Automated adaptive inference of coarse-grained dynamical models in systems biology
Cellular regulatory dynamics is driven by large and intricate networks of interactions at the molecular scale, whose sheer size obfuscates understanding. In light of limited experimental data, many parameters of such dynamics are unknown, and thus mo…
Authors: Bryan C. Daniels, Ilya Nemenman
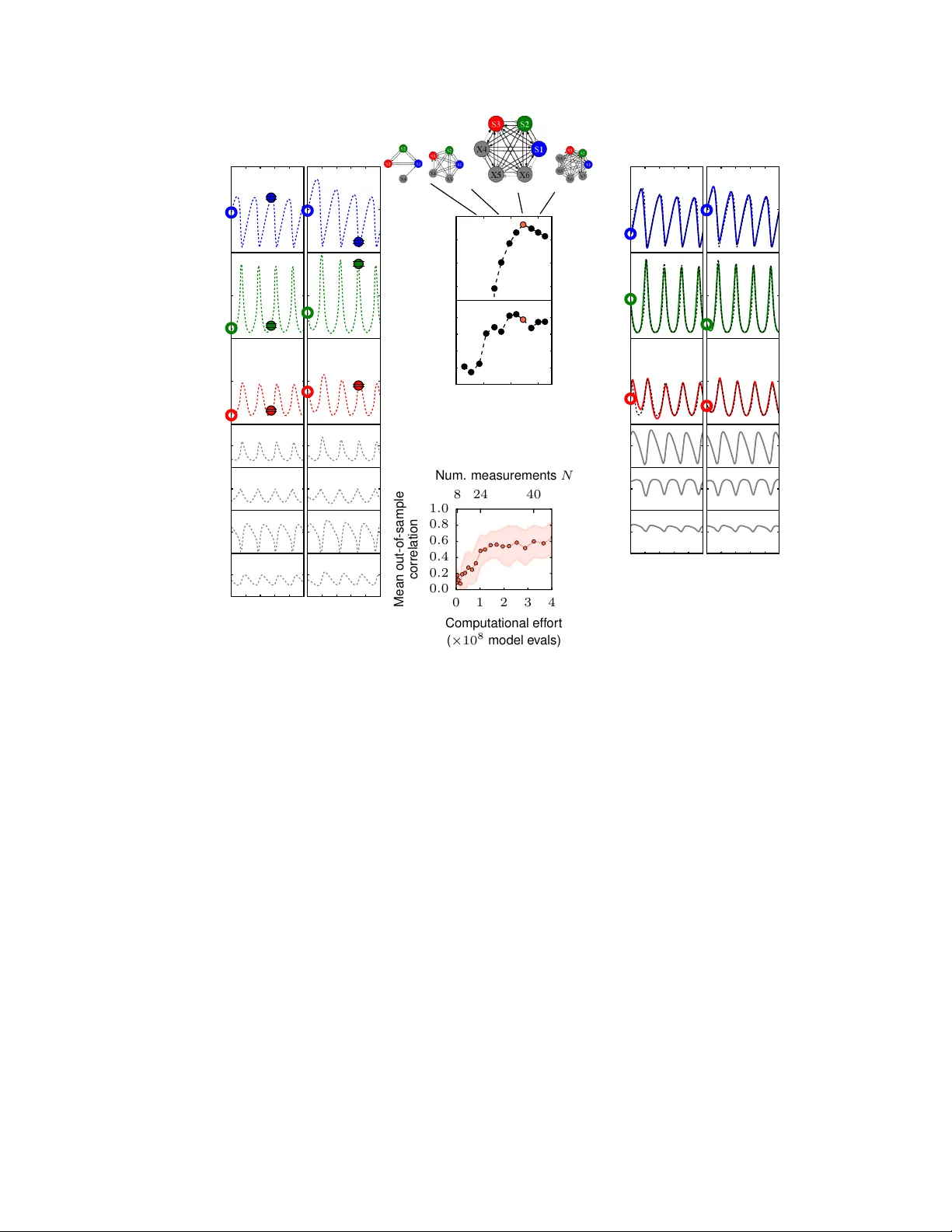
A utomated adaptiv e infer ence of coarse-grained dynamical models in systems biology Bryan C. Daniels ∗ Center for Comple xity and Collective Computation, W isconsin Institute for Discovery , Univer sity of W isconsin, Madison, WI 53715, USA Ilya Nemenman ∗ Departments of Physics and Biology Emory University , Atlanta, GA 30322, USA Abstract Cellular regulatory dynamics is driv en by large and intricate networks of interactions at the molecular scale, whose sheer size obfuscates understanding. In light of limited experimental data, many parameters of such dynamics are unknown, and thus models built on the detailed, mechanistic viewpoint ov erfit and are not predictiv e. At the other extreme, simple ad hoc models of complex processes often miss defining features of the underlying systems. Here we propose an approach that instead constructs phenomenolo gical , coarse-grained models of network dynamics that automatically adapt their complexity to the amount of av ailable data. Such adaptiv e models lead to accurate predictions ev en when microscopic details of the studied systems are unkno wn due to insuf ficient data. The approach is computationally tractable, e ven for a relati vely lar ge number of dynamical v ariables, allowing its software realization, named Sir Isaac , to make successful predictions ev en when important dynamic variables are unobserv ed. For e xample, it matches the kno wn phase space structure for simulated planetary motion data, av oids ov erfitting in a complex biological signaling system, and produces accurate predictions for a yeast glycolysis model with only tens of data points and ov er half of the interacting species unobserved. ∗ E-mail: bdaniels@discovery .wisc.edu, ilya.nemenman@emory .edu 1 I. INTR ODUCTION Systems biology is a field of complicated models — and rightfully so: the vast amount of ex- perimental data has clearly demonstrated that cellular networks hav e a degree of complexity that is far greater than what is normally encountered in the physical world [1]. Mathematical models of these data are often as complicated as the data themselves, reflecting the humorous maxim that “the best material model of a cat is another , or preferably the same, cat” [2]. Howe v er , continued success of approaches that systematize all known details in a combinatorially large mathematical model is uncertain. Indeed, generalizing and generating insight from complex models is difficult. Further , specification of myriads of microscopic mechanistic parameters in such models demands v ast data sets and computational resources, and sometimes is impossible e v en from very large data sets due to widely v arying sensiti vities of predictions to the parameters [3]. Finally , the very struc- tures of these models are often unkno wn because they depend on many yet-unobserved players on the cellular , molecular , and sub-molecular le vels. Identification of these structural characteristics of the in v olved processes is labor intensiv e and does not scale up easily . With these challenges, it is unlikely that mathematical models based solely on a reductionist representation will be able to account accurately for the observed dynamics of cellular networks. More importantly , even if the y could, the resulting models would be too unwieldy to bring about understanding of the modeled systems. Because of these difficulties, the need to use systems biology data to predict responses of bio- logical systems to dynamical perturbations, such as drugs or disease agents, has led to a resurgence of research into automated inference of dynamical systems from time series data, which had been attempted since the early days of the field of nonlinear dynamics [4, 5]. Similar needs in other data-rich fields in natural and social sciences and engineering hav e resulted in successful algo- rithms for distilling continuous dynamics from time series data, using approaches such as linear dynamic models [6], recurrent neural networks [7], e v olv ed regulatory networks [8], and sym- bolic regression [9, 10]. The latter two approaches produce models that are more mechanistically interpretable in that they incorporate nonlinear interactions that are common in systems biology , and they activ ely prune unnecessary complexity . Y et these approaches are limited because, in a search through all possible microscopic dynamics, computational effort typically explodes with the growing number of dynamical variables. In general, this leads to very long search times [8, 10], especially if some underlying variables are unobserved, and dynamics are coupled and cannot be 2 inferred one v ariable at a time. T o move forward, we note that, while biological netw orks are comple x, they often realize rather simple input-output relations, at least in typical experimental setups. Indeed, acti v ation dynamics of a combinatorially complex receptor can be specified with only a handful of large- scale parameters, including the dynamic range, cooperati vity , and time delay [11 – 13]. Also, some microscopic structural complexity arises in order to guarantee that the macroscopic functional output remains simple and rob ust in the face of various perturbations [12, 14]. Thus one can hope that macroscopic prediction does not require microscopic accurac y [15], and hence seek phenomenological, coarse-grained models of cellular processes that are simple, inferable, and interpretable, and nonetheless useful in limited domains. In this report, we propose an adapti ve approach for dynamical inference that does not attempt to find the single best microscopically “correct” model, but rather a phenomenological model that remains mechanistically interpretable and is “as simple as possible, but not simpler” than needed to account for the experimental data. Relaxing the requirement for microscopic accuracy means that we do not hav e to search through all possible microscopic dynamics, and we instead restrict our search to a much smaller hierarchy of models. By choosing a hierarchy that is nested and complete, we gain theoretical guarantees of statistical consistency , meaning the approach is able to adapti v ely fit any smooth dynamics with enough data, yet is able to avoid problems with ov erfitting that can happen without restrictions on the search space [16]. While similar complexity control methods are well established in statistical inference [17], we believ e that they have not been used yet in the context of inferring complex, nonlinear dynamics . Importantly , this adaptiv e approach is typically much more ef ficient because there are far fe wer models to test. Instead of searching a super-exponentially large model space [9], our method tests a number of models that scales polynomially with the number of dynamical v ariables. Further , it uses computational resources that asymptotically scale linearly with the number of observ ations. This allows us to construct interpretable models with much smaller computational effort and fewer experimental measurements, ev en when many dynamical v ariables are unobserved. W e call the approach Sir Isaac due to its success in discov ering the law of univ ersal gravity from simulated data (see below). 3 II. METHODS AND RESUL TS A. Classes of phenomenological models used by Sir Isaac W e are seeking a phenomenological model of dynamics in the form d~ x dt = ~ F x ( ~ x, ~ y , ~ I ) , d~ y dt = ~ F y ( ~ x, ~ y , ~ I ) , (1) where ~ x are the observed variables, ~ y are the hidden variables, and ~ I are the inputs or other pa- rameters to the dynamics. W e neglect intrinsic stochasticity in the dynamics (either deterministic chaotic, or random thermal), and focus on systems where repeated observ ations with nearly the same initial conditions produce nearly the same time series, sa ve for measurement noise. The goal is then to find a phenomenological model of the force fields ~ F x , ~ F y [4]. The same dynamics may produce different classes of trajectories ~ x ( t ) dependent on initial conditions (e. g., elliptical vs. hy- perbolic trajectories in gra vitational motion). Thus the focus on dynamical inference rather than on more familiar statistical modeling of trajectories allo ws the representation of multiple functional forms within a single dynamical system. T o create a model, we would like to gradually increase the comple xity of F until we find the best tradeof f between good fit and suf ficient robustness, essentially e xtending traditional Bayesian model selection techniques to the realm of dynamical models. Ideally , this process should progress much like a T aylor series approximation to a function, adding terms one at a time in a hierarchy from simple to more complex, until a desired performance is obtained. T o guarantee that this is possible, the hierarchy of models must be nested (or ordered) and complete in the sense that any possible dynamics can be represented within the hierarchy [16] (see Supplementary Online Ma- terials (SOM) ). Any model hierarchy that fits these criteria may be used, but ordering dynamical models that can be made more complex along two dimensions (by adding either nonlinearities or unobserved variables) is nontri vial. Further , dif ferent model hierarchies may naturally perform dif ferently on the same data, depending on whether the studied dynamics can be represented suc- cinctly within a hierarchy . W e construct two classes of nested and complete model hierarchies, both well matched to properties of biochemistry that underlies cellular network dynamics. W e build the first with S- systems [18] and the second with continuous time sigmoidal networks [19] (see SOM ). The S- systems use production and degradation terms for each dynamical variable formed by products of powers of species concentrations; this is a natural generalization of biochemical mass-action 4 laws. The sigmoidal class represents interactions using linear combinations of saturating functions of species concentrations, similar to saturation in biochemical reaction rates. Both classes are complete and are able to represent any smooth dynamics with a sufficient number of (hidden) dynamical v ariables [18, 20, 21]. It is possible that both classes can be unified into power -law dynamical systems with algebraic power -law constraints among the dynamical variables [18], b ut this will not be explored in this report. B. Description of model selection procedur e T o perform adapti ve fitting within a model class, a specific ordered hierarchy of models is cho- sen a priori that simultaneously varies both the de gree of nonlinearity and the number of hidden v ariables (see FIG. S1 and SOM ). For each model in the hierarch y , its parameters are fit to the data and an estimate of the Bayesian log-likelihood Ł of the model is calculated. This estimate makes use of a generalized version of the Bayesian Information Criterion [22], which we hav e adopted, for the first time, for use with nonlinear dynamical systems inference. As models increase in com- plexity , Ł first grows as the quality of fit increases, but ev entually begins to decrease, signifying ov erfitting. Since, statistical fluctuations aside, there is just one peak in Ł [16], one can be certain that the global maximum has been observed once it has decreased suf ficiently . The search through the hierarchy is then stopped, and the model with maximum Ł is “selected” (see FIG. 4(b)). C. The law of gra vity Before applying the approach to complex biological dynamics, where the true model may not be expressible simply within the chosen search hierarchy , we test it on a simpler system with a kno wn exact solution. W e choose the iconic law of gravity , inferred by Newton based on empiri- cal observ ations of trajectories of planets, the Moon, and, apocryphally , a falling apple. Crucially , the in verse-squared-distance law of Newtonian gravity can be represented exactly within the S- systems power -la w hierarchy for elliptical and hyperbolic trajectories, which do not go to zero radius in finite time. It requires a hidden parameter , the velocity , to completely specify the dynam- ics of the distance of an object from the sun (see SOM for specification of the model). FIG. 1 displays the result of the adapti v e inference using the S-systems class. When gi ven data about distance of an object from the sun o ver time, we discover a model that reproduces the un- 5 0 50 100 150 200 Time (units GM /v 3 0 ) 0 20 40 60 80 100 120 Distance from sun (units GM /v 2 0 ) r 0 1 . 00 (circle) 1 . 25 (ellipse) 1 . 50 (ellipse) 1 . 75 (ellipse) 2 . 00 (parabola) 2 . 25 (hyperbola) 2 . 50 (hyperbola) A 0 1 2 X 2 2 Fit model ( N = 150 ) 1 0 1 dr /dt Tr u e m o d e l r 0 =1 . 0 (circle) 0 1 2 X 2 2 1 0 1 dr /dt r 0 =1 . 5 (ellipse) 0 2 4 6 8 r 0 1 2 X 2 2 0 2 4 6 8 r 1 0 1 dr /dt r 0 =2 . 5 (hyperbola) B FIG. 1: The law of gravity: an example of dynamical inference. A particle is released with v elocity v 0 perpendicular to the line connecting it to the sun, with varying initial distance r 0 from the sun. (a) W ith only N = 150 examples (each consisting of just a single noisy observation of r at a random time t after the release; see SOM ), we infer a single dynamical model in the S-systems class that reproduces the data. W ith no supervision, adapti ve dynamical inference produces bifurcations that lead to qualitati v ely dif ferent behavior: in this case, a single model produces both oscillations (corresponding to elliptical orbits) and monotonic growth (corresponding to hyperbolic trajectories). Inferred trajectories are shown with solid colored lines, and the corresponding true trajectories are sho wn with dashed lines. (b) Like the true model (left), the inferred model (right) contains a single hidden v ariable X 2 and w orks using a similar phase space structure. Specifically , the location of nullclines (green lines) and a single fixed point (green circle) as a function of r 0 are reco vered well by the fit. Note that the hidden variable is defined up to a power (see SOM ), and we choose to plot X 2 2 here. derlying dynamics, including the necessary hidden v ariable and the bifurcation points. Since the trajectories include hyperbolas and ellipses, this example emphasizes the importance of inferring a single set of dynamical equations of motion, rather than statistical fits to trajectories themselv es, which would be different for the two cases. FIG. S3 additionally sho ws fits for the law of gravity using the sigmoidal models class. While accurate, the fits are worse than those for the S-systems, illustrating importance of understanding of basic properties of the studied system when approach- 6 ing automated model inference. Empo wered by the success of the adapti ve inference approach for this problem, we chose to name it Sir Isaac . The software implementation can be found under the same name on GitHub . D. Multi-site phosphorylation model When inferring models for more general systems, we do not expect the true dynamics to be perfectly representable by any specific model class: e ven the simplest biological phenomena may in volve combinatorially man y interacting components. Y et for simple macroscopic behavior , we expect to be able to use a simple approximate model that can produce useful predictions. T o demonstrate this, we en vision a single immune receptor with n modification sites, which can exist in 2 n microscopic states [23], yet has simple macroscopic beha vior for man y underlying parameter combinations. Here, we test a model receptor that can be phosphorylated at each of n = 5 sites arranged in a linear chain. The rates of phosphorylation and dephosphorylation at each site are af fected by the phosphorylation states of its nearest neighboring sites. This produces a complicated model with 32 coupled ODEs specified by 52 parameters, which we assume are unkno wn to the experimenter . W e imagine an experimental setup in which we can control one of these parameters, and we are interested in its effects on the time ev olution of the total phosphorylation of all 5 sites. Here, we treat as input I the maximum rate of cooperative phosphorylation of site 2 due to site 3 being occupied, V , and measure the resulting time course of total phosphorylation starting from the unphosphorylated state. Experimental measurements are corrupted with noise at the scale of 10% of their v alues ( SOM ). A straightforward approach to modeling this system is to fit the 52 parameters of the kno wn model to the phosphorylation data. A second approach is to rely on intuition to manually dev elop a functional parameterization that captures the most salient features of the timecourse data. In this case, a simple 5 parameter model (see SOM ) captures exponential saturation in time with an asymptotic value that depends sigmoidally on the input V . A third approach, adv ocated here, is to use automated model selection to create a model with complexity that matches the amount and precision of the av ailable data. In FIG. 2, we compare these three approaches as the amount of av ailable data is varied, and FIG. 3(a) shows samples of fits done by different procedures. W ith limited and noisy data, fitting 7 10 1 10 2 Number of measurements N 10 − 3 10 − 2 10 − 1 Out-of-sample mean squared error Out-of-sample error Full model max. likelihood Sigmoidal adaptive model Simple model 2 5 10 20 50 Mean number of parameters in selected model Number of parameters Full model max. likelihood Sigmoidal adaptive model Simple model FIG. 2: Multi-site phosphorylation model selection as a function of the number of measurements N . The sizes of errors made by three models decrease as the amount of data increases. Adaptiv e sigmoidal models perform roughly as well as a custom-made simple 5-parameter model for small N , but outperform the simple model for large amounts of data. Although we expect that it will eventually outperform all other models as N → ∞ , a maximum likelihood fit to the full 52-parameter model (dark blue) performs worse in this range of N . The mean ov er 10 sets of input data are sho wn, with shaded regions indicating the standard de viation of the mean. On the right axis, the number of parameters in each model is indicated, with the sigmoidal model adapting to use more parameters when gi ven more data (red squares). the parameters of the full kno wn model risks ov erfitting, and in the regime we test, it is the worst performer on out-of-sample predictions. The simple model performs best when fitting to less than 100 data points, but for larger amounts of data it saturates in performance, as it cannot fit more subtle effects in the data. In contrast, an adapti ve model remains simple with limited data and then grows to accommodate more subtle behaviors once enough data is av ailable, e v entually outperforming the simple model. The multi-site phosphorylation example also demonstrates that dynamical phenomenological models found by Sir Isaac are more than fits to the existing data, but rather they uncover the true nature of the system in a precise sense: they can be used to make predictions of model responses to some classes of inputs that are qualitativ ely dif ferent from those used in the inference. For 8 0 2 4 6 8 10 10 − 3 10 0 10 3 Input V 0 . 0 0 . 5 1 . 0 1 . 5 2 . 0 2 . 5 3 . 0 T otal phosphor ylation 0 2 4 6 8 10 Time (minutes) 10 − 3 10 0 10 3 Input V Input V 0 . 0 0 . 5 1 . 0 1 . 5 2 . 0 2 . 5 3 . 0 T otal phosphor ylation A B T otal phosphor ylation output Full model Sigmoidal adaptive model Full model true parameters FIG. 3: Response (right axis) to (a) out-of-sample constant and (b) time-v arying input (left axis) in the models of multi-site phosphorylation. Fit to N = 300 constant input data points, the full known model (dark blue) produces erratic behavior typical of overfitting, while the adapti v e sigmoidal model (red) produces more stable out-of-sample predictions with median behavior that is closer to the true dynamics. Dark lines indicate the median behavior ov er 100 samples from each model’ s parameter posterior (see SOM ), and shaded regions indicate 90% confidence interv als. example, as seen in FIG. 3(b), an adaptiv e sigmoidal model inferred using temporally constant signals produces a reasonable extrapolated prediction for response to a time-varying signal. At the same time, overfitting is evident when using the full, detailed model, ev en when one av erages the model responses ov er the posterior distrib ution of the inferred model parameters. 9 E. Y east glycolysis model A more complicated system, for which there has been recent interest in automated inference, is the oscillatory dynamics of yeast glycolysis [10]. A recent model for the system [24, 25], informed by detailed knowledge of cellular metabolic pathways, consists of coupled ODEs for 7 species with concentrations that oscillate with a period of about 1 minute. The system dynamic is simpler than its structure in the sense that some of the complexity is used to stabilize the oscillations to external perturbations. On the other hand, the oscillations are not smooth (see FIG. 4) and hence are hard to fit with simple methods. These considerations make this model an ideal next test case for phenomenological inference with Sir Isaac . If we were giv en ab undant time series data from all 7 species and were confident that there were no other important hidden species, we may be in a position to infer a “true” model detailing interactions among them. If we are instead in the common situation of having limited data on a limited number of species, we may more modestly attempt to make predictions about the types of inputs and outputs that we have measured. This is conceptually harder since an unknown number of hidden v ariables may need to be introduced to account for the dynamics of the observed species. W e demonstrate our approach by constructing adapti v e models of the dynamics from data for only 3 of the 7 coupled chemical species, as their initial conditions are v aried. Depicted in FIG. 4 is the model selection procedure for this case. After selecting an adapti ve model fit to noisy data from N single timepoints, each starting from initial conditions sampled from specified ranges, we test the inferred model’ s ability to predict the timecourse resulting from out-of-sample initial conditions. W ith data from only N = 40 measurements, the selected model is able to predict beha vior with mean correlation of o v er 0.6 for initial conditions chosen from ranges twice as lar ge as those used as training data (shown in FIG. 4) and 0.9 for out-of-sample ranges equal to in-sample ranges (shown in FIG. S6). Previous work that inferred the exact equations of the original 7-dimensional model [10] used roughly 500 times as many measurements of all 7 v ariables and 200 times as many model e v aluations. This e xample also demonstrates that adapti v e modeling can hint at the complexity of the hidden dynamics beyond those measured: the best performing sigmoidal model requires three hidden v ariables, for a total of six chemical species — only one less than the true model. Crucially , the computational comple xity of Sir Isaac still scales linearly with the number of observations, ev en when a large fraction of variables remains hidden (see SOM and FIG. S7). 10 Sp ecies concen tration (mM) 0 1 2 3 4 Computational eff or t ( × 10 8 model e v als) 0 . 0 0 . 2 0 . 4 0 . 6 0 . 8 1 . 0 Mean out-of-sample correlation 8 24 40 Num. measurements N 0 . 0 1 . 3 2 . 6 S 1 Condition 1 ... Condition N 0 . 0 1 . 3 S 2 0 . 0 0 . 2 S 3 0 . 0 0 . 3 S 4 0 . 0 0 . 2 S 5 0 2 S 6 0 1 2 3 4 0 . 0 0 . 1 S 7 0 1 2 3 4 5 Time (minutes) − 450 − 400 − 350 − 300 L 0 20 40 60 Number of parameters 0 . 0 0 . 2 0 . 4 0 . 6 0 . 8 1 . 0 Out-of-sample correlation 0 . 0 1 . 3 2 . 6 S 1 Condition 1 ... Condition M 0 . 0 1 . 3 S 2 0 . 0 0 . 2 S 3 − 4 1 X 4 − 4 0 X 5 0 1 2 3 4 − 2 − 1 X 6 0 1 2 3 4 5 Time (minutes) A B D C FIG. 4: An example of the model selection process using measurements of timecourses of three metabolites in yeast glycolysis as their initial concentrations are v aried. (a) For each set of initial conditions (open circles), a noisy measurement of the three observable concentrations (filled circles) is made at a single random time. Hidden variables (in gray) are not measured. In this example, we fit to N = 40 in-sample conditions. (b) Models from an ordered class, with the illustrated connectivity , are fit and tested sequentially until Ł, an approximation of the relativ e log-likelihood, decreases suf ficiently from a maximum. (c) The selected model (large connectivity diagram) is used to make predictions about out-of-sample conditions. Here, we compare the output of the selected model (solid lines) to that of the model that created the synthetic data (dashed lines). (d) Performance versus computational and experimental effort. The mean out-of- sample correlation for 3 measured biochemical species from the range of initial conditions twice that used in training rises to over 0.6 using less than 5 × 10 8 model e v aluations and 40 in-sample measurements. In Ref. [10], inferring an exact match to the original 7-dimensional model used roughly 500 times as many measurements of all 7 species (with none hidden), which were chosen carefully to be informativ e. The approach also uses 200 times as many model ev aluations (see SOM ). Nonetheless, the accuracy of both approaches is comparable, and Sir Isaac additionally retains information about the phase of the oscillations. 11 III. DISCUSSION The three examples demonstrate the power of the adaptiv e, phenomenological modeling ap- proach. Sir Isaac models are inferred without an exponentially comple x search o ver model space, which w ould be impossible for systems with many v ariables. These dynamical models are as sim- ple or complex as warranted by data and are guaranteed not to overfit e v en for small data sets. Thus they require orders of magnitude less data and computational resources to achiev e the same pre- dicti ve accuracy as more traditional methods that infer a pre-defined, large number of mechanistic parameters in the true model describing the system. These advantages require that the inferred models are phenomenological, and are designed for ef ficiently predicting the system dynamics at a gi ven scale, determined by the av ailable data. While FIG. 1 sho ws that Sir Isaac will infer the true model if it falls within the searched model hierarchy , and enough data is a v ailable, more generally , the inferred dynamics may be quite distinct from the true microscopic, mechanistic processes, as shown by a different number of chemical species in the true and the inferred dynamics in FIG. 4. What is then the utility of the approach if it says little about the underlying mechanisms? First, there is the obvious advantage of being able to predict responses of systems to yet-unseen experimental conditions, including those qualitativ ely dif ferent from the ones used for inference. Second, some general mechanisms, such as the necessity of feedback loops or hidden variables, are easily uncov ered e ven in phenomenological models. Howe ver , more importantly , we dra w the follo wing analogy . When in the 17th century Robert Hooke studied the force-extension relations for springs, a linear model of the relation for a specific spring did not tell much about the mecha- nisms of force generation. Ho we ver , the observation that all springs exhibit such linear relations for small extensions allo wed him to combine the models into a law — Hooke’ s law , the first of many phenomenological physical laws that follo wed. It instantly became clear that e xperimentally measuring just one parameter , the Hookean stiffness, provided an exceptionally precise description of the spring’ s behavior . And yet the mechanistic understanding of how this Hooke’ s constant is related to atomic interactions within materials is only now starting to emerge. Similarly , by study- ing related phenomena across complex biological systems (e.g., chemotactic behavior in E. coli [26] and C. ele gans [27], or behavioral bet hedging, which can be done by a single cell [28] or a behaving rodent [29]), we hope to build enough models of specific systems, so that general laws describing ho w nature implements them become apparent. 12 If successful, our search for phenomenological, emergent dynamics should allay some of the most important skepticism regarding the utility of automated dynamical systems inference in sci- ence [30], namely that such methods typically start with known variables of interest and kno wn underlying physical laws, and hence cannot do transformativ e science and find ne w laws of nature. Indeed, we demonstrated that, for truly successful predictions, the model class used for automated phenomenological inference must match basic properties of the studied dynamics (contrast, for example, FIG. 1 to FIG. S3, and see FIG. S4). Thus fundamental understanding of some ke y prop- erties of the underlying mechanisms, such as the power -law structure of the law of gravity , or the saturation of biochemical kinetic rates, can be inferred from data ev en if unkno wn a priori . Finally , we can contrast our approach with a standard procedure for producing coarse-grained descriptions of inanimate systems: starting from a mechanistically accurate description of the dynamics, and then mapping them onto one of a small set of univ ersality classes [15, 31]. This procedure is possible due to symmetries of physical interactions that are not typically present in living systems. W ithout such symmetries, the power of universality is diminished, and microscopic models may result in similarly different macroscopic ones. Then specifying the microscopic model first in or- der to coarse-grain it later becomes an example of solving a harder problem to solv e a simpler one [32]. Thus for li ving systems, the direct inference of phenomenological dynamics, such as done by Sir Isaac , may be the optimal w ay to proceed. [1] W Hlav acek. How to deal with lar ge models? Mol Syst Biol , 5:240, 2009. [2] A Rosenblueth and N W iener . The role of models in science. Phil Science , 12:316–321, 1945. [3] R Gutenkunst, J W aterfall, F Casey , K Bro wn, C Myers, and J Sethna. Univ ersally sloppy parameter sensiti vities in systems biology models. PLoS Comput Biol , 3:1871–1878, 2007. [4] J Crutchfield and B McNamara. Equations of motion from a data series. Comple x Systems , 1:417, 1987. [5] NH Packard, JP Crutchfield, JD Farmer , and RS Shaw . Geometry from a T ime Series. Physical Review Letters , 45(9), 1980. [6] K.J. Friston, L. Harrison, and W . Penny . Dynamic causal modelling. Neur oImag e , 19(4):1273–1302, August 2003. [7] David Sussillo and L F Abbott. Generating coherent patterns of acti vity from chaotic neural netw orks. 13 Neur on , 63(4):544–57, August 2009. [8] Paul Franc ¸ ois, V incent Hakim, and Eric D Siggia. Deriving structure from e volution: metazoan se g- mentation. Molecular systems biolo gy , 3(154):154, January 2007. [9] M Schmidt and H Lipson. Distilling free-form natural laws from experimental data. Science , 324:81, 2009. [10] M Schmidt, R V allabhajosyula, J Jenkins, J Hood, A Soni, J W ikswo, and H Lipson. Automated refinement and inference of analytical models for metabolic networks. Phys Biol , 8:055011, 2011. [11] B Goldstein, J Faeder , and W Hla v acek. Mathematical and computational models of immune-receptor signalling. Nat Rev Immunol , 4:445–456, 2004. [12] G Bel, B Munsky , and I Nemenman. The simplicity of completion time distributions for common complex biochemical processes. Phys Biol , 7:016003, 2010. [13] R Cheong, A Rhee, W ang, I Nemenman, and A Levchenko. Information transduction capacity of noisy biochemical signaling networks. Science , 334:354–358, 2011. [14] A Lander . Pattern, gro wth, and control. Cell , 144:955–969, 2011. [15] Benjamin B Machta, Ricky Chachra, Mark K T ranstrum, and James P Sethna. Parameter space com- pression underlies emergent theories and predictive models. Science , 342(6158):604–7, November 2013. [16] I Nemenman. Fluctuation-dissipation theorem and models of learning. Neur al Comput , 17:2006, 2005. [17] D MacKay . Information theory , infer ence, and learning algorithms . Cambridge UP , 2003. [18] Michael A. Sav ageau and Eberhard O. V oit. Recasting Nonlinear Dif ferential Equations as S-Systems: A Canonical Nonlinear Form. Mathematical Biosciences , 115, 1987. [19] Randall D. Beer . Parameter space structure of continuous-time recurrent neural networks. Neural computation , 18(12):3009–51, December 2006. [20] Ken-Ichi Funahashi and Y uichi Nakamura. Approximation of Dynamical Systems by Continuous T ime Recurrent Neural Networks. Neural networks , 6:801–806, 1993. [21] T ommy W .S. Chow and Xiao-Dong Li. Modeling of continuous time dynamical systems with input by recurrent neural networks. IEEE T r ansactions on Cir cuits and Systems I: Fundamental Theory and Applications , 47(4):575–578, April 2000. [22] G Schwarz. Estimating the dimension of a model. The annals of statistics , 6(2):461, 1978. [23] W illiam S Hla vacek, James R Faeder , Michael L Blinov , Richard G Posner , Michael Hucka, and 14 W alter Fontana. Rules for modeling signal-transduction systems. Sci. STKE , 2006(344):re6, July 2006. [24] Jana W olf and Reinhart Heinrich. Ef fect of cellular interaction on glycolytic oscillations in yeast: a theoretical in v estigation. Biochem. J . , 334:321–334, 2000. [25] P Ruof f, M Christensen, J W olf, and R Heinrich. T emperature dependency and temperature compen- sation in a model of yeast glycolytic oscillations. Biophys Chem , 106:179, 2003. [26] H Berg. E. coli in Motion . Springer , 2004. [27] W Ryu and A Samuel. Thermotaxis in Caenorhabditis elegans analyzed by measuring responses to defined thermal stimuli. J Neur osci , 22:5727–5733, 2002. [28] E Kussell and S Leibler . Phenotypic diversi ty , population growth, and information in fluctuating en vironments. Science , 309:2075–2078, 2005. [29] CR Gallistel, T Mark, A King, and P Latham. The rat approximates an ideal detector of changes in rates of reward: implications for the law of effect. J Exp Psychol: Anim Behav Pr ocess , 27:354–372, 2001. [30] P . W . Anderson and E Abrahams. Machines fall short of rev olutionary science. Science , 324:1515– 1516, 2009. [31] K W ilson. Renormalization group and critical phenomena. I. Renormalization group and the Kadanof f scaling picture. Phys Rev B , 4:3174, 1971. [32] V V apnik. The natur e of statistical learning theory . Springer , Ne w Y ork, NY , 2nd edition, 2000. Acknowledgments W e thank W illiam Bialek and Michael Sav ageau for important discussions, Andre w Mugler , David Schwab, and Fereydoon Family for their critical comments, and the hospitality of the Center for Nonlinear Studies at Los Alamos National Laboratory . This research was supported in part by the James S. McDonnell foundation Grant No. 220020321 (I. N.), a grant from the John T empleton Foundation for the study of complexity (B. D.), the Los Alamos National Laboratory Directed Research and De velopment Program (I. N. and B. D.), and NSF Grant No. 0904863 (B. D.). 15 Supplementary Materials A. Materials and Methods 1. Hierar chical Bayesian model selection For consistent inference, we need a hierarchy of models that satisfies criteria laid out in Ref. [1]. First, we desire a model hierarch y that will produce a single maximum in Ł, up to statistical fluctuations, as we add complexity . For this, the hierarchy should be nested (but not necessarily regular or self-similar), meaning that once a part of the model is added, it is nev er taken away . Second, the hierarchy should be complete, meaning it is able to fit any data arbitrarily well with a sufficiently complex model. Intuiti v ely , instead of searching a lar ge multidimensional space of models, hierarchical model selection follo ws a single predefined path through model space (FIG. S1). While the predefined path may be suboptimal for a particular instance (that is, the true model may not fall on it), e ven then the completeness guarantees that we will still e ventually learn any dynamical system F giv en enough data, and nestedness assures that this will be done without ov erfitting along the way . 1 2. Adaptive model classes and hier ar chies Our first model class is the S-system po wer -la w class. The general form of the S-system rep- resentation consists of J dynamical variables x i and K inputs I k = x J + k , with each dynamical v ariable gov erned by an ordinary dif ferential equation: [2] dx i dt = G ( x ) i − H ( x ) i , (S1) with production G and de gradation H of the form G ( x ) i = α i J + K Y j =1 x g ij j (S2) H ( x ) i = β i J + K Y j =1 x h ij j . (S3) 1 In general, we are not guaranteed good predictive po wer until N → ∞ , but we can hope that the assumptions implicit in our priors (consisting of the specific form of the chosen model hierarchy and the priors on its parameters) will lead to good predictiv e po wer e ven for small N . S1 Space of all models Many params.; fit anything Few params. More nonlinearities More hidden variables FIG. S1: Hierarchical model selection follo ws a single predefined path through model space. In a process called “recasting, ” any set of differential equations written in terms of elementary functions can be rewritten in the power -la w form by defining new dynamical variables in the correct way [2]. Since any sufficiently smooth function can be represented in terms of a series of elementary functions (e. g., T aylor series), a power -la w network of suf ficient size can describe an y such deterministic dynamical system. Note that, since exponents are not constrained to be positiv e or integer -v alued, dynamics in this class are generally ill-defined when v ariables are not positi v e. W e find that the S-systems model class works well for planetary motion, which has an exact representation in the class; see Section S A 3. For our biological test examples, the S-systems class is outperformed by the sigmoidal class (see belo w). This may be indicating that beha vior common in the S-systems class is not common in typical biological systems (e. g., real production and degradation terms cannot grow without bounds). It may also stem from the positi vity constraint: since the condition that variables remain positiv e is not easily determined from parameter v alues, we are forced in our model selection process to simply discard any tested parameters that lead to zero or negati ve v alues. The second model hierarchy is the sigmoidal network class. In this class, we use the fact that the interactions among biological components often take the form of a sigmoidal function to define the follo wing system of ODEs: dx i dt = − x i /τ i + J X j =1 W ij ξ ( x j + θ j ) + N p X k =1 V ik I k , (S4) where the sigmoidal function ξ ( y ) = 1 / (1 + e y ) . This class of models has also been shown to approximate any smooth dynamics arbitrarily well with a sufficient number of dynamical v ariables [3 – 6]. Note that natural v ariations of this class to be explored in future work include rescaling S2 of the arguments of the sigmoids ξ or switching the order of operations to apply the sigmoidal function to a linear combination of state v ariables in order to more closely match traditional neural network models [7]. An advantage of the S-systems and sigmoidal representations is the existence of a natural scheme for creating a one-dimensional model hierarchy: simply adding dynamical variables x i . The most general network is fully connected, such that e very variable x i has an interaction term in ev ery other dx j /dt . Our hierarchy starts with a fully-connected network consisting of the nec- essary number of input and output variables, and adds “hidden” dynamical variables to add com- plexity . W ith each additional x i , we add parameters in a predetermined order . In the S-systems class, without connections, v ariable x i ’ s beha vior is specified by 5 parameters: x init i , α i , β i , g ii , and h ii . Each connection to and from x j is specified by 4 parameters: g ij , g j i , h ij , and h j i . When adding a new dynamic v ariable, we first fix its parameters (to zero for the expo- nential parameters and one for the multiplicative parameters), and then allow them to v ary one at a time in the follo wing order: g ii , g j i , h j i , g ij , h ij , β i , h ii , α i (adding connections to ev ery other x j one at a time). An e xample is sho wn in T able S1. The sigmoidal class is similar: without connections, variable x i ’ s behavior is specified by 4 parameters: x init i , W ii , τ i , and θ i . Each connection to and from x j is specified by 2 parameters: W ij and W j i . When adding a new dynamic v ariable, we first fix its parameters (to zero for W and θ and one for τ ), and then allo w them to vary one at a time in the follo wing order: W ij , W j i , W ii , τ i , θ i (adding connections to e very other x j one at a time). An e xample is sho wn in T able S2. For e very adapti ve fit model and the full multi-site phosphorylation model, 2 we use the same prior for ev ery parameter α k , which we choose as a normal distribution N (0 , 10 2 ) with mean 0 and standard de viation ς = 10 . 3 2 For the simple model fit to the phosphorylation data, parameters are always well-constrained and priors are unim- portant, and we therefore do not use explicit priors. 3 Some parameters ( α and β in the S-systems model class, τ in the sigmoidal model class, and k and K parameters in the full phosphorylation model) are restricted to be positi v e, which we accomplish by optimizing o ver the log of each parameter . The priors are still applied in non-log space, effecti vely creating a prior that is zero for negati v e parameter values and 2 N (0 , 10) for positiv e parameter values. S3 Model No. i Num. parameters N p Form of po wer -law ODEs 0 3 x 1 (0) = x init 1 dx 1 dt = x g 10 I x g 11 1 − β 1 1 4 x 1 (0) = x init 1 dx 1 dt = x g 10 I x g 11 1 − β 1 x h 10 I 2 5 x 1 (0) = x init 1 dx 1 dt = x g 10 I x g 11 1 − β 1 x h 10 I x h 11 1 3 6 x 1 (0) = x init 1 dx 1 dt = α 1 x g 10 I x g 11 1 − β 1 x h 10 I x h 11 1 4 8 x 1 (0) = x init 1 x 2 (0) = x init 2 dx 1 dt = α 1 x g 10 I x g 11 1 x g 12 2 − β 1 x h 10 I x h 11 1 dx 2 dt = x g 22 2 − 1 5 9 x 1 (0) = x init 1 x 2 (0) = x init 2 dx 1 dt = α 1 x g 10 I x g 11 1 x g 12 2 − β 1 x h 10 I x h 11 1 x h 12 2 dx 2 dt = x g 22 2 − 1 6 10 x 1 (0) = x init 1 x 2 (0) = x init 2 dx 1 dt = α 1 x g 10 I x g 11 1 x g 12 2 − β 1 x h 10 I x h 11 1 x h 12 2 dx 2 dt = x g 21 1 x g 22 2 − 1 T ABLE S1: The first sev en models of an example hierarchy in the S-systems class with one input x I and fixed initial conditions x init 1 and x init 2 . S4 Model No. i Num. parameters N p Form of sigmoidal ODEs 0 3 x 1 (0) = x init 1 dx 1 dt = − x 1 /τ 1 + W 11 ξ ( x 1 ) + W 10 x I 1 4 x 1 (0) = x init 1 dx 1 dt = − x 1 /τ 1 + W 11 ξ ( x 1 + θ 1 ) + W 10 x I 2 6 x 1 (0) = x init 1 x 2 (0) = x init 2 dx 1 dt = − x 1 /τ 1 + W 11 ξ ( x 1 + θ 1 ) + W 12 ξ ( x 2 ) + W 10 x I dx 2 dt = − x 2 3 7 x 1 (0) = x init 1 x 2 (0) = x init 2 dx 1 dt = − x 1 /τ 1 + W 11 ξ ( x 1 + θ 1 ) + W 12 ξ ( x 2 ) + W 10 x I dx 2 dt = − x 2 + W 20 x I 4 8 x 1 (0) = x init 1 x 2 (0) = x init 2 dx 1 dt = − x 1 /τ 1 + W 11 ξ ( x 1 + θ 1 ) + W 12 ξ ( x 2 ) + W 10 x I dx 2 dt = − x 2 + W 21 ξ ( x 1 + θ 1 ) + W 20 x I 5 9 x 1 (0) = x init 1 x 2 (0) = x init 2 dx 1 dt = − x 1 /τ 1 + W 11 ξ ( x 1 + θ 1 ) + W 12 ξ ( x 2 ) + W 10 x I dx 2 dt = − x 2 + W 22 ξ ( x 2 ) + W 21 ξ ( x 1 + θ 1 ) + W 20 x I T ABLE S2: The first six models of an example model hierarchy in the sigmoidal class with one input x I and fixed x init 1 and x init 2 . S5 3. The law of gravity model For a mass m in motion under the influence of the gravitational field of a mass M m , the distance r between the two ev olves as [8] d 2 r dt 2 = h 2 r 3 − GM r 2 , (S5) where h = ( ~ v 0 · ˆ θ ) r 0 is the specific angular momentum, ~ v 0 is the initial velocity , r 0 is the initial distance, ˆ θ is the unit vector perpendicular to the line connecting the two masses, and G is the gravitational constant. Setting the initial velocity parallel to ˆ θ and measuring distance in units of GM v 2 0 and time in units of GM v 3 0 , the dynamics become 4 d 2 r dt 2 = 1 r 2 r 2 0 r − 1 . (S6) When written as two first-order differential equations, we see that this system can be repre- sented exactly in the S-systems class if the particle does not f all onto the Sun: dr dt = χ − 1 dχ dt = r 2 0 r − 3 − r − 2 , (S7) where we use the variable χ = dr dt + 1 , so that the resulting system’ s variables are nev er negati v e, a requirement of the S-systems class. T o illustrate constructing an adaptive model for planetary motion, we consider as input the initial distance from the sun r 0 . W e sample r 0 uniformly between 1 and 3 (in units of GM /v 2 0 ), which co vers the possible types of dynamics: at r 0 = 1 , the orbit is circular; when 1 < r 0 < 2 the orbit is elliptical; when r 0 = 2 the orbit is parabolic; and when r 0 > 2 the orbit is hyperbolic. In this and later examples, to best determine the minimum number of measurements needed for a gi ven le vel of performance, we sample the system at a single time point for each initial condition (FIG. S2), rather than sampling a whole trajectory per condition. This ensures that samples are independent, which would not be the case for subsequent data points of the same trajectory , and hence allo ws us to estimate the data requirements of the algorithm more reliably . Further , this is similar to the sampling procedure already used in the literature [9]. In the planetary motion case, we assume only the distance r is measured, meaning the total number of of datapoints N D = N , 4 Note that r 0 sets the (conserved) angular momentum: h = GM v 0 r 0 with r 0 in rescaled units. S6 where N is the number of initial conditions sampled. W e choose the time of the observation as a random time uniformly chosen between 0 and 100 , with time measured in units of GM /v 3 0 . T o each measurement we add Gaussian noise with standard deviation equal to 5% of the maximum v alue of r between t = 0 and t = 100 GM /v 3 0 . T ypical training data for the model can be seen in FIG. S2. Fits to N = 150 data points are sho wn in FIG. 1. Here our adaptiv e fitting algorithm selects a model of the correct dimension, with one hidden v ariable. The selected model ODEs in this case are dr dt = e − 3 . 405 r 3 . 428 0 r 0 . 049 X 7 . 372 2 − e − 2 . 980 r 2 . 936 0 r 0 . 046 X 2 − 4 . 925 dX 2 dt = r − 0 . 651 0 r − 3 . 435 X − 0 . 014 2 − e − 0 . 006 r − 4 . 288 0 r − 1 . 595 . (S8) Note that certain transformations of the hidden v ariable and parameters can leav e the output be- havior unchanged while remaining in the S-systems class. First, the initial condition of hidden parameters can be rescaled to 1 without loss of generality , so we remov e this de gree of freedom and set X 2 (0) = 1 . Second, we hav e the freedom to let the hidden variable X 2 → X γ 2 for any γ 6 = 0 with appropriate shifts in parameters. T o more easily compare the fit model with the perfect model, in the rightmost column of FIG. 1 we plot X 2 2 on the vertical axes instead of X 2 when comparing it to the dynamics of the true hidden v ariable χ . Finally , we may compare performance when we fit the gra vitation data using sigmoidal models, a model class that we kno w is not representati ve of the underlying mechanics. The results are sho wn in FIG. S3; the selected sigmoidal network, which contains three hidden variables, still provides a good fit to the data, as expected, but it does not generalize as well when r 0 is near the edge of the range contained in the data and timepoints are outside of the range of data to which they were fit. This is expected since forces can div er ge in the true law of gravity , and they are necessarily limited in the sigmoidal model. 4. Multi-site phosphorylation model T o explore a complicated biological system with relati vely simple output beha vior , we imagine a situation in which an immune receptor can be phosphorylated at each of fi ve sites arranged in a linear chain. The rates of phosphorylation and dephosphorylation at each site are affected by the phosphorylation states of its nearest neighboring sites. A site can be unphosphorylated ( U ) or phosphorylated ( P ), and its state can change via one of two processes. The first process does not S7 depend on states of neighboring sites: U i * ) P i , (S9) with on-rate k on i ([ U i ]) and of f-rate k off i ([ P i ]) that depend on the concentration of the corresponding substrate. The second, cooperativ e process happens only when a neighboring site j is phosphory- lated: U i P j * ) P i P j (S10) with on- and off-rates k on ij ([ U i P j ]) and k off ij ([ P i P j ]) . All rates k are modeled as Michaelis-Menten reactions: k ([ S ]) = V [ S ] K m +[ S ] . W ith each reaction specified by two parameters ( V and K m ) and 26 possible reactions, the phosphorylation model has a total of 52 parameters. T o more easily generate the differential equations that gov ern the multi-site phosphorylation model, we use the BioNetGen package [10, 11]. When fitting this phosphorylation model, we use as input the parameter V on 23 , which is chosen from a uniform distribution in log-space between 10 − 3 and 10 3 min − 1 . The remaining 51 V and K m parameters we sample randomly from our priors on these parameters. As output, we measure the total phosphorylation of the 5 sites P tot at a single random time uniformly chosen between 0 and 10 minutes. T o each measurement we add Gaussian noise with standard deviation equal to 10% of the P tot v alue at t = 10 min. T ypical training data for the model is sho wn in FIG. S2. The out-of-sample mean squared error , as plotted in FIG. 2, is measured o ver 100 ne w input v alues selected from the same distrib ution as the in-sample values, each of which is compared to the true model at 100 timepoints evenly spaced from 0 to 10 minutes. As a simple guess to the functional form of the total phosphorylation timecourse as a function of our control parameter V = V on 23 (the “simple model” in FIG. 2), we use an exponential saturation starting at 0 and ending at a v alue P ∞ that depends sigmoidally on V : P tot = P ∞ ( V ) 1 − exp t t 0 , (S11) where P ∞ ( V ) = a + b 2 1 + tanh log( V ) − d c (S12) and a , b , c , d , and t 0 are parameters fit to the data. FIG. 2 sho ws that this simple ad hoc model can fit the data quite well. S8 For the e xample sho wn in FIG. 3, the selected sigmoidal model consists of the ODEs dP tot dt = − P tot e − 1 . 219 + 0 . 409 1 + exp( P tot − 4 . 469) + 7 . 087 1 + exp( X 2 ) + 0 . 0005 V dX 2 dt = − X 2 − 2 . 303 1 + exp( P tot − 4 . 469) − 0 . 071 V (S13) X 2 (0) = 0 . 101 , with P tot (0) = 0 . In this multi-site phosphorylation example, the sigmoidal model class is a better performer than the S-systems class. A typical example of performance is depicted in FIG. S4. Though the S-systems class makes predictions that are still qualitativ ely correct, and its predictions steadily improv e as N increases, the sigmoidal class comes closer to the true underlying model with an equal amount of data. The confidence intervals on the dynamics in FIG. 3 correspond to samples from the poste- rior over parameters giv en N = 300 data points. In the notation of section S B, this posterior P ( α | data) ∝ exp [ − ˜ χ 2 ( α ) / 2] . T o generate samples from this distribution, we use Metropolis Monte Carlo as implemented in SloppyCell [12, 13]. As a starting point, we use the best-fit pa- rameters from the model selection procedure, and we sample candidate steps in parameter space from a multidimensional Gaussian corresponding to the Hessian at the best-fit parameters. 5 From 10 4 Monte Carlo steps, the first half are remov ed to av oid bias from the initial condition, and ev ery 50 of the remaining steps are used as 100 approximately independent samples from the parameter posterior . W e note that the median behavior over the Bayesian posterior is less extreme than the behavior at the maximum lik elihood parameters (not sho wn), but still has fast-timescale dynamics indicati ve of o v erfitting. 5. Y east glycolysis model As an example of inference of more complicated dynamics, we use a model of oscillations in yeast glycolysis, originally studied in terms of temperature compensation [14] and since used as a test system for automated inference [9]. The model’ s behavior is defined by ODEs describing the 5 Unconstrained parameter directions in the proposal distribution, corresponding to singular values smaller than λ cut = λ max / 10 , where λ max is the largest singular val ue, are cut off to λ cut to produce reasonable acceptance ratios (near 0.5). S9 dynamics of the concentrations of sev en molecular species (the biological meaning of the species is not important here): dS 1 dt = J 0 − k 1 S 1 S 6 1 + ( S 6 /K 1 ) q dS 2 dt = 2 k 1 S 1 S 6 1 + ( S 6 /K 1 ) q − k 2 S 2 ( N − S 5 ) − k 6 S 2 S 5 dS 3 dt = k 2 S 2 ( N − S 5 ) − k 3 S 3 ( A − S 6 ) dS 4 dt = k 3 S 3 ( A − S 6 ) − k 4 S 4 S 5 − κ ( S 4 − S 5 ) (S14) dS 5 dt = k 2 S 2 ( N − S 5 ) − k 4 S 4 S 5 − k 6 S 2 S 5 dS 6 dt = − 2 k 1 S 1 S 6 1 + ( S 6 /K 1 ) q + 2 k 3 S 3 ( A − S 6 ) − k 5 S 6 dS 7 dt = ψ κ ( S 4 − S 5 ) − k S 5 . Parameter values, listed in T able S3, are set to match with those used in Ref. [9] and T able 1 of Ref. [14], where our S 5 = N 2 , our S 6 = A 3 , and our S 7 = S ex 4 . For the yeast glycolysis model, we use as input the initial conditions for the visible species S 1 , S 2 , and S 3 . These are each chosen uniformly from ranges listed in the “In-sample IC” column of T able S4. Each of the three visible species are then measured at a random time uniformly chosen from 0 to 5 minutes, meaning the total number of datapoints N D = 3 N for this system, where N is the number of initial conditions sampled. Gaussian noise is added to each measurement with standard deviations giv en in T able S4. T o ev aluate the model’ s performance, we test it using 100 ne w input values selected uniformly from the ranges listed in the “Out-of-sample IC” column of T able S4, each of which is compared to the true model at 100 timepoints ev enly spaced from 0 to 5 min. The correlation between the adaptiv e fit model and the actual model ov er these 100 timepoints is calculated separately for each visible species, set of initial conditions, and in-sample data, and the average is plotted as the “mean out-of-sample correlation” in FIG. 4. The topology of the selected network model is illustrated in FIG. S5. Note that our model fitting approach assumes that the model timecourse is fully determined (aside from measurement error) by the concentrations of measured species. T o be consistent with this assumption we do not v ary the initial conditions of the four hidden v ariables. In future work it may be possible to relax this assumption, allo wing the current state of intrinsic v ariations in hidden variables to be learned as well. In Ref. [9], the EUREQa engine is used to infer the same yeast glycolysis model that we use S10 J 0 2.5 mM min − 1 k 1 100. mM − 1 min − 1 k 2 6. mM − 1 min − 1 k 3 16. mM − 1 min − 1 k 4 100. mM − 1 min − 1 k 5 1.28 min − 1 k 6 12. mM − 1 min − 1 k 1.8 min − 1 κ 13. min − 1 q 4 K 1 0.52 mM ψ 0.1 N 1. mM A 4. mM T ABLE S3: Parameters for the yeast glycolysis model defined in Eqns. (S14). here. W e can roughly compare performance as a function of computational and experimental ef fort by measuring the number of required model ev aluations and measurements (FIG. 4). Here we compare the two approaches in more detail. First, Ref. [9] attempts to match time deriv ati v es of species concentrations as a function of species concentrations, instead of species concentrations as a function of time as we do. This means that each model ev aluation 6 is more computationally costly for us, since it requires an 6 In our setup, we define a model ev aluation as a single integration of the model ODEs (see Section S D). S11 V ariable In-sample IC (mM) Out-of-sample IC (mM) In-sample σ (mM) S 1 [0.15, 1.60] [0.15, 3.05] 0.04872 S 2 [0.19, 2.16] [0.19, 4.13] 0.06263 S 3 [0.04, 0.20] [0.04, 0.36] 0.00503 S 4 0.115 0.115 N/A S 5 0.077 0.077 N/A S 6 2.475 2.475 N/A S 7 0.077 0.077 N/A T ABLE S4: Initial conditions (IC) and standard de viations of experimental noise ( σ ) used in the yeast glycolysis model. Initial conditions for visible species S 1 , S 2 , and S 3 are chosen uniformly from the giv en ranges, chosen to match Ref. [9]. Out-of-sample ranges are each twice as lar ge as in-sample ranges. Initial conditions for the remaining hidden species are fixed at reference initial conditions from Refs. [9] and [14]. In-sample noise is set at 10% of the standard deviation of each v ariable’ s concentration in the limit cycle, as quoted in Ref. [9]. integration of the ODEs over time. It also means, howe v er , that we are able to match well the phases of oscillations, which remain unconstrained in Ref. [9]. The fitting of timecourses instead of deri v ati ves also makes our method focus on the fitting of dynamics near the attractor , rather than attempting to constrain dynamics through the entire phase space. T o consistently infer exact equations for the full 7-dimensional model, Ref. [9] used 20 , 000 datapoints and roughly 10 11 model e v aluations. W e contrast this with our method that produces reasonable inferred models using 40 datapoints and less than 5 × 10 8 model e v aluations (FIG. 4). Finally , in the main text we test the performance of our yeast glycolysis models for out-of- sample ranges of initial conditions that are twice as large as the in-sample ranges from which data is taken, as in Ref. [9], in order to more directly test their ability to extrapolate to regimes that were not tested in training. In FIG. S6, we compare this to performance when out-of-sample initial conditions are chosen from the same ranges as in-sample data (note that, nonetheless, none S12 of the test examples has appeared in the training set). Here we see that the mean correlation can reach 0.9 using N = 40 measurements. B. Derivation of Bay esian log-likelihood estimate Ł The deriv ation here lar gely follows Refs. [15, 16], but can be traced to the 1970s [17]. For a gi ven model M that depends on parameters α , our model selection algorithm requires an estimate of the probability that M is the model that produced a gi v en set of data { y i } with corresponding error estimates { σ i } (measured at a set of timepoints { t i } ), and i = 1 , . . . , N , so that there are N measurements. Since the parameters α are unknown aside from a prior distribution P ( α ) , we must integrate o v er all possible v alues: P ( M | data) = P ( M | { y i , σ i , t i } ) (S15) = Z − 1 α Z d N p α P ( M | { y i , σ i , t i } ; α ) P ( α ) , (S16) where the normalization constant Z α = R d N p α P ( α ) and N p is the number of parameters. In terms of the output gi ven the model, Bayes rule states P ( M | { y i , σ i , t i } ; α ) = P ( M ) P ( { y i } ) P ( { y i } | M ( α ); { σ i , t i } ) . (S17) Assuming that the model output has normally distributed measurement errors, P ( { y i } | M ( α ); { σ i , t i } ) = N Y i =1 P ( y i | M ( α ); σ i ; t i ) (S18) = N Y i =1 1 p 2 π σ 2 i exp " − 1 2 y i − M ( t i , α ) σ i 2 # = Z − 1 σ exp " − 1 2 N X i =1 y i − M ( t i , α ) σ i 2 # = Z − 1 σ exp − 1 2 χ 2 ( M ( α ) , { y i , σ i , t i } ) , where χ 2 is the usual goodness-of-fit measure consisting of the sum of squared residuals, and Z σ is the normalization constant Q N i =1 p 2 π σ 2 i . Thus we ha ve: 7 P ( M | data) = C Z − 1 α Z d N p α exp − 1 2 ˜ χ 2 ( α ) , (S19) 7 W e simplify notation by letting χ 2 ( α ) = χ 2 ( M ( α ) , { y i , σ i , t i } ) . S13 where C ≡ 2 P ( M ) / Z σ P ( { y i } ) and ˜ χ 2 ( α ) = χ 2 ( α ) − 2 log P ( α ) . Since we will be comparing models fitting the same data, and we assume all models hav e the same prior probability P ( M ) , C will be assumed constant in all further comparisons (but see Ref. [18] for the discussion of this assumption). If there are enough data to sufficiently constrain the parameters (as is the case for ideal data in the limit N → ∞ ), then the integral will be dominated by the parameters near the single set of best-fit parameters α best . T o lo west order in 1 / N , we can approximate the integral using a saddle-point approximation [16]: P ( M | data) ≈ C Z − 1 α exp − 1 2 ˜ χ 2 ( α best ) Z d N p α exp [ − ( α − α best ) H ( α − α best )] , (S20) where H is the Hessian: 8 H k` = 1 2 ∂ 2 ˜ χ 2 ( α ) ∂ α k dα ` α best . (S21) If we assume normally distrib uted priors on parameters with v ariances ς 2 k , the log posterior proba- bility becomes log P ( M | data) ≈ const − 1 2 ˜ χ 2 ( α best ) − 1 2 N p X µ =1 log λ µ − 1 2 N p X k =1 log ς 2 k , (S22) where λ µ are the eigen v alues of H , and the last term comes from Z α . W e thus use as our measure of model quality Ł ≡ − 1 2 ˜ χ 2 ( α best ) − 1 2 X µ log λ µ − 1 2 X k log ς 2 k . (S23) Eq. (S23) is a generalization of the Bayesian Information Criterion (BIC) [17] when parameter sensiti vities and priors are explicitly included. 9 The first term is the familiar χ 2 “goodness of fit, ” and the last two terms constitute the fluctuation “penalty” for overfitting or complexity . Note that here the goodness of fit and the complexity penalty are both functions of the entire dynamics, rather than indi vidual samples, which is not a common application of Bayesian model selection techniques. 8 Near the best-fit parameters where residuals are small, and when priors are Gaussian, H is approximated by the Fisher Information Matrix, which depends only on first deri v ati ves of model beha vior: H ≈ J T J + Σ − 2 , where the Jacobian J i` = 1 σ i ∂ M i ∂ α ` and the diagonal matrix Σ − 2 k` = δ k` ς − 2 k expresses the ef fects of parameter priors. 9 For well-constrained parameters, we expect, to lowest order in 1 / N , our result to be equal to the BIC result of − 1 2 ˜ χ 2 ( α best ) + 1 2 N p log N . S14 C. Fitting algorithm W e are gi v en N data points x i at kno wn times t i and kno wn exogenous parameters I i , and with kno wn or estimated v ariances σ 2 i . W e are approximating the functions ~ F X and ~ F Y in Eq. (1), where y are hidden dynamic model v ariables, and x = x ( t, I ) and y = y ( t, I ) in general depend on time t and inputs I . As described in Section S B, we fit to the data x i using a combination of squared residuals from the data and priors P ( α ) on parameters α , which we assume to be Gaussian and centered at zero: ˜ χ 2 = N X i =1 x i − x ( t i , I i ) σ i 2 + 2 N p X k =1 α k ς k 2 , (S24) where F ’ s are integrated to produce the model v alues x and y : x ( t, I ) = x 0 ( I ) + Z t 0 ~ F X ( x ( s, I ) , y ( s, I )) ds (S25) y ( t, I ) = y 0 ( I ) + Z t 0 ~ F Y ( x ( s, I ) , y ( s, I )) ds. (S26) T o fit parameters, we use a two step process akin to simulated annealing that uses samples from a “high temperature” Monte Carlo ensemble as the starting points for local optimization performed using a Lev enber g-Marquardt routine. The phenomenological models are implemented using SloppyCell [12, 13] in order to mak e use of its parameter estimation and sampling routines. Follo wing is a high-le vel description of the fitting algorithm, with choices of parameters for the examples in the main te xt listed in T able S5. 1. Choose a model class, consisting of a sequence of nested models index ed by i , where the number of parameters N p monotonically increases with i . Choose a step size ∆ p . 2. Giv en data at N total timepoints, fit to data from the first N timepoints, where N is increased to N total in steps of ∆ N . 3. At each N , test models of increasing number of parameters N p (stepping by ∆ p ) until Ł consistently decreases (stopping when the last i ov ersho ot models tested hav e smaller Ł than the maximum). F or each model, to calculate Ł: (a) Generate an ensemble of starting points in parameter space using Metropolis-Hastings Monte Carlo to sample from P ( α ) ∝ exp( − ˜ χ 2 ( α ) / 2 T N D ) with ˜ χ 2 from (S24). The temperature T is set large to encourage exploration of large regions of parameter space, S15 but if set too large can result in a small acceptance ratio. Infinities and other inte gration errors are treated as ˜ χ 2 = ∞ . i. Use as a starting point the best-fit parameters from a smaller N p if a smaller model has been pre viously fit, or else default parameters. ii. As a proposal distrib ution for candidate steps in parameter space, use an isotropic Gaussian with standard deviation √ T N D /λ max , where N D is the total number of data residuals and λ max is the largest singular v alue of the Hessian [Eq. (S21)] at the starting parameters. iii. If this model has previously been fit to less data, use those parameters as an addi- tional member of the ensemble. (b) Starting from each member of the ensemble, perform a local parameter fit, using Le venber g-Marquardt to minimize ˜ χ 2 from (S24). Stop when con ver gence is detected (when the L1 norm of the gradient per parameter is less than avegtol ) or when the number of minimization steps reaches maxiter . The best-fit parameters α ∗ are taken from the member of the ensemble with the smallest resulting fitted ˜ χ 2 . (c) At α ∗ , calculate Ł from (S23). 4. For each N , the model with lar gest log-likelihood Ł is selected as the best-fit model. D. Scaling of computational effort In FIG. S7, we plot the number of model ev aluations used in each search for the best-fit phe- nomenological model. W e define a model ev aluation as a single integration of a system of ODEs. 10 This includes both integration of model ODEs and the deri v ati ves of model ODEs, used in gradient calculations. 11 Note that in FIG. 4, to indicate the total number of ev aluations used as N is grad- ually increased to its final value, we plot the cumulativ e sum of the number of model ev aluations depicted in FIG. S7. W e see that the number of model ev aluations scales superlinearly with N if the selected model size is gro wing with N , as is the case in the yeast glycolysis model below 10 Note that the amount of necessary CPU time per integration is dependent on the size and stif fness of the system. 11 The number of integrations per gradient calculation is proportional to the number of parameters. This means that the computational effort used to fit lar ge models is dominated by gradient calculations. S16 ∆ p (gravitation and phosphorylation e xamples) 2 ∆ p (yeast example) 5 i ov ersho ot 3 Ensemble temperature T (full phosphorylation model) a 10 Ensemble temperature T (all other models) 10 3 T otal number of Monte Carlo steps (full phosphorylation model) a 10 2 T otal number of Monte Carlo steps (all other models) 10 4 Number of ensemble members used 10 avegtol 10 − 2 maxiter 10 2 T ABLE S5: Adapti ve inference algorithm parameters. 1 In the full phosphorylation model, we fit parame- ters in log-space since the y are kno wn to be positi v e. This makes the model more sensiti v e to lar ge changes in parameters, meaning that we are forced to be more conservati v e with taking large steps in parameter space to achie ve reasonable acceptance ratios. about N = 20 (FIG. S8). When the model size saturates, the number of model ev aluations scales roughly linearly with N . [1] I Nemenman. Fluctuation-dissipation theorem and models of learning. Neur al Comput , 17:2006, 2005. [2] Michael A. Sav ageau and Eberhard O. V oit. Recasting Nonlinear Dif ferential Equations as S-Systems: A Canonical Nonlinear Form. Mathematical Biosciences , 115, 1987. [3] Randall D. Beer . Parameter space structure of continuous-time recurrent neural networks. Neural computation , 18(12):3009–51, December 2006. S17 [4] Randall D. Beer and Bryan Daniels. Saturation Probabilities of Continuous-T ime Sigmoidal Net- works. arXiv pr eprint arXiv:1010.1714 , (812):856–873, 2010. [5] Ken-Ichi Funahashi and Y uichi Nakamura. Approximation of Dynamical Systems by Continuous T ime Recurrent Neural Networks. Neural networks , 6:801–806, 1993. [6] T ommy W .S. Chow and Xiao-Dong Li. Modeling of continuous time dynamical systems with input by recurrent neural networks. IEEE T r ansactions on Cir cuits and Systems I: Fundamental Theory and Applications , 47(4):575–578, April 2000. [7] DE Rumelhart, GE Hinton, and RJ Williams. Learning representations by back-propagating errors. Natur e , 323:533, 1986. [8] L Landau and E Lifshitz. Mechanics . Butterworth-Heinemann, 3rd edition, 1976. [9] M Schmidt, R V allabhajosyula, J Jenkins, J Hood, A Soni, J W ikswo, and H Lipson. Automated refinement and inference of analytical models for metabolic networks. Phys Biol , 8:055011, 2011. [10] W illiam S Hla vacek, James R Faeder , Michael L Blinov , Richard G Posner , Michael Hucka, and W alter Fontana. Rules for modeling signal-transduction systems. Sci. STKE , 2006(344):re6, July 2006. [11] Bionetgen. http://bionetgen.org. [12] Christopher R Myers, Ryan N Gutenkunst, and James P Sethna. Python unleashed on systems biology . Computing in Science and Engineering , 9(3):34, 2007. [13] Ryan N Gutenkunst, Jordan C Atlas, Fer gal P Casey , Robert S K uczenski, Joshua J W aterf all, Chris R Myers, and James P Sethna. Sloppycell. http://sloppycell.sourcefor ge.net. [14] P Ruof f, M Christensen, J W olf, and R Heinrich. T emperature dependency and temperature compen- sation in a model of yeast glycolytic oscillations. Biophys Chem , 106:179, 2003. [15] V Balasubramanian. Statistical inference, occam’ s razor , and statistical mechanics on the space of probability distributions. Neural Comput , 9:349, 1997. [16] W Bialek, I Nemenman, and N T ishby . Predictability , complexity , and learning. Neural Comput , 13:2409, 2001. [17] G Schwarz. Estimating the dimension of a model. The annals of statistics , 6(2):461, 1978. [18] D.H. W olpert and W .G. Macready . No free lunch theorems for optimization. IEEE T ransactions on Evolutionary Computation , 1(1):67–82, April 1997. 18 0 20 40 r Condition 1 Condition 2 ... Condition N 0 50 1 0 dr dt 0 50 0 50 100 Time (units GM /v 3 0 ) 0 2 4 6 8 0 . 0 1 . 5 3 . 0 To t a l phosphorylation Condition 1 V =0 . 327 0 2 4 6 8 Condition 2 V = 12 . 92 0 2 4 6 8 10 ... Condition N V =0 . 017 Time (minutes) I n -sa mp l e d a t a Planetary motion Multi-site phosphorylation FIG. S2: T ypical in-sample data points for the planetary motion and multi-site phosphorylation model examples. For the planetary motion, r 0 is treated as input, and for each in-sample r 0 , r is measured, with added noise, at a single randomly chosen time between 0 and 100. For multi-site phosphorylation, the single parameter V is treated as input, and the total phosphorylation is measured, with added noise, at a single randomly chosen time between 0 and 10 minutes. Dotted lines sho w the original model behavior , filled circles with error bars show the in-sample data, and unfilled circles sho w the v arying initial conditions in the planetary motion case. The original planetary motion model includes a single hidden variable X 2 corresponding to the time deriv ati v e of r . (For the yeast glycolysis e xample, a similar depiction of typical in-sample data is sho wn in the left panel of FIG. 4.) 19 0 2 4 6 8 1 0 1 Fit model ( N = 180 ) 0 2 4 6 8 1 0 1 dr /dt Tr u e m o d e l r 0 =1 . 0 (circle) 0 2 4 6 8 1 0 1 0 2 4 6 8 1 0 1 dr /dt r 0 =1 . 5 (ellipse) 0 10 20 30 40 50 60 r 1 0 1 0 10 20 30 40 50 60 r 1 0 1 dr /dt r 0 =2 . 5 (hyperbola) 0 50 100 150 200 Time (units GM /v 3 0 ) 0 20 40 60 80 100 120 Distance from sun (units GM /v 2 0 ) r 0 1 . 00 (circle) 1 . 25 (ellipse) 1 . 50 (ellipse) 1 . 75 (ellipse) 2 . 00 (parabola) 2 . 25 (hyperbola) 2 . 50 (hyperbola) A B FIG. S3: Fit of sigmoidal model to planetary data. W e kno w that the sigmoidal network model class is not likely to perform as well for the planetary data case because gravitational interactions do not saturate. Here we show the performance of a model fit to N = 180 data points, which contains three hidden variables. The model still fits well in the time region where data is gi v en (between 0 and 100 GM /v 3 0 , corresponding to the left half of A and the dark blue part of the trajectories in B), but has a larger div ergence from the expected behavior at the extremes of the range of giv en r 0 s in the extrapolated time region (corresponding to the right half of A and the light blue part of the trajectories in B). 20 0 1 2 3 4 5 10 − 3 10 0 10 3 Input V Input V 0 . 0 1 . 2 2 . 4 T otal phosphor ylation T otal phosphor ylation output Full model (best fit) Sigmoidal best fit S-systems best fit Full model true parameters FIG. S4: A typical example of out-of-sample performance in the multi-site phosphorylation e xample. Here, each model is fit using N = 50 datapoints. W ith this small amount of data, the differences between model classes are more apparent, with the sigmoidal model class clearly better predicting the dynamics than the S-systems model class and the full phosphorylation model. 21 S1 S2 A3 S3 N2 S4 S4ex 1 3 1 1 1 1 _ co mp l e xExa mp l e _ n e t w o rk_ sma l l F o n t _ Mo d e l 9 . p d f ye a st _ n e t w o rkF i g u re _ o ri g i n a l . p d f FIG. S5: (Left) Network depicting the yeast glycolysis model defined by Eqns. (S14). Solid arrows represent excitation, solid lines with circles represent inhibition, and dashed arrows represent other types of interaction terms. (Right) Selected sigmoidal network fit to N = 40 noisy measurements from the yeast glycolysis model, as shown in FIG. 4. Again, arro ws represent excitation and circles inhibition, with the thickness of arro ws indicating interaction strength. For clarity , self-inhibitory terms for each v ariable are not sho wn. 0 10 20 30 40 Number of measurements N 0 . 0 0 . 2 0 . 4 0 . 6 0 . 8 1 . 0 Mean out-of-sample correlation Sigmoidal, wide out-of-sample range Sigmoidal, narro w out-of-sample range FIG. S6: Performance of inferred models of yeast glycolysis as a function of the number of measurements N . Here we compare mean correlations produced for out-of-sample initial conditions chosen from ranges twice as large as in-sample ranges (“wide ranges, ” plotted in red) to when out-of-sample conditions are chosen from the same ranges as in-sample ranges (“narrow ranges, ” plotted in orange). The mean and standard de viation ov er 5 realizations of in-sample data are sho wn by filled symbols and shaded regions. 22 Sca l i n g o f co mp u t a t i o n a l e f f o rt 10 1 Number of measurements N 10 5 10 6 10 7 10 8 Computational eff or t for single N (model ev aluations) Y east glycolysis Sigmoidal / N 10 1 10 2 Number of measurements N 10 5 10 6 10 7 10 8 Computational eff or t for single N (model ev aluations) Multi-site phosphorlyation Sigmoidal / N FIG. S7: The number of model ev aluations (integrations) used at each N , for the multi-site phosphorylation and yeast glycolysis examples. Once the size of model has saturated, we expect the number of e v aluations to scale linearly with N (black lines). If the selected model size is growing with N , as in the yeast glycolysis example belo w N = 20 (see FIG. S8), we expect faster than linear gro wth. 0 5 10 15 20 25 30 35 40 45 Number of measurements N 0 20 40 60 80 100 120 Number of parameters in selected model Sigmoidal Sigmoidal, eff ectiv e Number of data points N D FIG. S8: Fitting sigmoidal models to the yeast glycolysis oscillation data, the number of total parameters in the selected model, plotted in red, saturates to roughly 65. The solid line compares the number of parameters in the selected model to the number of data points N D used to infer the model. In orange, we plot the ef fecti ve number of parameters, which we define as the number of directions in parameter space that are constrained by the data such that the corresponding Hessian eigen v alue λ > 1 (compared to parameter priors with eigen v alue 10 − 2 ). W e expect the optimal effecti v e number of parameters to stay below N D . Sho wn are the median and full range of v alues ov er 5 data realizations. 23
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment