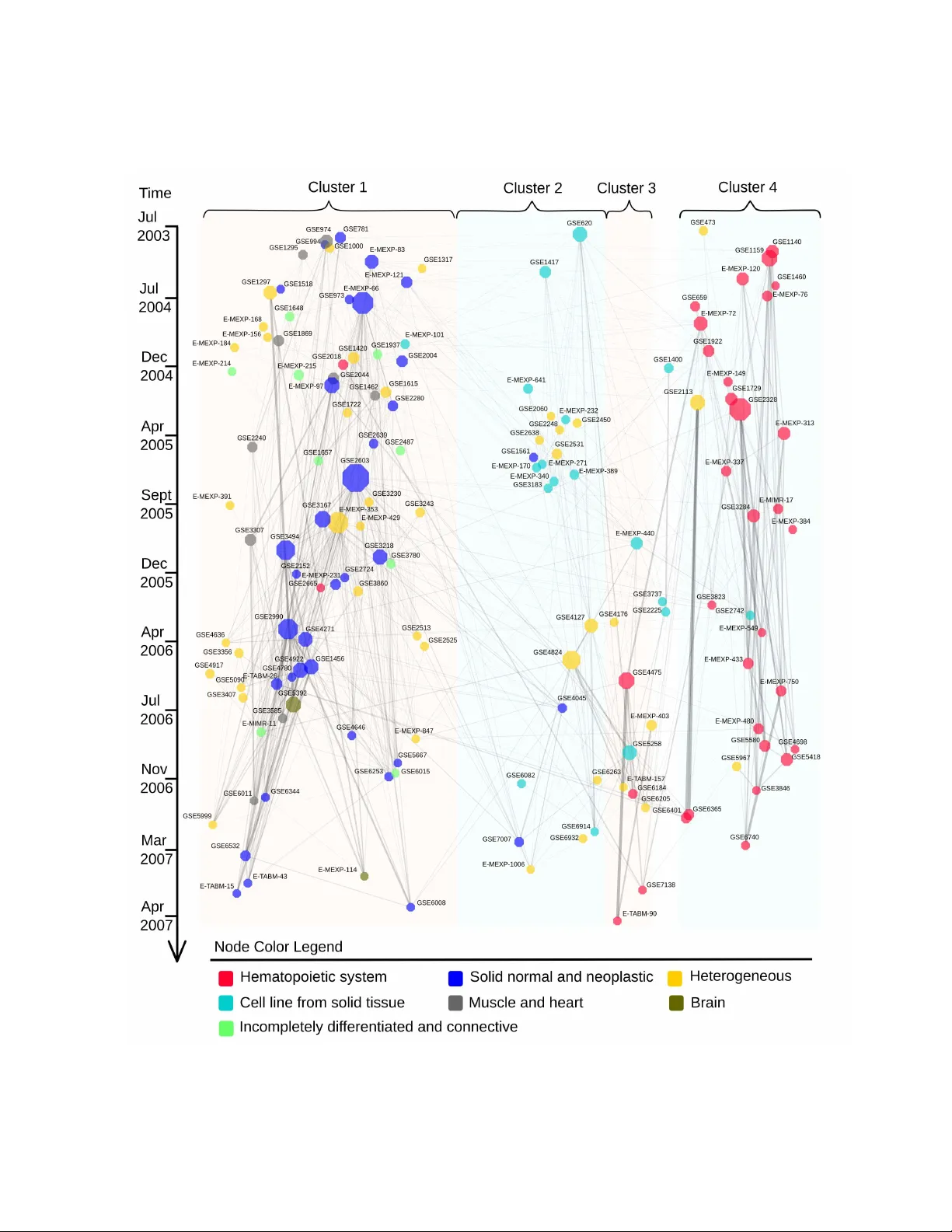
T o war d computational cum ulative biolo gy b y combinin g models of biolo gical datasets Ali Faisal ∗ , Jaak ko Peltonen ∗ , Elisab eth Geor gii ∗ , Johan Rung † and Samuel Kaski ∗ ‡ ∗ Helsinki Institute f or Inf ormation T echnology HIIT , Department of Inf ormation and Compu ter Science , PO Bo x 154 00, Aalto Unive rsity , 00076 Aalt o, Finland, † European Molecular Biology Laboratory , European Bioinf ormatics Institute (EMBL-EBI), Wellcome T rust Genome Campus Hinxton, CB10 1SD , UK, and ‡ Helsinki Institute f or Inf ormation T echnology HIIT , Department of Computer Science, PO Box 68, 00014 Univer sity of Helsinki, Finland A main challenge of data-driven science s is how to make maxi- mal use of the progressively expanding da tabases of experimental datasets in order to keep research cumulative. We introduce the idea of a modeling-b ased dataset retrieval en gine design ed for relating a researcher’ s experimental dataset to earlier work in the field. The searc h is (i) data-dr iven to en able new fi ndings, g oing bey ond the state of the ar t o f keyword searches in annotations, (ii) modeling-driven, to both i nclude biological knowledge and in- sights learn ed from data, and (iii) scalable, as it is accomp lished without building one unified grand model of all data. Assuming each dataset has b een modeled beforehan d, by the researchers or by datab ase ma nagers, we ap ply a rapidly computable and o pti- mizable combination model to decomp ose a n ew dataset into co n- tributions from earlier relev ant models. By using the d ata-driven decomposition w e identify a network of interr elated datasets from a larg e an notated human g ene expression atlas. While tissue type and disease were major d riving forces for de termining rele- vant datasets, the foun d relati onships were richer and the model- based search was more accurate than keyword search; it mo re- over recovered biologically meaningful relationsh ips that are no t straightforwardly vi sible from annotation s, for instance , between cells in d ifferent d evelopmental stages such as thymocytes and T -cells. Data-driven links and citations matched to a large extent; the data-driven links even uncovered co rrections to the publica- tion data, as two of the most li nked datasets wer e not highly cited and turned out to have w rong p ublication entries in the database . bioinf ormatics | gener ative mode ls | inf ormation retriev al | machine learn- ing Signific ance Measurement data sets of molecular biology and other experimental sciences are being collected comprehensi vely to openly accessible databases. W e demonstrate that it is no w possible to relate ne w re- sults to earlier science by searching wit h the actual data, instead of only in the textual annotations which are restricted to kno wn findings and are the current state of the art. In a gene exp ression database, the data-driv en relationships between datasets matched well with ci- tations between the correspon ding research papers, and e ven found mistakes in the databas e. Molecular biology , historically driv en by the pursuit of experi- mentally characterizing each component of the li ving cell, has been transformed into a data-driv en science [1, 2, 3, 4, 5, 6] with just as much importance given t o the computational and statisti cal analysis as to experimental design and assay technology . This has brought to the fore new computational challenges, such as the processing of massi ve new sequen cing data, and ne w statistical challenges arising from t he problem of ha ving relatively few ( n ) samples characterized for rel ativ ely many ( p ) variables—the “large p , small n ” problem. High t hroughp ut technologies often are de veloped t o assay man y pa r- allel variables for a single sample in a run, rather t han many parallel samples for a single variable, whereas the statistical powe r to infer properties of biolog ical conditions increases with l arger sample sizes. For cost reasons, most labs are restricted to generating datasets wit h the statistical power to detect only the strongest effects. In combina- tion w ith the penalties of multiple hypothesis testing, the limitati ons of “large p , small n ” datasets are ob vious. It is therefore not surpris- ing that much work has been de voted to add ress this problem. Some of the most successful methods r ely on increasing the ef- fectiv e number of samples by combining with data fr om other , sim- ilarly designed, experiments, in a large meta-analysis [7]. Unfortu- nately , t his is not straightforward either . A lthough public data repos- itories, such as the ones at NCBI i n the Unit ed States and the EBI i n Europe, serv e the research community with e ver-gro wi ng amo unts of experimen tal data, the y lar gely rely on annotation and meta-data pro- vided by the submitter . Database curators and semantic tools such as ontologies provide some help in harmonizing and standardizing the annotation, but the user who wan ts to find datasets t hat are com- binable with her own, most often must resort t o searches in free text or in controlled vocab ularies which would need much downstream curation and data analysis before any meta-analysis can be done [8]. Ideally , we would l ike to let the data speak for themselves. In- stead of searching for datasets that have been described simil arly , which may not correspond to a statistical similarity in the datasets themselves, we would like to conduct that search in a data-driv en way , using the dataset itself as t he query , or a statistical (rather than a semantic) description of it. T his is implicitly done for example in multi-task learning, a method from the machine learning field [9, 1 0], where sev eral related estimation task s are pursued together , assuming shared pro perties acro ss tasks. Multi-task learning is a form of global analysis, which builds a si ngle unified model of the datasets. But as the number of datasets keep s increasing and the amount of quanti- tativ e biological kno wledge keeps accumulating, the complex ity of building an accurate unified model beco mes increasingly prohibiti ve. Addressing the “large p , small n ” problem requires taking into account both the uncertainty in the data and the existing biological kno wl edge. W e no w consider t he hypothesized scena rio where future researchers i ncreasingly dev elop hypothe ses in terms of (probabilis- tic) models of their data. Although far from realistic today , a similar trend exists for sequence motif data, which are often published as Hidden Marko v models, for instance in the Pfam database [11]. In this paper we report on a feasibility study to wards the scenario where a l arge number of experiments have been modeled beforehand, potentially by the resea rcher generating the data or the database stor- ing the model together with the data. W e ask what could be done with these models towar ds cumulatively building knowledge from data i n molecular biology . Spea king about models generally and assum- ing the many practical issues can be solved technically , we arrive at our answer: a modeling-driven dataset re trieval engine , which a researcher can use for positioning her o wn measurement data into the context of the earlier biology . The engine will point out relation- ships between experiments in the form of the retriev al results, which is a naturally understandable interface. The retriev al will be based on data i nstead of the state of the art of using keyw ords and ontolo- gies, which will make unexpec ted and previously unkno wn findings possible. The r etriev al wil l use the models of the datasets which, by our assumption abov e, incorporate what the researchers producing the data thought was important, but the retriev al will be designed to be more scalable than building one unified grand model of all data. This also implies that the way the models are utilized needs to be approximate. Compared to existing data-driv en retriev al methods 1 – 10 [5, 3 ], whole datasets, incorporating the experimental designs, will be matched instead of individual observations. The r emaining ques- tion is how to design the retriev al so that it b oth re veals t he interesting and important relationships and is fast to compu te. The model we present is a first step to wards this goal. W e assu me a new dataset can be ex plained by a combination of the models for the earlier datasets and a nov elt y term. This is a mixture modeling or regression task, in which the weights can be computed rapidly; the resulting method scales well to large numbers of datasets, and the speed of the mixture modeling does not depend on the sizes of the earlier datasets. T he large st weights in the mixture model point at the most rel e v ant earlier datasets. The method is applicable t o se veral types of measurement datasets, assuming s uitable models e x- ist. Unlike traditional mixture modeling we do not li mit the form of the mi xture components, t hus we bring in the knowledg e built into the stored models of each dataset. W e apply this approach to a large set of experiments from EBI’ s ArrayExpress gene expres- sion database [12], treating each e xperiment in turn as a ne w dataset, queried against all earlier datasets. Under our assumptions the r e- triev al results can be i nterpreted as studies that the authors of the study generating the query set could have cited, and we sho w t hat the actual cit ations ove rlap with the retriev al results. T he discov ered links between datasets additionally enable forming a “hall of fame” of gene expression studies, containing the studies that would have been i nfluential assuming the retriev al system would hav e existed. The links in the “hall of fame” v erify and complement the citation links: in our study they revealed corrections to the citation data, as two frequently retriev ed studies were not highly cit ed and turned out to have erroneous publication entries in the database. W e provide an online resource for exploring and searching t his “hall of fame”: http://res earch.ics.a alto.fi/mi/setretrieval . Earlier work on relating datasets has provided partial solutions along this line, with the major limitation of being r estricted to pair- wise dataset comparisons, in contrast to the proposed approach o f de- composing a dataset into contributions from a set of earlier datasets. Russ and Futschik [13] represented each datase t by pairwise correla- tions of genes, and used them t o compute dataset similarities. This dataset representation is ill-suited for typical functional genomics ex- periments as a large number of samples is required to sensibly esti- mate gene co rrelation matrices. In addition, it makes the dataset com- parison computationally expensi ve, as the representation is bulkier than the original dataset. In oth er w orks s pecific case-control designs [14] or kno wn biological processes [15] are assumed; we generalize to decompositions ov er arbitrary models. Combination of stored models for dataset retriev al Our goal is to i nfer data-driv en relati onships between a new “query” dataset q and earlier datasets. The query is a dataset of N q samples { x q i } N q i = 1 ; in the Arr ayExpress study the samples are gene ex pression profiles, with the element x q i j being expressio n of the gene set j in the sample i of the query q , but the setup is gen eral and applicable to other experimental data as well. Assume further a dataset repository of N S earlier datasets, and assume that each dataset s j , j = 1 , . . . , N S , has already been modeled w ith a model denoted by M s j , later called a base model. The base models are assumed t o be probabilistic generati ve models, i.e. , principled data descriptions capturing prior kno wl edge and data-driven discov eries under specific distributional assumptions. Base models for different datasets may come fr om dif- ferent model families, as chosen by the researchers who authored each dataset. In this paper we use two types of base models, which are discrete varian ts of principal component analysis ( Results ), but any pro babilistic generativ e models can be applied. Assume tentative ly that t he dataset repository contains a library of “base experiments”, carefully selected to induce all important kno wn biological effects with suitable design factors. In the special examp le case of metagenomics with known constituent organisms, an obvio us set of base experimen ts wo uld b e the set of genomes of those organ isms [16]. A ne w experimen t could then be expressed as a com- bination of the base experiments, and potential nov el effe cts. More generally , for instance in a broad gene expression atlas, it would be hard if not impossible to settle on a clean, well-defined and up-to- date base set of experiments to correspon d to each known ef fect, so we choose to use the compr ehensive collection of experiments in the curr ent databases as the base e xperiments . The problem setting then changes, f rom searching for a unique explanation of the ne w exp eri- ment, to the do wn-to-earth and realistic task of data-driv en retriev al of a set of relev ant earlier experimen ts, rele v ant in the sense of having induced one or more of the known or as-of-yet unkno wn biological effe cts. W e combine t he earlier datasets by a method that is probabilis- tic but si mple and fast. W e build a combination model for the query dataset as a mixture model of base distrib utions p ( x | M s j ) , which hav e been estimated beforehand. In our scenario, generativ e models M s j are av ailable in the repository a long wi th datasets s j ; note that the M s j need not a ll ha ve the same form. In th e mixture mod el parameterized by Θ q = { θ q j } N S + 1 j = 1 , the likelihood of observ ing the query is p ( { x q i } N q i = 1 ; Θ q ) = N q ∏ i = 1 h N S ∑ j = 1 θ q j p ( x q i | M s j ) + θ q N S + 1 p ( x q i | ψ ) i [ 1 ] where θ q j is the mixture proportion or weight of the j th base distri - bution ( model of dataset s j ) and θ q N S + 1 is the weight for the nov- elty term. The nov elty is modeled by a “background model” ψ , a broad nonspecific distribution covering overall gene-set activity across the whole dataset repository . All weights are non-ne gativ e and ∑ N S + 1 j = 1 θ q j = 1. In essence, this representation assumes that bio- logical acti vity in the query dataset can be approximately ex plained as a combination of earlier datasets and a nov elty term. The remaining task is t o i nfer t he combination mod el Θ q for each query q given the kno wn models M s j of datasets in the repository . W e infer a maximum a posteriori (MAP) estimate of the weights Θ q = { θ q j } N S + 1 j = 1 . Alternativ ely we could sample over the posterior , but MAP inference already yielded good results. W e optimize the combination weights to maximize their (log) posterior probability log p ( { θ q j }|{ x q i } , { M s j } ) ∝ log p ( { x q i }|{ M s j } , { θ q j } ) + log p ( { θ q j } ) ∝ ∑ i log h N S ∑ j = 1 θ q j p ( x q i | M s j ) + θ q N S + 1 p ( x q i | ψ ) i − λ N S + 1 ∑ j = 1 θ q j 2 [ 2 ] where p ( { θ q j } ) = N ( 0 , λ − 1 I ) is a naturally non-sparse L 2 prior f or the weights with a regu larization term λ . The cost function [ 2 ] is strictly conca ve ( SI T ext ), and standard c onstrained con vex optimiza- tion techniques can be used to find the optimized weights. Algo- rithmic details for t he F rank-W olfe algorithm and a proof of con ver- gence are provided in SI T ext . A fter compu ting the MAP estimate, we rank t he datasets for retrie val according to decreasing combina- tion weights. This modeling-dri ven approach has se veral advantages: 1) the approximations become more accurate as more datasets are submit- ted to the repository , increasing naturally t he number of base distribu- tions; 2) it is fast, since only the models of the datasets are needed, no t the large d atasets themselv es; 3) any m odel types can be included, as long as likelihoods of an observed sample can be co mputed, hence all expert kn owled ge built into the models in the repository can b e used; 4) relev ant datasets are not assumed to be “similar” to t he query i n any na¨ ıve sense, they only need to explain a part of the query set; 5) the relev ance scores of datasets hav e a natural quantitative meaning as weights in the probabilistic combination model. Scalability . As the size of repositories such as ArrayExpress doubles e very two years or ev en faster [17], fast computation with respect 2 0.0 0.2 0.4 0.6 0.8 1.0 0.0 0.2 0.4 0.6 0.8 1.0 Recall Precision Combination model Random K e yw ord: 96 classes K e yw ord: disease Fig. 1. Data-driven retrieval outperfo rms the state of the ar t of ke yword search on the human gen e e xpression atlas [12]. Blue: T radition al precision-r ecall curve where progr essively more d atasets are retrieve d from left to r ight. A ll e xperiments sharing one or more of the 96 biological categories of the atlas were considered rele vant . In ke yword retrie val, either the categor y nam es (“K eywo rd: 96 classes”) or the disease annotation s (“K eywo rd: disease”) were used as keyw ords. All datasets having at least ten samples were used as query datasets , and the curves are ave rages ov er all queries. to the number N S of background datasets is crucial for future-proof search methods. Already the first method abo ve ha s a fast linear com- putation time in N S ( SI T ext ), and an approximate variant can be run in sublinear time. For that, t he model combination will be optimized only ov er t he k background datasets most similar to the query , which can be found in time O ( N 1 / ( 1 + ε ) S ) where ε ≥ 0 is an approximation parameter [18], by suitable hashing functions. Results Data-driven retrieval of experiments is more accurate than stan- dard keywor d search. W e benchmarked the combin ation mod el against state-of-the-art dataset retriev al by keyw ord search, in the scenario where a user queries with a ne w dataset against a database of earlier released datasets represented by models. The data were from a large human g ene e xpression atlas [12], containing 206 public datasets with 5372 samples in total that hav e been systematically an- notated and con sistently normalized. T o mak e use of prior b iological kno wl edge, we preprocessed the data by gene set enrichment anal- ysis [19], representing each sample by an integer vector telling for each gene set the number of leading edge activ e genes [20] ( Meth- ods ). As base models we used t wo model types pre viously applied in gene expression analysis [6, 20 , 21, 3 ]: a discrete principal com- ponent analysis method called L atent Di richlet Allocation [22, 23], and a simpler variant called mixture of unigrams [24] ( SI T ext ). Of the tw o t ypes, for each dataset we chose the model yielding the larger predicti ve likelihood ( SI T ext ). For each query ( q ), the earlier da tasets ( s j ) were ranked in descending order of the combination proportion ( θ q j ; estimated f rom Eq. [ 2 ] ). That is, base models which explained a larger proportion of the gene set activity in the query were rank ed higher . The approach yields good retri e va l: the retr iev al result was consistently better than with key word searches applied to the tit les and textual descriptions of the datasets (Fig. 1), which is a st andard approach for dataset retriev al from repositories [25]. W e checked that the result is not o nly du e to laboratory effects by discarding, i n a follow-u p st udy , all retri e ved results from the same laboratory . The mean average precision decreased slightly (f rom 0 . 44 to 0 . 42; precision-recall curve in Fig. S2) but still supports the same conclusion. Network of comp utationally recommend ed dataset conn ections reveals b iological relationships. When each dataset in turn is used as a q uery , the estimated combina tion weights form a “rele vance ne t- work” between datasets (Fig. 2 left), where each dataset is linked to the relev ant earlier datasets (for details, see Methods ; a larger fi gure in F ig. S5; and an interactiv e searchable version in the online r e- source). The network structure is dominated but not fully exp lained by the tissue type. Normal an d neoplastic solid tissues (cluster 1) are clearly separate from cell li nes (cluster 2) and from hematopoietic tissue (clus ter 4); the same main cl usters were observ ed in [12]. Note that the model has not seen the t issue types but has found them fr om the data. In closer inspection of the clusters, some finer structure is e vident. The m uscle an d heart datasets (g ray) form an interco nnected subnetwo rk in the left edge of the image: nodes near the bottom of the image ( do wnstream) are explained by earlier (upstream) nodes, which in turn are explained by nodes even f urther upstream. As an- other example, in cluster 4 myeloma and leuke mia datasets are con- centrated on the left side of the cluster , whereas the right side mostly contains normal or infected mononuclear cells. There is a substantial number of li nks both across clusters and across tissue categories. Among the top thirty cross-cate gory links, 25 in volve heterogeneous datasets containing samples from dive rse tissue o rigins. The strongest link connects GSE6365, a stud y o n mu l- tile myelom a, with GSE2113, a larger study from the same lab which largely includes the GSE6365 samples. T he dataset E-MEXP-66 is a hub connected to all the clusters and to nodes in its own cluster having different tissue labels. It contains samples studying K aposi sarcoma, and includes control samples from skin endothelial cells from blood vessels and t he lymph system. Blood vessels and cells belonging to the lymph system are ex pected to be present in almost any solid tissue biopsy as well as in samples based on blood sam- ples. The st rongest link betwee n tw o homogeneo us datasets of dif f er- ent tissue types connects GSE3307 (which co mpares skeletal muscle samples from healthy individu als wit h 12 groups of patients affected by various muscle diseases) to GS E5392, which measures tr anscrip- tome profiles of normal brain and brain bipolar disorder . Interest- ingly , shortening of telomeres has been associated both with bipolar disorder [28] and muscular disorder [26]. Treatment of bipolar dis- order has been found to also slow down the onset of skeletal muscle disorder [27]. Next we in vestigated “outlier” datasets where the tissue type does not match the main tissue types of a cluster, implying that t hey might revea l commonalities between cellular conditions across tis- sues. Cluster 1 contained three outlier datasets: t wo hematopoietic datasets and one cell line dataset. The two hematopoietic outlier datasets are studies related to macrophages and are both strongly connected t o GSE2004, which contains samples from kidne y , liv er , and spleen, sites of l ong-li ved macrophages. The first hematopoi- etic outlier , GSE2018 studies broncho alveolar lav age cells from lung transplant receipts; the majority of these cells are macrophages. The dataset has strong links to solid ti ssue datasets including GSE2004, and the diverse dataset E-MEXP -66. The second hematopoietic out- lier , GSE2665, is also strongly connected to GSE2004 and measures express ion of lymphatic organs (sentinel lymph node) that contain sinusoidal macrophage s and sinusoidal endothe lial cell s. The third outlier , E-MEXP-101, studies a colon carcinoma cell line and has connections to other cancer datasets in cluster 1. T op dataset links overlap well with citation graph . W e compared the model-dri ven network to the actual citation links (Fig. 2, right) to find out to what extent the c itation practice in t he research community matches t he data-driv en relationships. Of the top two hundred data- driv en edges, 50% overlapp ed with direct or indirect citation links (see Methods and SI T ext ). Most of the direct citations appear wi thin 3 Fig. 2. Relev ance network of datasets in the human gene expr ession atlas; data-drive n links from the model (left) and citation links (right). Left: each dataset was used as a query to retriev e earlier datasets; a link from an earlier dataset to a later one means the earlier dataset is releva nt as a par tial model of activity in the later dataset. Link width is propo r tional to the normalized relev ance weight (combination weight θ q j ; only links with θ q j ≥ 0 . 025 are shown, and dataset s without links have been discarded) . Right: links are direct (gr ay) and i ndirect (purple) citat ions. Node size is prop ortional to the estimated influe nce, i.e . , the to tal o utgoing w eight. Colors: tissue types (six meta tissue types [12]). The node la yout was comp uted from the data-driven netwo rk (details in Methods ). the four tissue clusters (Fig. 2 , right). The two cro ss-cluster citations are not due to biological similarity of the datasets. The publication for GSE 1869 cites the publication for GSE1159 regarding the method of d ifferential express ion detection. The GSE7007, a study on Ewing sarcoma samples, cites the study on human mesenchymal stem cells (E-MEXP-168) for stating that the ov erall gene expressio n profiles differ be tween those samples. W e additionally compared the densely connected sets of ex- periments between the two networks. In the cit ation graph the breast cancer datasets GS E2603, GSE3494, GSE2990, GS E4922, and GSE1456 form an interconnected clique in cluster 1, while the three leukocyte datasets GSE2328, GSE 3284, and GSE5580 form an interconnected module in cluster 4. In the rel e va nce network the corresponding edges for both cliques are among the str ongest links for those datasets, and some of them are among the top 20 strongest edges in the network (see SI T ext for t he list of top 20 edges). There are also densely connected modules in t he relev ance network t hat are not strongly connected in the citation graph; when we systematically sought cliques associated to each of the top 20 edges, the most strong edges constitute a clique among E-MEXP- 750, GSE6740 and GSE473, all three studying CD4+ T helper cells which are an essential part of the human i mmune system. Another interesting set is among three T -cell relat ed datasets i n cluster 3. T wo of the datasets contain T lymphoblastic leuk emia samples (E- MEXP-313 and E-MEXP -549), whereas E -MEXP-337 reports thy- mocyte profiles. Thymocytes are dev el oping T lymphocytes that are matured in thymus, so this con nection is biologically meaningful but not straightforward to find f rom dataset annotations. O ther strongly connected cliques are discussed in the SI T ext . Analysis of n etwork hu bs discovers d atasets deser ving more ci- tations. Datasets that have high weights in explaining other datasets hav e a large weighted outdegree in the data-driven relev ance net- work, and are expected to be useful for many other studies. W e check ed wh ether the publications corresponding to these central hubs are highly cited in the research community . There is a lo w but sta- tistically significant correlation between the weighted outdegree of datasets and their citation counts (Fig. 3; Spe arman ρ ( 169 ) = 0 . 2656, p < 0 . 001). Both quantities were normalized t o avoid bias due to differe nt release ti mes of the datasets ( Methods ). W e further exam- ined whether the prestige of the publication venue (measured by im- pact factor) and the senior author (h-index of the last author) biased the cit ation counts, which could explain t he low correlation between the outdegree and the citation count, and the answer was affirmati ve ( Methods ). W e i nspected more closely the datasets where the recommended or the actual citati on counts were high (Fig. 3): (A) datasets having lo w citation counts but high ou tdegrees, (B) both high citation coun ts and high outde grees and (C) high citation coun ts but lo w outdegrees. W e manually checked the publication records of region A in Gene Expression Omnibus (GEO) [33 ] and ArrayExpress [17], to fi nd out why the datasets ha d low citation counts despite their high outde gree (data-dri ven citation recommendation s). T wo of the eight datasets had an inconsistent publication record. The blue arrows in Fig. 3 point from their original position to the corrected position confirmed by GEO and ArrayExpress. Thus the data-driven network rev ealed the inconsistency , and the new positions, corresponding to higher ci- tation coun ts, v alidate the model-based finding that these datasets are good explainers for other datasets. In region B, most of the papers hav e been published in high impact journals and hav e relativ ely high number of samples (average sample size of 154) compared to region 4 A (average sample size of 75). One of the eight datasets in the col- lection is the well known Connecti vity Map experiment (GSE5258). Lastly the set C mostly contains unique targeted studies; there are fiv e studies in the set, which are about leuk ocytes of injured patients, Polycomb group (PcG) proteins, senescence, Alzheimer’ s disease, and effect of cAMP agonist forskolin, a traditional Indian medicine. The studies have been published in high impact forums, and a possi- ble reason of their low outdegree is t heir specific cellular responses, which are not very common in the atlas. Discuss ion Our main goal was to test the feasibility of the scenario where re- searchers l et the data speak for themselves when relating new re- search to earlier studies. The conclusion is positive: even a rela- tiv ely strai ghtforward and scalable mixture mo deling a pproach fo und both expected relationships such as tissue types, and relationships not easily found with ke yword searches, including cells in diffe rent de- velopm ental st ages or treatments resembling conditions in other cell types. While biologists could fi nd such connections by bringing ex- pert kno w ledge into keyw ord searches, the ultimate advantag e of the data-dri ven approach is that it also yields connections beyond cur- rent kno wledge, giving rise to new hypotheses and follo w-up stud- ies. For examp le, i t seems surprising that the skeletal muscle dataset GSE6011 is linked also to kidne y and brain datasets. Closer inspec- tion yielded possible partial e xplanations. Some kidn ey areas are rich in blood vessels, lined by smooth muscle. S tudies have sho wn com- mon gene signatures between skeletal muscle and brain. Abnormal express ion of the protein d ystrophin leads to Duchenne muscular dys- trophy , exhibited by a majority of samples in GSE6011; the brain is another major exp ression site for dystrophin [30]. Interestingly the top three potentially novel datasets, where only less than 50% of the express ion pattern is modelled by earlier datasets ( i.e. θ q N S + 1 > 0 . 5), are GSE260 3 (a central breast cancer set), the Conn ectivity Map data (GSE5258) and the Burkitt’ s L ymphoma set (GS E4475; a cancer fun- damentally disti nct from other types of lymphoma). The first two are also recov ered by the citation data (have relati vely high citation counts and appear in region B in Fig. 3), unlike the third (which is part of region A in Fig. 3 ). Our case study focused on global analysis of the relev ance net- work obtained for a representativ e dataset collection, allowing for Fig. 3. Data-driven pr ediction of usefulne ss of datasets vs. their citation coun ts. Manu al checks comparing sets for which the two scor es differ ed rev ealed incon- sistent database records for two datasets; the blue ar rows point to their corrected locations , which are more in li ne with the data-driven model. Regions A, B, and C: see te xt. comparisons with the citation graph. The data-driv en relationships corresponde d to actual citations when available, but were ri cher and were able to spot out errors in citation links. Anothe r intended use of the retrie val method is to support researchers in fi nding relev ant data on a particular topic of interest. W e performed a study to obtain insights into rel ationships among skeletal muscle datasets as well as between skeletal muscle and other datasets, and showed that the re- triev al metho d lessens the need for laborious manual searches ( SI T ext and Fig. S4). In this work we made simplifying assumptions: we only em- ployed two model families, included biological knowledg e only as pre-chosen gene sets, and assumed all new experiments to be mix- tures of earlier ones, instead of sums of effects in them. W e ex- pect results to impro ve considerably wi th more adv anced future al- ternativ es, with t he research challenge being to maintain scalability . Generalizability of the search acro ss measurement batches, laborato- ries, and measureme nt platforms is a ch allenge. Our feasibility study sho wed that f or carefully preprocessed d atasets (of the microarray at- las [12]), data-driv en retriev al is useful e ven across laborato ries. Our method is generally applicable to any single platform, and takes into account the expert kno wledge built into models of datasets for that platform; abstraction-based data representations, such as the gene set enrichment representation we used, have potential to facilitate cross-platform analysis. As data integration approaches dev elop fur- ther [34, 35], i t may be possible to do searches even across different omics types; here, i ntegration of meta data (pioneered in a specific semi-supervised framewo rk [36]), several ontologies ( MGED ontol- ogy , experimental factor ontology and ontology of biomedical in ves- tigations [37]) and text mining results [38, 39] are obviously useful first steps. Materials and Methods Gene expression data. We used the human gene expressio n atlas [12] avail- able at Arra yE xpress under accession num ber E-MT A B-62. The data were pre- processed by gene set enrichment ana lysis (GSEA) using the canonical path wa y collection (C2-CP) from the Molecular Signatures Database [19]. Each sample was repre sented by its top enriched gene sets [20] ( SI T ext ). Node layout and n ormalized r elev an ce weight. The weight matrix contains a weight vector for each query dataset, encoding the amount of variation in that query e xplained by each earli er dataset. A s query datasets from early year s hav e on ly fe w ev en e arlier sets a vailab le, there is a bias to wards the edg es being stronger for the datasets from earl y years . T o remove the bias we normalized, f or the visualizations, the edge strengths of each query data set by the numb er of earlier datasets . T o visualize the relationship network ov er time in F ig. 2, we needed a layou t algorithm that positions the datasets on the horizontal axis highlighting structure and avoiding tangling. We used a cluster-emph asizing Sammon’ s mapping [40]; Sammon’ s mapping is a nonlinear projection method or Multidime nsional Scaling algorithm which aims at preserving the i nterpoint distances (here 1 − θ q j ). By clustering the network (with unsupervised Markov clustering [41]) and increasing between-cluster distances by adding a constant ( c = 1 ) to them, the mapping was made to emphasize clusters and hence un- tangle the lay out. Citation g raph. Direct citations between dataset-link ed publicatio ns were e x- tracte d from the Web of S cience (26 Jul 2012) and PubMed (17 Oct 2012). We additionally considered two types of indirect edges. F irstly , we introduced l inks betwee n datasets w hose pub lications share common refer ences. This covers f or instance related datasets whose pub lications appeared close in time, making direct citation un likely . A natural measure of e dge strength is giv en by the n umber of shared ref erences. Secondly , we connect datasets whose articles are cited together , because co-citation is a sign that the community perceives the ar ticles as related. Here, the edge strength was taken to be the number of articles co- citing the tw o datase t publications; these edg es dominate the indirect links in the citation g raph . F or this an alysis we used cit ation data, a vailable for 17 1 datasets and provided by Thom son Reuters as of 13 September 2012. Normalization o f citation cou nts and weighted o utdegrees. As early datasets ha ve man y more papers which can cite them, and many more later datasets which they can help model, both the citation counts and estimated 5 weighte d outdeg rees are exp ected to be upwar ds biased for them. For Fig. 3 we normalized the quantities; for each dataset we normalized the outdegr ee by the number of newe r datasets , and the citation count by the time diff erence be- tween publishing the data and the newest dataset in the atlas. T o make sure the normalization did not introduce side effe cts we additionally check ed that the same conclusions were reached without the citation count normalization (Fig. S1; plotted as str atified subfigures f or each 1-ye ar time window) . The citation counts were e xtracte d from PubMed on 16 May 201 2. Citation counts are strongly influenced by external esteem of the pu blica- tion forum and the senior author. We stratified the data sets according to the numbers of data-driven citation recommendat ions, and studied whether the impact facto r of the forum or the h-inde x of the last author were predictiv e of the actual citation count in each stratum. The strata were the top and bottom quartiles, and f or each we compared the top and bottom quartiles of the actual citation counts (resulting in comparing the fou r corners of Fig. 3). For low out- degr ee (low recomme nded citation count), the h-inde x was lower fo r less cited datasets ( t 11 = 2 . 78 , p = 0 . 0086 ; mean value 24 . 20 vs 54 . 62 ) , and also the impact factor was low er ( t 7 = 2 . 6 , p = 0 . 016 ; mean value 4 . 38 vs 21 . 13 ). Similarly , for high recom mended citation count th e impact f actor f or the little-cited datasets was lower ( t 19 = 3 . 99 , p = 4 . 0 − 4 ; mean value 6 . 45 vs 2 1 . 91 ), wh ile the diff erence in h-inde x was not significant. All t statistics and p-va lues wer e computed by one-sided independent sample Welch’ s t-tests. The h-indices and impact factors were collected from Thomson Reuters Web of Knowledg e and Journal Citation Reports 2011 respective ly on 23rd July 2012 . ACKNO WLEDGMENTS. We thank Matti Nelimarkka and T uukka Ruotsalo for helping with citation data. Certain data included herein are derived from the f ollowing indices: Science Ci tation Inde x Expanded, Social Science Citation In- de x and Arts & Humanities Citation Inde x, prepared by Thomson Reuters R , Philadelphia, P ennsylvania, USA, c Cop yright Thomson Reu ters R , 201 1. This work was financially supported by the Academy of Finland (Finnish Centre of Excellence in Computationa l Inferen ce Research COIN, gra nt no 251170). 1. Greene C-S, T ro yanskaya O-G (2011) PILGRM: An interactive data-driven discovery platform fo r expert biologists. Nucleic Acids Res 39: W368–74. 2. T a nay A, Ste infeld I, Ku piec M, Shamir R (2005) Integrative anal ysis of genome-wide experiments in the context o f a large high-thro ughput data compendium. Mol Syst Biol 1:e1–10 . 3. Caldas J, et a l. (201 2) Data-driven information retriev al in heterog eneous collections of transcriptomics dat a links SIM2s to malignant pleural mesothelioma. Bioinfor- matics 28:i246–i2 53. 4. Adler P , et al. (2 009) Mining for coexpression a cross hundreds of dataset s using novel rank aggreg ation and visualizati on m ethods. Genome Biol 10:R139 . 5. Schmid P-R, Palmer N-P , K ohane I-S, Ber ger B (2012) Making sense out o f massive data by going beyond differential expression. Proc Natl Acad Sci U S A 109: 5594– 5599. 6. Gerber G -K, Dowell R-D, Jaakkola T -S, Gifford D- K (2007) A utomated discovery of functional generality of human gene expression programs. PLoS Comput. Biol 3:e148. 7. Tseng G-C, Ghosh D, Feingold E (2012) Comprehensive literature review and statis- tical considerations fo r micro array meta-analysis. Nu cleic Acids Res 40 :3785–3 799. 8. Rung J, Brazma A (2012) Reuse of public genome-wide gene expression data. Nat Rev Genet 14:89–99. 9. Bax ter J (1997) A Bayes ian/information theoretic model of le arning t o learn via multiple task sampling. Machine Learning 28:7–39 . 10. Ca ruana R (1997) Multitask learning. Machine Learning 28:41–75. 11. Punta M, et al. ( 2012) The Pfam pro tein families database. Nuc leic Ac ids Re s 40:D290– D301. 12. Lukk M, et al. (20 10) A global map of human gene expression. Nat Bi otechnol 28:322– 324. 13. Russ J, Futschik M-E (2010) Comparison and consolidation o f micr oarray data sets of human tissue expression. BMC Genomics 11:30 5. 14. Suthram S, et al. (201 0) Network-based e lucidation of human disease si milarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput Biol 6:e1000 662. 15. Hutt enhower C, T roy anskaya O-G (200 8) Assess ing the functional s tructure of ge- nomic data. Bioinformatics 24:i330– 8. 16. Meinicke P , Asshauer K-P , Lingner T (2011) Mixture models for analysis of the taxonomic composition of metagenomes. Bioinformatics 27:1618 –24. 17. Parkinson H, et al. (2009) ArrayExpress update–f rom an archive of functional ge- nomics e xperiments t o the atlas of gene expression. Nucleic Aci ds Res 37:D86 8–72. 18. Gi onis A, Indyk P , Motwani R (1999) Similarit y search in high dimensions v ia hashing. Proceedings of the T wenty -Fifth Inte rnational Conference on Very Large Databases eds Atk inson M, Orlowwska M, V a lduriez P , Zdonik S, Brodie M ( Edin- bur gh, Scotland) pp 518–5 29. 19. Subramanian A, et al. (2005) Gene set enrichment analysis: A knowledge-based appro ach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:155 45–1555 0. 20. Ca ldas J, Gehlenborg N, Faisal A, Brazma A, Kaski S (2009 ) Probabilistic retrieval and vi sualization of biologically relevant microarra y experiments. Bioinform atics 25:i145 –i153. 21. Engreitz J- M, et al . (2010) Content-based microarray se arch using differential ex- pression profi les. BMC Bioinformatics 11:603 . 22. Pritc hard J-K, Ste phens M, Donnelly P (2000) Inference of popu lation structure using multilocus genotype data. Genetic s 155:945–959. 23. Ble i D-M, Ng A-Y , Jordan M-I, Laffe rty J ( 2003) Late nt Dirichlet allocation. J Mach Learn Res 3:993– 1022. 24. Nigam K, McCallum A, Thrun S, Mitchell T (2000) T ext classification from labeled and unlabeled documents using EM. Machine Learning 39: 103–134 . 25. Zhu Y , Davis S, Stephens R, Meltzer P- S, Chen Y (2008 ) GEOmetadb: power- ful alte rnative se arch engine for the Gene Expression O mnibu s. Bioinformatics 24:2798 –2800. 26. Mourkioti F , et al. (2013) Role of t elomere d ysfunction in cardiac fa ilure in Duchenne muscular d ystroph y . Nature Cell Bio 15:895–904 . 27. Kit azawa M, T rinh D-N, LaFerla F-M (200 8) Inflammation induces t au pat hology i n inclusion body myositis model via glycogen synthase kinase-3 beta. Ann Neurol 64:15–2 4. 28. Mar tinsson L, et al. (2013 ) Long-term lithium treatment in bipolar disor der i s asso- ciated with longer leukocyte telomeres. T ransl Psyc hiatry 3:e261. 29. Kimura K, et al. (1 995) Diversity a nd variability of smooth muscle phenotypes of renal arterioles as revealed by my osin isofor m expression. Kidney Int 48:3 72–382. 30. Culligan K, Glover L, Do wling P , Ohlendieck K (2001) Brain dystrop hin-glycopr otein complex: Persistent expression of beta-dystroglycan, impaired oligomerization of Dp71 and up -regulation of utrop hins in animal models o f m uscular d ys troph y. BMC Cell Biol 2:2. 31. Kirchner T , et al. (1988) Pathogenesis of my asthenia gravis. acetylc holine rece ptor- related antigenic determinants in tumor-free thym uses and thymic e pithelial tu- mor s. Am J Pathol 130:268 –280. 32. Holliday D-L, Speirs V (2011) Choosing the right cell line fo r breast cancer research. Breast Cancer Res 13:215 . 33. Ba rrett T , et al. (2011) NCBI GEO: archive for functional genomics dat a sets-1 0 years on. Nucleic Acids Res 39:D1005– D1010. 34. T ripathi A, Klami A, Ore ˇ si ˇ c M, Ka ski S ( 2011) Matching samples of multiple views. Data Min Knowl Discov 23:300–321. 35. Vir tanen S, Klami A, Khan S-A, Kaski S (2012) Bayesian group factor analysis. JMLR W orkshop Conf Pr oc 22 :1269–1 277. 36. Wis e A, Oltvai Z, Bar-Joseph Z (2012) Matching experiments ac ross sp ecies using expression values and textual information. Bioinformatics 28:i258– i264. 37. Zheng J, et al. ( 2011) Annotcompute: annotation-based exploration and meta - analysis of genomics experiments. Database (Oxford) . 38. J ensen L-J, Saric J, Bork P (2006 ) Literature mining for t he biologist: from infor- mation retrieval to b iological discovery . Nat Rev Genet 7:119–12 9. 39. Rzhet sky A, Seringhaus M, Gerstein M (20 08) Seeking a new biology throu gh text mining. Ce ll 134:9–13 . 40. Sammon J- W (19 69) A nonlinear mapping for dat a st ructure analysis. IEEE T rans Comput 18:401– 409. 41. van Dongen S ( 2000) Graph Clustering by Flow Simulation. PhD thesis , University of Utrecht . 6 Suppor ting Inf o rmation Appendi x: T owar d computat ional cum u lative biology b y combin ing models of biologic al datasets Ali Faisal ∗ , Jaak ko Peltonen ∗ , Elisab eth Geor gii ∗ , Johan Rung † and Samuel Kaski ∗ ‡ ∗ Helsinki Institute f or Inf ormation T echnology HIIT , Department of Inf ormation and Compu ter Science , PO Box 15 400, Aalto University , 00076 Aalto , Finland, † European Molecular Biology Laboratory , European Bioinf ormatics Institute (EMBL-EBI), Wellcome T rust Genome Campus Hinxton, CB10 1SD , UK, and ‡ Helsinki Institute f or Inf ormation T echnology HIIT , Department of Computer Science, PO Box 68, 00014 Univer sity of Helsinki, Finland Methods Gene Set Enrichmen t Analysis. W e u sed GSEA [1] to bring in bi- ological knowledge in the form of pre-defined gene sets. GSEA starts by sorting genes with respect to their normalized ex pression le vels. GSEA essentially consists of comp uting a running sum on the sorted list for each gene set; this running sum (enrichment score) increases when a gene belongs to the gene set and decreases otherwise; the final statistic is the maximum of this running sum. The procedure essentially amounts to computing a weighted Kolmogo rov-Smirno v (KS) statist ic. For each sample the KS statisti c i s normalized by di- viding it by the mean of ran dom KS statistics computed o n randomly generated gene sets whose size is matched with the actual gene set; the 5 0 top-scoring g ene se ts are selected acco rding to this no rmalized score. This simple thresholding ignores significance v alues but has been successfully used in earlier meta-analysis studies [2, 3, 4]; we earlier inv estigated the alternativ e of selecting gene sets based on a standard q-v alue cut-of f (with q < 0 . 05), but it produced an exces- siv ely sparse encoding where more than 80% samples had no activ e gene sets [4]. The activity of each gene set is fi nally expressed as it s core set or leading edge subset , consisting of genes found before the running KS score reac hed its maximum. W e quantify the activity or a set simply b y the size of the leading edge su bset. It can be u sed anal- ogously to the so c alled bag-of-wor ds rep resentations in text analysis, and we use it for the subsequent modeling of each dataset with the base models. Base models. Latent Di richlet allocation (LDA) [5, 6, 7] and mix- tur e of unigr ams [8] are probabilistic unsupervised models that giv e insight to datasets by describing them in terms of latent components. Each data sample, in o ur case quantified as gen e set acti vities, is rep- resented by a probability distribution ov er components (sometimes also called topics). The compon ents are shared by all samples, but with different degrees of activ ation for each, and each component produces a charac teristic distrib ution over the ge ne sets. In LD A each sample may be produced by multi ple hidden components whil e the mixture of unigrams is a simplified version where each sample is assumed to come from a single comp onent. The computationa l prob- lem is to estimate the latent component structure (for each sample, the distribution over components and for each componen t, it s distri- bution ov er gene sets) that has m ost likely generated the observed set of samples. W e use standard inference solutions for the models: collapsed Gibbs sampler for the LD A [7] and Expectation Maximization for the mixture of unigrams [8] . In LDA, the hyperparameters, the prior probability of each component, were optimized with Minka’ s sta- ble fixed point iteration scheme [10], interleaved with the collapsed Gibbs sampling of the other parameters. The number of Gibbs i tera- tions was 2500, found to yield go od p erformance also earlier [6, 7, 9]. For the mixture of unigrams model, the maximum a posterior solu- tion is estimated with the Expectation M aximization (EM) algorithm, using Laplace smoothing for the prior probabilities of the mixture componen ts [8]. Model selection for base models. For each dataset we estimated the two base models an d selected t he one that best models t he dataset. W e split the dataset into two parts where 90% of the dataset samples were used for training the two models and the remaining 10% sam- ples to compute the test-set predicti ve likelihood, a measure of how well the model fits the data. W e repeated this procedure in a 10-fold cross-v alidation setup for each dataset and each of the two models. The model that performed better on av erage across the 10 folds was chosen to represent the dataset. Mo st datasets ( 74 out of 112) pre- ferred the more expressi ve LDA mod el. The number of components was selected with cross-validation for both models. W e again used 10-fold cross-validation, separately for each dataset, to choose the number of components that lead to best predictiv e likelihoods on t est samples from the same dataset. W e observed that the optimal number of total topics could roughly be summarized by ⌈ N D / 3 ⌉ for LD A and ⌊ √ N D ⌋ for mixture of uni- grams, where N D is the total number of samples in a dataset. All pre- dictiv e likelihood s ( both for model selection and for r etriev al) were computed using an empirical likeliho od method (see [11]). For very small datasets ( N D < 10) within-set cross-validation is v ery noisy and we chose LDA which had been selected for the majorit y of datasets with 10 ≤ N D ≤ 15. Strict Conca vity of the objectiv e function. For each query dataset q , there are multiple samples i = 1 , . . . , N q . For each sample, each background dataset giv es a probability . Let ~ x i = [ p ( x q i | M s 1 ) , p ( x q i | M s 2 ) , . . . , p ( x q i | M s N S ) , p ( x q i | Ψ )] ⊤ be the col- umn v ector of prob abilities for sam ple i from a ll b ackground datasets and from the no velty mo del, where ⊤ denotes v ector transpose. Then the probability given by a mixture of background datasets is ~ θ ⊤ ~ x i where ~ θ is the column vector of mixture weights (mixing propor- tions) ov er the backg round datasets and the no velty model. Since the mixing weights are mixture probabilities, they must lie in the canon- ical simplex deno ted as ∆ = ( θ | θ j ≥ 0 , N S + 1 ∑ j = 1 θ j = 1 ) . [ 1 ] Our optimization takes the form max ~ θ ∈ ∆ log exp ( − λ || ~ θ || 2 ) ∏ i ~ θ ⊤ ~ x i ! = max ~ θ ∈ ∆ ∑ i log ( ~ θ ⊤ ~ x i ) − λ || ~ θ || 2 = max ~ θ ∈ ∆ f ( ~ θ ) [ 2 ] where the objectiv e function is f ( ~ θ ) = ∑ i log ( ~ θ ⊤ ~ x i ) − λ || ~ θ || 2 . The objectiv e f ( ~ θ ) is a stri ctly concav e function wit h respect to the multiv ariate parameter ~ θ . T his can be shown since the second 1 – 10 deri va tiv e of the function is negati ve. In detail, the function’ s gradi- ent is ∇ f ( ~ θ ) = ∑ i ~ x i ~ θ ⊤ ~ x i − 2 λ ~ θ [ 3 ] and the matrix of second-ord er partial deriv atives (Hessian matrix) is ∇ 2 f ( ~ θ ) = − ∑ i ~ x i ~ x ⊤ i ( ~ θ ⊤ ~ x i ) 2 − 2 λ I [ 4 ] where I denotes the identity matrix. S ince all elements of ~ x i are non- negati ve, it is easy to see the Hessian matrix is negativ e definite for all ~ θ where θ i ≥ 0 as long as λ > 0. Therefore t he objectiv e function is strictly concave. A local maximum of a strictly concav e function on a con vex feasible region i s the unique global maximum [12 ]; there- fore maximizing the objecti ve function to a local maximum by any algorithm yields the unique global maximum. Maximizing th e Obj ectiv e Fu nction. Max imization of concave functions on t he unit si mplex ∆ can be done by the F rank-W olfe al- gorithm . The algorithm performs the follo wing steps. Step 1: initialization. T he algorithm initializes a solution as the verte x of the si mplex having the l argest objecti ve value. A vertex of the simplex has θ j = 1 for some j and all other elements of ~ θ are zero. At verte x j , the objectiv e function simplifi es to ∑ i log ( x i j ) − λ where x i j = p ( sam pl e i | d at ase t j ) , which takes O ( N q ) t ime to ev alu- ate per verte x. Thus creating the initialization take s linear O ( N q N S ) time ev en wit h a simple brute-force e v aluation of all vertices. Step 2: Iteration. In each iteration, the algorithm improves the solution by two steps: (1) Find the maximal element j of the gra- dient. (2) Find the point along the line ~ θ + α ( ~ e ( j ) − ~ θ ) , α ∈ [ 0 , 1 ] , which maximizes the objectiv e function. Here ~ e ( j ) means the vector where the element j i s one and the others are zero. Computation of the gradient and finding the maximal element takes O ( N q N S ) time. Proof of Con ve rgence and Scala bility Con ver gence Analysis. Since each iteration takes linear time with respect to the number of query samples and b ackground datasets, the only remaining issue is t he number of iterations required for good enough con ver gence. W e no w analyze t he con verge nce properties of this iteration in two cases. Case 1: optimal α . At fi rst, we consider the case where the best v alue of α can be found along the line to a sufficient accuracy in a fixed amount of time, for example by restricting ev aluations along the line to a fixed nu mber . Define a proportional regret function h ( ~ θ ) = ( f ( ~ θ ∗ ) − f ( ~ θ )) / 4 C f where ~ θ ∗ is the optimal parameter value maximizing the objecti ve function, and C f ≥ 0 is a measur e of curvatur e of f . In detail, C f is defined as the largest quantity such that for all ~ θ A ∈ ∆ , ~ θ B ∈ ∆ , ~ θ C where ~ θ C = ~ θ A + α ( ~ θ B − ~ θ A ) for some α , we hav e f ( ~ θ C ) ≥ f ( ~ θ A ) + ( ~ θ C − ~ θ A ) ⊤ ∇ f ( ~ θ A ) − α 2 C f . W ith this notation, it can be sho w n ([13], Theorem 2.2) that at iteration k of the Frank-W olfe algorithm the current solution ~ θ k has regret h ( ~ θ k ) ≤ 1 / ( k + 3 ) and thus f ( ~ θ ∗ ) − f ( ~ θ k ) ≤ 4 C f / ( k + 3 ) . Thus, to achiev e a desired regret ε , at most 4 C f / ε + 3 iterations are needed independently of t he number of backgro und datasets; the amount of iterations needed depends only on the curva ture. Case 2: fixed α . It can ev en be shown that using a fixed value α k = 2 / ( k + 3 ) at each iteration k suffices to yield bounds for per- formance: wit h this choice of α k the regret boun d at i teration k + 1 becomes ([13], Section 7) h ( ~ θ k + 1 ) ≤ 1 / ( k + 4 ) and thus f ( ~ θ ∗ ) − f ( ~ θ k + 1 ) ≤ 4 C f / ( k + 4 ) which again shows the number of iterations to achiev e a desired r e- gret does not depend on the number of background datasets, only on the c urv ature. It is enough to sh o w the curvature is finite an d d oes n ot depend on the number of backgroun d datasets; we now sho w this. Analysis o f the Curvatur e: As seen above, the smaller the c urv a- ture C f , the better the bounds for the regret f ( ~ θ ∗ ) − f ( ~ θ k ) are. In our case the function f is twice differentiable and it can be shown ([13], Section 4.1) that C f ≤ sup ~ θ A ∈ ∆ , ~ θ B ∈ ∆ , α ∈ [ 0 , 1 ] − 1 2 ( ~ θ B − ~ θ A ) ⊤ ∇ 2 f ( ~ θ α )( ~ θ B − ~ θ A ) . where ~ θ α = ~ θ A + α ( ~ θ B − ~ θ A ) . For our cost function this becom es C f ≤ sup ~ θ A ∈ ∆ , ~ θ B ∈ ∆ , α ∈ [ 0 , 1 ] 1 2 ( ~ θ B − ~ θ A ) ⊤ ∑ i ~ x i ~ x ⊤ i ( ~ θ ⊤ α ~ x i ) 2 + 2 λ I ! ( ~ θ B − ~ θ A ) = sup ~ θ A ∈ ∆ , ~ θ B ∈ ∆ , α ∈ [ 0 , 1 ] 1 2 ∑ i (( ~ θ B − ~ θ A ) ⊤ ~ x i ) 2 ( ~ θ ⊤ α ~ x i ) 2 + 2 λ || ~ θ B − ~ θ A || 2 ! ≤ sup ~ θ A ∈ ∆ , ~ θ B ∈ ∆ , α ∈ [ 0 , 1 ] , 1 ≤ i ≤ N q 1 2 N q (( ~ θ B − ~ θ A ) ⊤ ~ x i ) 2 ( ~ θ ⊤ α ~ x i ) 2 + 4 λ ≤ sup ~ θ A ∈ ∆ , ~ θ B ∈ ∆ , α ∈ [ 0 , 1 ] , 1 ≤ i ≤ N q 1 2 N q ( ~ θ B ~ x i ) 2 + ( ~ θ ⊤ A ~ x i ) 2 ( ~ θ ⊤ α ~ x i ) 2 + 4 λ ≤ sup ~ θ A ∈ ∆ , ~ θ B ∈ ∆ , α ∈ [ 0 , 1 ] , 1 ≤ i ≤ N q 1 2 N q 2 ( max j x i j ) 2 ( min j x i j ) 2 + 4 λ [ 5 ] where for bre vity we denoted x i j = p ( x q i | M s j ) for j = 1 , . . . , N S and x i , N S + 1 = p ( x q i | Ψ ) . Notice that t he right-hand side only depends on the maximal and minimal values that background datasets give to query samples, not on the number of such background datasets. Thus, as long as we ensure the maximal va lue is below some finite constant and the minimal va lue is above some small nonzero con- stant, the curvature C f is finite and the con ver gence bounds of the Frank-W olfe algorithm therefore do not depend on the number of backgroun d datasets. This condition is simple to ensure by suitable regularization of the models of back ground datasets. Therefore under the simple condition that probabilities giv en t o query samples are upp er bounded and lo wer bounde d above zero, the algorithm con verges (tow ards the unique global maximum) to a de- sired tolerance of the regret in a fi nite number of iterations I , which can dep end on the nu mber of query sam ples b ut is indep endent of the size of number of background datasets. 2 Computational complexity . Our model needs to perform two main computation tasks for each n ew dataset: optimization of the objecti ve function and computation of the predictiv e likelihoods. The opti- mization step needs to ev aluate the function value, the gradient and maximal element of the gradient; the computation of the function v alue h as co mplexity O ( N q ∗ N S ∗ O ( computing p ( x i q | M s j )) while, a s discussed in the p rev ious section, co mputing the gradient and finding the maximal element tak es linear O ( N q ∗ N S ) time in each iteration if a fixed step size is used (or if the number of li ne search ev aluations is restricted belo w some fixed maximum). Thus the complete algo- rithm takes O ( I ∗ N q ∗ N S ∗ O ( computing p ( x i q | M s j )) + O ( I ∗ N q ∗ N S ) . Clearly the dominating factor is the computation of the function v alue: O ( I ∗ N q ∗ N S ∗ O ( com puting p ( x i q | M s j )) . The p redictiv e like lihood for the query sample is co mputed as its av erage probability from V = 1000 multinomial distributions, esti- mated from randomly generated samples coming from the generati ve process of the earli er-trained model M s j ; this is the standard empir- ical likelihood scheme discussed in [11]. The complexity of com- puting the predictiv e likelihood for t he query dataset with G features (in our case gene-sets), giv en a model with T latent components 1 is O ( G ∗ V ∗ T ) . The total computational complexity is then simply t he complex ity for predicti ve like lihoods times the o ptimization scheme: O ( I ∗ N q ∗ N S ∗ G ∗ V ∗ T ) . Since the number of iterations is inde- pendent of the number of back ground datasets, N S , the comple xity is linear with respect to it, O ( N S ) , and th erefore the model is reaso nably tolerant to the fast gro w th of public repositories. In our implementation a single query dataset took about 31 i ter- ations and 0 . 15 seconds on average on an Intel R Core (TM) i7 CPU @ 2.93GHz, to find the optimal weight vector . Results Normalization of citation counts. Older datasets tend to have higher citation cou nts and outde grees; in Fig. 3 we remov ed this bias by a norma lization technique, and identified interesting datasets hav- ing very low citation counts and very high outde grees or vice ve rsa. T o v erify the analysis results (identified datasets) are not artifacts of the normalization, we reanalyzed the original data without applying citation count normalization. Fig. S1 shows the result as stratified subfigures plotted for each year separately . The datasets identified using the original citation counts (top-left and bottom-right corners in each subfigure of Fig. S1) are an exact match with the datasets identified after normalization. Retriev al performance after discounti ng for the laboratory effect. In mi croarray experiments laboratory ef fects are known to be strong [14]. The 206 datasets studied were generated from 163 labo ratories. The top laboratory was responsib le for 7 datasets, whereas 142 labo- raties only contributed a single set. T o test how much the laboratory effe cts have affected our results, w e discarded all retriev ed results from the same laboratory as the query set. The original precision- recall curve and the corrected curve are i n Figure S2; the mean aver - age precision dropp ed to 0 . 44 from 0 . 42. The small change in p erfor- mance shows t hat our result is mainly due to other ef fects captured by the model than the laboratory effe ct. Quantitative comparison of data-driven results against th e ci- tation patterns. Of the 23 direct citati on links, elev en were also found by our model as having a non-zero edge weight. Six links could not hav e been found; fiv e of them are citations by papers having very small datasets ( < 10), which we had considered t o be too small to act as queries while one citation li nk is between datasets released on the same date. Of the remaining six links not observed in our model, two are the cross-cluster citations that are not due to biolog- ical similarity of t he datasets as discussed in the paper; two cell line datasets about mu ltiple myeloma and large cell lymphoma (G SE6205 and GSE6184 respecti vely) cite a leukemia study (GSE2113) where plasma cells are profiled; one dataset measuring HIV i nfection from T cells (GS E6740) cites a very small dataset about t hymoc yte, the limited size of the set reducing its corresponding model’ s relative ca- pability to explain othe r sets c ompared to large datasets in the collec- tion. Finally , E-T ABM-26, a prostate cancer study , cites E-MEXP- 156 (a st udy about t umorigenic and nontumorigenic Human Embry- onic Kidney Cells) in the context of cell apoptos is of cancer cells i n general. V ice versa, we ev aluated the top -weight data-drive n edges against direct and indirect citati on links and f ound a favorab le, non- random ov erlap. For this we use a modern standard metric in in- formation retriev al called precision @ k which measures precision at each position k in the rank ed list of t op retrie val results from a sea rch engine. In our case t he results are the ranke d list of inferred edges in descending order of t heir edge strengths. The citation patterns were used as the gold standard for existence of an edge between two datasets using their correspon ding publication information. Fig. S3 sho ws that t he precision @ k i s reasonably high; for the top two hun- dred data-driv en edges ( i.e. , k = 200) the v alue is 0.5. Densely conn ected set of experiments in the relev ance n etwork. For each o f the top 20 strongest edges in the relev ance ne twork (li sted in T able S1) w e searched for associated cliques, i. e. , connections within neighbors of the edge, where the clique size is at least three. W e found sev en cliques; four of them (breast cancer, leuko cytes, hu- man immun e T helper cells and dev elopmental stages of thym ocytes) are described i n the main text. The remaining three are an adenocar- cinoma, a brain tissue and a skeletal muscle clique. T he first cli que is among GSE4824, GSE5258 and GSE6914; all three datasets are heterogeneo us collections of different cancerous tissues where ma- jority of the samples are cell line profiles from either lung or breast adenocarcino ma. The second clique is among GS E1297, GSE5392 and E-MEXP-114 that profil e normal and diseased brain tissue. The last clique is among all sk eletal muscle experiments of the collection where the strongest edge is between GSE3307 and GSE6011 that both measure Duchenne muscular dystrophy sampled from quadri- ceps muscle tissues, the former also containing samples form other skeletal muscle diseases. Skeletal muscle retriev al case study . W e lastly present a case study t o illustrate how the model retriev al can support a researcher in finding rel e v ant data on a specific topic, lessening the need for laborious manual searches. The topic of interest was gene expres- sion in skeletal muscle and our database consisted o f the human gen e express ion atlas [15], which includes eight skeletal muscle datasets among a total of 206 datasets, plus additional 16 skeletal muscle datasets extracted manually from ArrayExp ress (T able S2). First, we tested how well the data-driv en method retri e ved other skeletal mus- cle datasets when querying wit h an y si ngle skeletal muscle dataset. The retri e va l performance across all 16 query datasets was close to optimal, whe reas ke yword searches were not as good in the s ame task (Fig. S4). The reason for that was the lack of a consistent annotation vocab ulary in t he dataset descriptions; in particular , different lev els of specificity had been used. Next we looked in more detail i nto the retriev al results of the indi vidual queries. For all of them, t he retrie va l result was sparse, i.e. , less than 10% of the datasets were found to be relev ant to the query (by a non-zero weight). W e further inv esti gated the ranking of results provid ed by the data-driven retriev al model. For 12 queries all retrieved results were other skeletal muscle datasets; results for the remaining queries contained at l east one false positi ve, and they are summarized in T able S3. The top retrieved non-skeletal muscle dataset is the brain tissue dataset GSE 5392, followed by the kidney dataset GSE781. Kidney does no t ha ve a direct biological con nection 1 T is upper bounded by the maximum number of samples in a background dataset. 3 to skeletal muscle. It is kno wn t hat some areas of kidney are ri ch in blood vessels, which are lined by smooth muscle. Skeletal muscle, smooth muscle and cardiac muscle are the three main muscle t ypes in the human body . Interestingly , one skeletal muscle datasets, E- MEXP-216, was ne ver retrie ved by an y skeletal mu scle query , which suggests that it is an outlier in some respect. The dataset contains a combination of human and macacque liver and skeletal muscle sam- ples. It contains only four human skeletal muscle samples. Finally , the internal r anking of skeletal muscle datasets in re- sponse to a particular query dataset seems to be guided to a large extent by health cond itions: • E-GEOD-9397 contains samples annotated with disease status FSHD (Facioscapulohume ral muscular dystrophy). T he top re- triev ed dataset for that query is E-GEOD-10760, the only other dataset in the collection annotated with FSHD. • The top retrie ved r esult for E-GEOD-12648 i s E-GEOD-11686 and vice v ersa. Both of them are neuromuscu lar disorders, hered- itary inclusion body myopathy (HI BM) and cerebral palsy , and they are the only datasets with these specific diseases in our data collection. • E-GEOD-1786 partiall y contains samples from C OPD (Chronic Obstructi ve Pulmonary Disease) subjects; the disease has a mus- cle wasting ef fect. The top retriev ed result is E-GEOD-10760; it contains samples fr om the same muscle type, but another disease (FSHD) which also leads to muscle wasting. • E-GEOD-1295 contains samples of the trained and untrained muscle of non-young overweight people with prediabetic metabolic syndrom e. H alf of the samples in E-GEOD-1786 are also from trained muscles (of COPD patients and controls, old ov erweight men). E -GEOD-1786 appears at rank 6, and it is the only background dataset known to contain trained muscle sam- ples. Among th e top 5 datasets, 3 are annotated to contain disease samples related to weakening of muscles, another one is known to contain old ov erweight samples ( E-GEOD-8441 ). In summary , disease conditions and health states of tissue seem to determine the ranking within datasets of the same tissue (sub)type. 1. Subramanian A, et al. ( 2005) Gene set enrichment analysis: A k nowledge-based appro ach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:155 45–1555 0. 2. Segal E, et al. (2004) A module map s howing conditional activ ity of expression modules in cancer . Nat Genet 36:10 90–1098. 3. Caldas J, Gehlenborg N, Faisal A , Brazma A, Ka ski S ( 2009) Pr obabilistic retrieval and vi sualization of biologically relevant microarra y experiments. Bioinform atics 25:i145 –i153. 4. Caldas J, et a l. (201 2) Data-driven information retriev al in heterog eneous collections of transcriptomics dat a links SIM2s to malignant pleural mesothelioma. Bioinfor- matics 28:i246–i2 53. 5. Pritchard J-K, Stephens M, Donnelly P (2000) Inference of popu lation structure using mu ltilocus genotype data . Genet ics 155:945–959 . 6. Blei D-M, Ng A-Y , Jordan M-I, Laf ferty J ( 2003) Late nt Dirichlet allocat ion. J Mach Learn Res 3:993–102 2. 7. Griffiths T -L, Steyvers M (2004) Finding scientific topics. Proc Natl Acad Sci U S A 101:522 8–5235. 8. Nigam K, Mccallum A-K, Thrun S, Mitchell T (2000) T ext classificat ion fr om labeled and unlabeled documen ts using EM. Machine Learning 39:103–134. 9. Teh Y-W , Jordan M-I, Beal M-J, Blei D-M (200 6) Hierarchical Dirichlet Processes. J Am Stat Assoc 101:1566 –1581. 10. Minka T -P (200 0) Estimati ng a Dirichlet dist ribution . T echnical report available at http://research.microsoft.com/en-us/um/peop le/minka/papers/dirichlet/. 11. Li W , McCallum A (20 06) Pachinko allocati on: DA G-s tructured mixt ure models of topic correlations. Proceedings of the T wenty-Third international conference on Machine learning (A CM New Y ork, USA) pp 577 –584. 12. Bradley S, Hax A, Magn anti T (1977) Applied Mathematical Prog ramming . Addison- Wesley . 13. Cla rkson, K L (2 010) Coresets , sparse greedy appr oximation, and t he Frank-W olfe algorithm. ACM T ransac tions on Algorithms 6(4): Ar ticle 63. 14. Zilliox M-J, Irizarry R-A (2007) A gene exp ression bar code f or micr oarray data. Nat Methods 4:911–913 . 15. Lukk M, et al. (20 10) A global map of human gene expression. Nat Bi otechnol 28:322– 324. 16. Shannon P , et a l. (2003) Cyt oscape: a software envir onment for integrated models of biomolecular interaction networks. Genome Res 13:2498 -2504. 4 20 30 40 50 0 1 2 3 4 Y ear 2003 Citation count Normalized weighted outdegree 0 50 100 150 200 250 0 1 2 3 4 Y ear 2004 Citation count Normalized weighted outdegree 0 20 40 60 80 100 120 140 0 1 2 3 4 Y ear 2005 Citation count Normalized weighted outdegree 0 50 100 150 200 250 0 1 2 3 4 Y ear 2006 Citation count Normalized weighted outdegree 0 50 100 150 200 250 0 1 2 3 4 Y ear 2007 Citation count Normalized weighted outdegree 10 20 30 40 50 ImpF ac Fig. S1: Stratified d ata-driv en prediction of usefulness of da tasets vs. their citation c ounts. Black solid lines mark the bo undary for po tentially interesting datasets; the bou ndaries are set to hold the same percentiles of da ta as in Fig. 3 in the main pap er . ImpF ac stands for Impact Factor of the publication venu e. 5 0.0 0.2 0.4 0.6 0.8 1.0 0.0 0.2 0.4 0.6 0.8 1.0 Recall Precision Original Lab . effects remo ved Fig. S 2: Remov al of l aboratory effects changes the retriev al performance only slightly , as measured by the precision-recall curves. Original: Replicated from Fig. 1 of the main paper; Lab. effects r emoved: all retriev al results from the same laboratory as the query data have been discarded. 6 0.0 0.2 0.4 0.6 0.8 1.0 "k" − P osition in the ranked list of decreasing edge strength Precision at "k" 1 50 100 150 200 250 300 350 Combination model Random expectation Fig. S3: Overlap of data-dri ven recommendations wi th the actual citation graph: Precision @ k for top edges that explain more than 2 . 5% v ariation. The gold standard is the extended citation g raph which is built as the un ion of edges from 1) the original directed graph, 2) between any tw o articles that are cited together by some other article and 3) between any two articles that ha ve at least one common reference. 7 0.0 0.2 0.4 0.6 0.8 1.0 0.0 0.2 0.4 0.6 0.8 1.0 Recall Precision Combination model Random K eyword: sk eletal musc Fig. S4: R etriev al performance ev aluation of the data-dri ven model against k eywo rd search in the skeletal muscle case study . The precision- recall curves are a veraged across the 16 sk eletal muscle datasets having at least 10 samples. 8 Fig. S5: E nlarged visualization of the rele va nce network of datasets in the human gene expression atlas (Fig. 2 in t he main pap er); each da taset was used as a query to retrieve earlier datasets. A link from an earlier dataset to a later one means the earlier dataset is rele va nt as a partial model of activity in the later dataset. The link width is proportional to the normalized rele va nce weight (combination weight θ q j ; only links with θ q j ≥ 0 . 025 are shown, and datasets without links have be en discarded). The node size is proportional to the estimated influence, i .e. , the total ou tgoing weight. Colors: tissue type s (the six meta tissue typ es [15]). The node layout was comp uted from the d ata-driv en network using the cluster-emph asizing Sammon mapping scheme. Cytoscape [16] was used to generate the figure. 9 T able S1: T op 20 strongest edges in the relev ance network. Edge Clique membersh ip GSE4475 - E-T A BM-90 Does not f or m a clique GSE2113 - GSE636 5 Does not f or m a clique GSE2113 - GSE640 1 Does not f or m a clique E-MEXP -750 - G SE674 0 Part of the CD4+ T helper cell clique GSE1297 - GSE539 2 Part of the brain tissue clique GSE2328 - E-MEX P-433 Extends the leukocyte clique GSE2990 - GSE653 2 Part of the breast cancer clique GSE2328 - GSE558 0 Part of the leukocyte clique GSE2990 - GSE145 6 Part of the breast cancer clique GSE3494 - GSE299 0 Part of the breast tissue clique GSE2990 - GSE600 8 Does not f or m a clique GSE4824 - GSE525 8 Part of the adenocarcin oma cliqu e E-MEXP -72 - GSE4 475 Does not f or m a clique E-MEXP -403 - E-T ABM-90 Does not f or m a clique GSE3494 - GSE145 6 Part of the breast tissue clique E-MEXP -337 - E-MEXP-549 Part of the dev e lopmental stage of lymphocytes clique GSE3284 - E-MEX P-433 Part of the leukocyte clique GSE3307 - GSE601 1 Part of the skeletal muscle datase ts clique GSE2990 - GSE492 2 Part of the breast cancer clique GSE2044 - GSE330 7 Part of the skeletal muscle datase ts clique T able S2: ArrayExpress accession numbers of 16 skeletal muscle datasets used in the retriev al case study in addition to the human gene express ion atlas [15]. All datasets w ere measured with the human genome platform HG-U133A, the same used in the atlas. E-CBIL-30 E-GEOD-1076 0 E-GEOD-1168 6 E-GEOD-119 71 E-GEOD-1264 8 E-GEOD-1295 E-GEOD-1786 E-GEOD-3307 E-GEOD-4667 E-GEOD-474 E -GEOD-6011 E-GEOD-7146 E-GEOD-8441 E-GEOD-9105 E-GEOD-9397 E-GEOD-9676 T able S3: S keletal muscle que ries wit h at least one retrie ved no n-skeletal muscle dataset, sorted according to decreasing precision. Quer y No. o f retriev ed datasets No. o f retriev ed skeletal m uscle datasets T op false positiv e dataset (rank) E-GEOD-4667 20 18 GSE5392 (15) E-GEOD-1168 6 26 22 GSE5392 (22) E-GEOD-7146 25 21 GSE781 (22) E-GEOD-1197 1 25 19 GSE5392 (15) 10
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment