A Unifying Model for the Analysis of Phenotypic, Genetic and Geographic Data
Recognition of evolutionary units (species, populations) requires integrating several kinds of data such as genetic or phenotypic markers or spatial information, in order to get a comprehensive view concerning the differentiation of the units. We pro…
Authors: Gilles Guillot, Sabrina Renaud, Ronan Ledevin
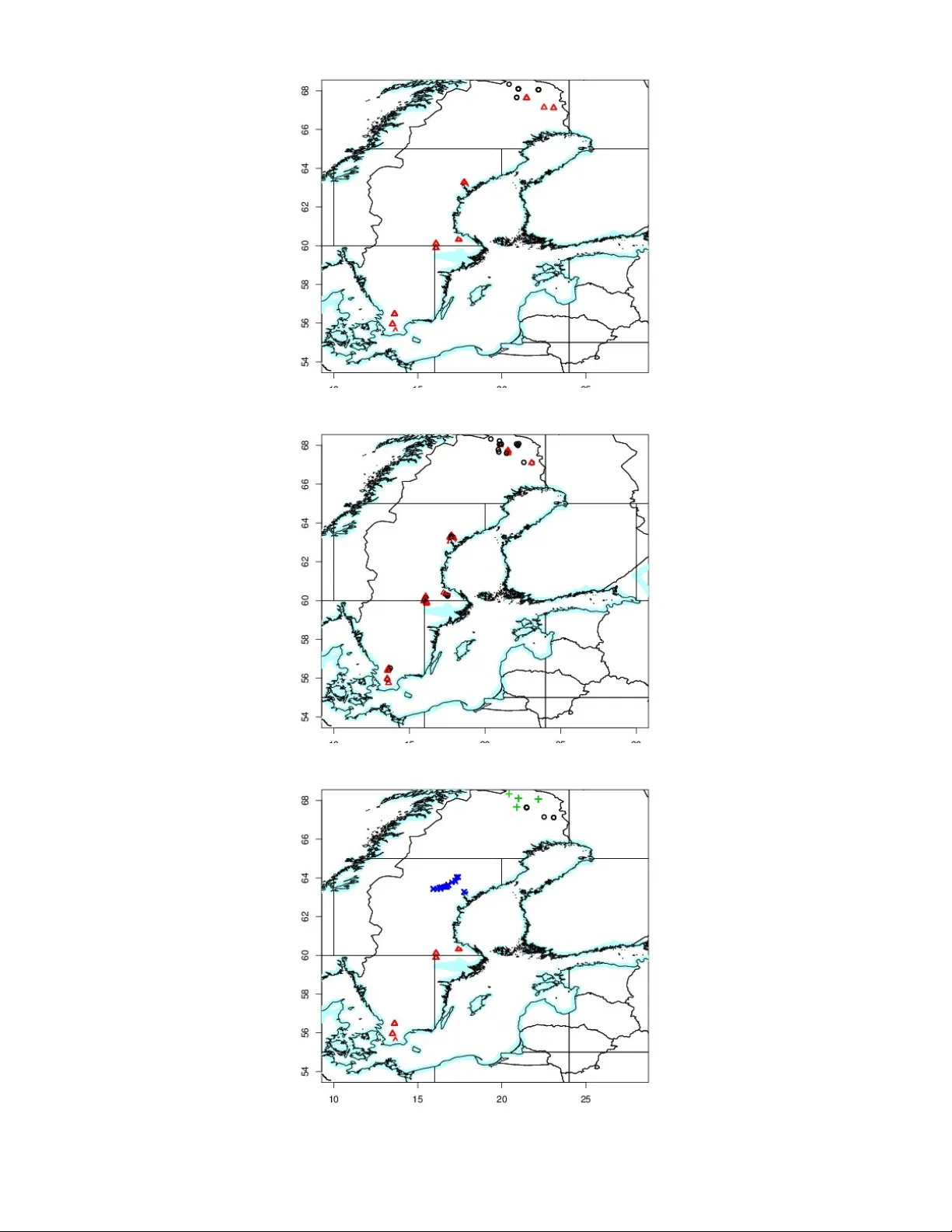
A Unifying Model f or the Anal ysis of Phenotypic, Genetic and Geographic D a t a Gilles Guillot ∗ † , Sabrina Renaud ‡ , Ronan Ledevin ‡ , Johan Mic haux § , Julien Claude ¶ No vem ber 4, 2018 ∗ Corresp onding author; gigu [at] imm.dtu.dk. † Informatics Departmen t, T ec hnical Univ ersity of Denmark, Copenhagen, Denmark. ‡ Lab oratoire de Biom´ etrie et Biologie Ev olutive UMR 5558, CNRS, Universit ´ e Lyon 1, Universit ´ e de Ly on, 69622 Villeurbanne, F rance, § CBGP , INRA/IRD/CIRAD/SupAgro, Mon tferrier-sur-Lez cedex, F rance. ¶ Lab oratoire de Morphom ´ etrie, ISE-M, UMR5554 CNRS/UM2/IRD, Univ ersit´ e de Mon tp ellier I I, 34095 F rance. 1 Abstract Recognition of evolutionary units (sp ecies, p opulations) requires integrating sev eral kinds of data such as genetic or phenotypic markers or spatial information, in order to get a comprehensive view concerning the differentiation of the units. W e prop ose a statistical mo del with a double original adv antage: (i) it incorp orates information ab out the spatial distribution of the samples, with the aim to increase inference p o wer and to relate more explicitly observ ed patterns to geography; and (ii) it allows one to analyze genetic and phenotypic data within a unified mo del and inference framew ork, thus op ening the wa y to robust comparisons b etw een markers and p ossibly com bined analyzes. W e sho w from simulated data as w ell are real data from the literature that our method estimates parameters accurately and improv es alternativ e approac hes in man y situations. The in terest of this metho d is exemplified using an in tricate case of inter- and intra-species differentiation based on an original data-set of georeferenced genetic and morphometric markers obtained on Myo des v oles from Sweden. A computer program is made a v ailable as an extension of the R pack age Geneland. Keyw ords Clustering, spatial data, bio-geography , Ba yesian mo del, Marko v c hain Monte Carlo, R pack age, mor- phometrics, molecular markers, Myo des . 2 Sp ecies delimitation are of interest in conserv ation biology (identification and management of endangered species), epidemiology (detection of new pathogens) but also from a purely cognitive p oin t of view to describe, quantify and understand mechanisms of speciation. Metho dological adv ances in evolutionary biology ha v e led to methods for sp ecies identification solely based on the v ariation of k ey genetic markers (e.g. DNA barcoding, Luo et al., 2011). Limits of these single-mark er approac hes are more and more evidenced b y conflicts betw een differen t genes in a m ulti-marker approac h (Ro dr ´ ıguez et al., 2010; T urmelle et al., 2011) or b etw een genetic and phenot ypic mark ers (Nesi et al., 2011). In this con text of sp ecies or p opulation iden tification, phenot ypic data still emerge of in terest together with genetic markers. Phenot ypic data such as size and/or shap e of morphological structures are the pro duct of numer- ous in teracting nuclear genes (Klingen b erg et al., 2001) and as such can pro vide a global estimate of the div ergence betw een units. F urthermore, by b eing the target of the screening b y selection, morphological v ariation can pro vide precious insigh ts on the selection pattern con tributing to shap e the units. In the case of fossil lineages, it may ev en b e the only information a v ailable to iden tify evolutionary and systematic units (N´ eraudeau, 2011; Girard and Renaud, 2011). A rich toolb ox is a v ailable to tackle these questions. Man y metho ds work as partition cluster- ing, and aim at defining how man y groups are represen ted in a sample of individuals, and assign these individuals to these groups follo wing some optimality principles. These metho ds w ere ini- tially developed to d eal with con tin uous quantitativ e measuremen ts. These classical clustering metho ds ha ve b een implemen ted in programs such as Emmix (McLac hlan et al., 1999) or Mclust (F raley, 1999) or Mixmod (Biernacki et al., 2006). The methods ab o ve did not received a strong in terest in Systematics until recen t Population Genetics extensions to deal with molecular data suc h as the widely used computer program Structure (Pritchard et al., 2000) and related work (review ed e.g. b y Excoffier and Heck el, 2006). More recen tly , Hausdorf and Hennig (2010) and Y ang and Rannala (2010) developed metho ds for delimiting sp ecies based on multi-locus data. While the approach of Hausdorf and Hennig (2010) metho d hinges on Gaussian clustering, the metho d of Y ang and Rannala (2010) is based on the coalescen t and makes use of a user-sp ecified guide tree. Metho ds for genetic data ha v e b een also extended to incorp orate information about the spatial location of eac h sample - an information rarely used although commonly av ailable in data analysis in ev olutionary biology - with the aim of increasing p o wer of inferences and of relating more explicitly observ ed patterns to geography (Guillot et al., 2005, 2009). 3 These to ols hav e been developed by differen t comm unities (evolutionists, population geneti- cists, statisticians). Therefore, one still lac ks a unified framew ork, and this constitutes a ma jor dra wback for combining v arious kinds of data. This is esp ecially true for morphological mark- ers that did not receiv ed as muc h attention as genetic mark ers for recognizing p opulations and sp ecies. There are therefore a few ma jor gaps in the to olb ox av ailable to iden tify ev olutionary units, namely there is to date: no metho d to analyze genetic data and phenot ypic data under the same general paradigm (model and inference framework), and no metho d to incorp orate spatial information in suc h phenotypic/genetic analysis. The goal of the presen t paper is to fill these gaps. W e prop ose a mo del to deal in an integrated w ay with georeferenced phenotypic and genetic data and we provide a computer program freely a v ailable that implemen ts this model and should ease data analysis in man y resp ects. Giv en the complexit y of the mo deling and inferen tial task, our metho d is not based on an explicit ev olutionary mo del (for example based on the coalescent) but on a statistical mo del. This mo del is a parametrization whic h is general enough to capture some essential features in the data v ariation, but also simple enough to be sub ject to a rigorous and accurate inference method. Briefly , our mo del assumes the existence of sev eral clusters which displa y some kind of homogeneity . This mo del mimics more or less what would be expected from a p opulation: homogeneit y in terms of genetic and phenotypic v ariation and some geographical con tinuit y . The existence of homogeneous clusters corresp onds to the fact that some individuals ha v e shared some asp ects of their recen t ecological or ev olutionary history . This shared history is summarized by cluster- sp ecific parameters whic h are allele frequencies and means and v ariances of phenotypic traits. Because it is not based on an explicit ev olutionary mo del, it do es not require prior information (as for instance a guide tree in the case of Y ang and Rannala’s metho d). The statistical challenge in this con text is to estimate the n um b er of clusters and these cluster-sp ecific parameters. This article is organized as follo ws. First we provide a description of the model and inference mac hinery . Next we illustrate our method and test its accuracy on a large set of simulated data as w ell as on tw o published real data-sets. Then w e implemen t our method on an original data-set of georeferenced genetic and morphometric markers to decipher the complex inter-and in tra-sp ecific structure of red-bac k ed and bank v oles Myo des rutilus and M. glar e olus in Sweden. W e conclude b y discussing p oten tial applications in a more general context. 4 Method Ov erview W e assume that we hav e a data-set consisting of n individuals sampled at sites s = ( s i ) i =1,..., n (where s i is the t w o-dimensional spatial co ordinate of individual i ), observed at some pheno- t ypic v ariables denoted y = ( y ij ) i =1,..., n j =1,..., q and/or some genetic mark ers denoted z = ( z ij ) i =1,..., n j =1,..., l . Our approac h is able to deal with an y com bination of phenotypic and genetic data, including situations where only phenotypic or only genetic data are av ailable and situations when eac h individual is observ ed through its o wn combination of phenotypic and genetic markers. As it will b e shown b elow, our approach also encompasses the case where sampling lo cations are missing (or considered to be irrelev an t). The only constrain t that w e imp ose at this stage is that if spatial co ordinates are used, they m ust b e av ailable for all individuals. W e assume that eac h individual sampled b elongs to one of K differen t clusters and that v ariation in the data can b e captured b y cluster-sp ecific lo cation and scale parameters. Prior and Lik eliho o d Model for Phenot ypic V ariables Denoting b y p i the cluster mem b ership of individual i ( p i ∈ { 1, ..., K } ), we assume that condi- tionally on p i = k , y ij is drawn from a parametric distribution with cluster-sp ecific parameters. Indep endence is assumed within and across clusters conditionally on cluster mem b ership. This means in particular that there is no residual dep endence b etw een v ariables not captured b y cluster mem b erships. Implications of this assumption are discussed later. Although most of the analysis that follo ws w ould b e v alid for all families of contin uous distribution, w e assume in the following that the y v alues arise from a normal distribution. Eac h cluster is therefore c haracterized b y a mean µ kj and a v ariance σ 2 kj and our mo del is a mixture of multiv ariate indep endent normal distributions (F r ¨ uh wirth-Schnatter, 2006). F ollo wing a common practice in Ba y esian analysis (Gelman et al., 2004), we use the natural conjugate prior family on ( µ kj , 1 /σ 2 kj ) for each cluster k and v ariable j . Namely , we assume that the precision 1 /σ 2 kj (i.e. in v erse v ariance) follows a Gamma distribution G ( α , β ) ( α shap e, β rate parameter) and that conditionally on σ kj , the mean µ kj has a normal distribution with mean ξ and v ariance σ 2 kj /κ . In the sp ecification ab o ve, α , β , ξ and κ are h yp er-parameters. Details ab out their c hoice are discussed in the app endix and in the supplemen tary material. 5 Prior and Lik eliho o d Model for Genetic Data W e assume here a mixture of multinomial distributions. This is the model previously in tro duced b y Pritchard et al. (2000) to mo del individuals with pure ancestries. Denoting frequency of allele a at lo cus l in cluster k b y f kla , for diploid genot yp e data w e assume that π ( z ij = { a , b }| p i = k ) = 2 f kla f klb whenev er a 6 = b (1) and π ( z ij = { a , a }| p i = k ) = f 2 kla . (2) While for haploid data, we ha ve π ( z ij = a | p i = k ) = f kla (3) W e also deal with dominant mark ers for diploid organisms with a mo dified likelihoo d (see Guillot and San tos, 2010; Guillot and Carp entier-Sk andalis, 2011, for details). W e assume indep endence of the v arious lo ci within and across clusters conditionally on cluster mem b erships. In particu- lar, as with all other p opulation genetic clustering mo dels (including Structure ), we do not attempt to mo del background link age disequilibrium (LD). Therefore, our mo del can handle non- recom bining DNA sequences (suc h as data obtained from mito c hondrial DNA, Y c hromosomes or tigh tly linked autosomal n uclear markers) pro vided data are reformatted in such a wa y that the v arious haplot yp es are recoded as alleles of a single lo cus, but see also discussion. W e assume that allele frequencies f kl . ha ve a Diric hlet distribution. Indep endence of the v ectors f kl . is assumed across lo ci. Regarding the dep endence structure across clusters, we consider either indep endence (referred to as Uncorrelated F requency Mo del or UFM) or an alternativ e mo del (referred to as Correlated F requency Mo del or CFM) introduced b y Balding and Nic hols (1995, 1997). In this second mo del, allele frequencies also follo w a Diric hlet distribution but no w depending on some cluster-sp ecific drift parameters. In this mo del, f kl . are assumed to follo w a Diric hlet distribution D ( ˜ f la (1 − d k ) / d k , ..., ˜ f lA (1 − d k ) / d k ) where d k s parametrize the sp eed of divergence of the v arious clusters and the ˜ f la s represent the allele frequency in an hypothetical ancestral p opulation. This mo del can be view ed as a heuristic and computationally con venien t appro ximation of a scenario in which presen t time clusters result from the split of an ancestral cluster some generations ago. It is also a Ba y esian wa y of introducing correlation betw een clusters at the allele frequency level and hence to infer subtle differentiations that would hav e b een missed b y a model assuming in- dep endence of allele frequencies across clusters (F alush et al., 2003; Guillot, 2008; Sir´ en et al., 6 2011) . Prior Models for Cluster Mem b ership Spatial mo del W e consider a statistical model kno wn as colored Poisson-V oronoi tessellation. Lo osely speaking, this model assumes that eac h cluster area in the geographic domain can b e appro ximated b y the union of a few p olygons. Most of the mo deling ideas can b e grasp ed from the examples sho wn in figure 1. The p olygons are assumed to b e cen tered around some p oin ts that are generated b y a homogeneous Poisson process (i.e. points located completely at random in the geographic domain). F ormally , we denote by ( u 1 , ..., u m ) the realization of this P oisson pro cess. These p oin ts in R 2 induce a V oronoi tessellation in to m subsets ∆ 1 , ..., ∆ m . The V oronoi tile asso ciated with p oint u i is defined as ∆ i = { s ∈ R 2 , dist ( s , u i ) < dist ( s , u j ) ∀ j 6 = i } . Each tile receiv es a cluster mem b ership c i (co ded graphically as a color hence the terminology) at random sampled indep enden tly from a uniform distribution on { 1, ..., K } . Denoting b y D k the union of tiles with color k , the set ( D 1 , ..., D K ) defines a tessellation in K subsets. This mo del is controlled b y the in tensity of the Poisson pro cess λ (the av erage num ber of points p er unit area) and the n um b er of clusters K . W e place a uniform prior on [0, λ max ] and on { 0, ..., K max } resp ectiv ely . This mo del is a flexible tool widely used in engineering to fit arbitrary shap es in a non-parametric w ay (Møller and Sto y an, 2009). It offers a go o d trade-off b etw een mo del complexity , realism and computational efficiency . It is presumably most useful in situations of incipien t allopatric sp eciation but examples of applications in other contexts can be found e.g. in the studies of Coulon et al. (2006); F on taine et al. (2007); W asser et al. (2007); Hannelius et al. (2008); Joseph et al. (2008); Sacks et al. (2008); Galarza et al. (2009); Beadell et al. (2010). See also Guillot et al. (2009) for review and additional references. Lastly , we note that our approac h relates to that of Hausdorf and Hennig (2003) who propose a test for clustering of areas of distribution. Ho w ever, rather than testing clusteredness, our approac h estimates these areas of distribution. T o do that, w e assume some clusteredness but without making strong assumptions ab out its intensit y . Non-spatial mo del If spatial co ordinates are not a v ailable or though t to b e irrelev ant to the sp ecies at the spatial scale considered, then a non-spatial mo del can b e used. The non-spatial mo deling option considered 7 Figure 1: Examples of spatial clusters sim ulated from our prior mo del. The square represents the geographic study area. Mem b ership of a geographical site to one of the K clusters is coded b y a color. F rom left to righ t: K = 2, 3 and 4 . A given clustering dep ends on K , and on the n um b er, lo cations and colors (cluster memberships) of eac h p olygon. If the prior placed on the num b er of p olygons tends to fav ors low v alues, then each cluster tends to be made of one or only a few large areas. This is in sharp contrast with non-spatial Bay esian mo dels which t ypically assume that clusterings with highly fragmented cluster areas are not unlikely . here do es not require to in tro duce an y auxiliary p oin t process as ab ov e but for the sak e of consistency , w e use the same setting as in the paragraph ab ov e. W e set m = n and imp ose ( u 1 , ..., u n ) = ( s 1 , ..., s n ) . Here the s i s are some kno wn spatial co ordinates or dummy p oin ts if this piece of information is missing. This mo del does not imp ose any spatial structure and corresp onds to the model implemented in most non-spatial cluster programs, including the genetic clustering programs Baps (Corander et al., 2003, 2004) and Structure (with the exception of the latest mo del presented b y Hubisz et al. (2009). Summary of Prop osed Mo del The parameters in our mo del are as follo ws: num b er of clusters K , rate of P oisson process λ , n umber of even ts (p oints) of the P oisson pro cess m , even ts of Poisson pro cess u = ( u 1 , ..., u m ) , color of tiles (i.e. cluster membership of spatial partitioning sub-domains) c = ( c 1 , ..., c m ) , allele frequencies f = ( f kla ) (frequency of allele a at locus l in cluster k ), genetic drift parameters d = ( d 1 , ..., d K ) , allele frequencies in the ancestral p opulation ˜ f = ( ˜ f la ) , exp ectations of phenotypic v ariables µ = ( µ kj ) , standard deviations of phenot ypic v ariables σ = ( σ kj ) (note that σ is not a v ariance-cov ariance matrix (the phenot ypic v ariables are assumed to be indep endent) but rather 8 a set of scalar v ariances stored in a t wo-dimensional arra y . On top of this, we place a uniform prior on [0, λ max ] on λ , a uniform prior on { 0, ..., K max } on K , a Beta B ( δ k , δ k )) prior on d k and a Gamma distribution G ( g , h ) on β . The vector of unknown parameters is therefore θ = ( K , λ , m , u , c , f , ˜ f , d , µ , σ , β ) . W e also denote by θ S = ( λ , m , u , c ) , θ G = ( f , ˜ f , d ) and θ P = ( µ , σ , β ) the parameters of the spatial, genetic and phenot ypic parts of the mo del respectively . The hierarc hical structure of the mo del is summarized on the graph shown in figure 2. There are three blo c ks of parameters relativ e to the genetic, phenotypic and geographic comp onent of the mo del. Information propagates from data to higher lev els of the mo del across the v arious no des of the graph through probabilistic relationships sp ecified b etw een neighboring nodes. The structure of the global mo del can b e summarized by the join t distribution of θ and ( y , z ) . By the conditional indep endence assumptions, w e get π ( θ , y , z ) = π ( θ ) π ( y , z | θ ) = π ( θ ) π ( y | θ ) π ( z | θ ) = π ( θ ) π ( y | θ P ) π ( z | θ G ) (4) Eac h genetic or phenotypic marker brings one factor in the lik eliho o d. Whether the clustering is driven b y the genetic or the phenotypic data dep ends on the respective differentiation and on the num ber of markers of each kind. Estimation of P arameters Ba y esian estimation and Mark o v chain Mon te Carlo inference W e are in terested in the p osterior distribution π ( θ | y , z ) . Note that this notation do es not re- fer explicitly to the sample lo cations b ecause, unlike genetic markers and phenotypic v ariables, lo cations are not considered as random quantities in our model. The mo del do es in fact implic- itly accoun t for spatial information. The distribution π ( θ | y , z ) is defined on a high dimensional space and deriving prop erties analytically about this distribution is out of reach. W e implemen t a Marko v c hain Monte Carlo strategy . This amounts to generating a sample of N correlated replicates ( θ 1 , ..., θ N ) from the p osterior distribution π ( θ | y , z ) . The initial state θ 1 is simulated at random from a distribution that do es not matter in principle, a fact that has to b e c hec ked in practice b y con vergence monitoring to ols (Gilks et al., 1996; Rob ert and Casella, 2004). W e 9 alw ays sample θ 1 from the prior and we c hec k that starting from v arious random states do es not affect the ov erall result provided a suitable num b er of burn-in iterations are discarded. In analyzes rep orted below, the order of magnitude of N was 50000-100000 iterations with 20000 burn-in iterations. See app endix for detail on the MCMC algorithm. Estimation of the num b er of clusters Eac h simulated state θ i includes a sim ulated num ber of clusters K i . The n umber of clusters is estimated as the most frequent v alue among the N sim ulated v alues K 1 , ..., K N and we denote it b y ˆ K . Estimating cluster mem b erships A model assuming that individuals i and j b elong resp ectively to clusters 1 and 2 c haracterized b y a mean phenot ypic trait equal to 5 and 7 is essen tially the same as a mo del assuming that individuals i and j b elong resp ectiv ely to clusters 2 and 1 c haracterized by a mean phenot ypic trait equal to 7 and 5. This trivial fact is due to the in v ariance of the likelihoo d under p erm utation of cluster lab els and brings up a n um b er of computational difficulties in the p ost-pro cessing of MCMC algorithm outputs known as the lab el switching issue (Stephens, 1997). In particular, it does not make sense to a verage v alues across the MCMC iterations. T o deal with this, w e implemen t the strategy described b y Marin et al. (2005) and Guillot (2008). W e consider the set of sim ulated θ v alues restricted to the set of states suc h that K = ˆ K . Then w orking on this restricted set, we relab el each state in suc h a w ay that they “b est lo ok like” the mo dal state of the p osterior distribution. Cluster memberships of each individual are estimated as the modal v alue in this relab eled sample. Then w e estimate all cluster-specific parameters (mean phenot ypic v alues and allele frequencies) b y taking the a verage sim ulated v alue ov er the relab eled sample. Anal ysis of Simula ted D a t a W e inv estigate here t w o new asp ects of the mo del, namely its abilit y to cluster phenot ypic data only and phenot ypic and genetic data jointly together with some spatial information. 10 Inference from Phenot ypic Data Only In this section, w e present new results on the model for phenotypic data and fo cus on the spatial mo del option. W e carried out simulations from our prior mo del and p erformed inferences as describ ed in section “Estimation of parameters” ab ov e. W e pro duced data-sets consisting of n = 200 individuals with q = 5, 10, 20 and 50 phenot ypic v ariables. F or eac h v alue of q , w e pro duced 500 data-sets with a uniform prior U ( { 1, ..., 5 } ) on K . In real-life, the range of v alue of the putative true K is largely unkno wn. T o b e as close as p ossible to this situation, w e carried out inference under a uniform U ( { 1, ..., 10 } ) prior for K . W e assessed the accuracy of inferences b y computing the classification error whic h is displa y ed in figure 3. F urther details are provided in Supp orting Material. W e also wished to assess ho w our metho d p erforms compared to other computer programs implemen ting state-of-the-art metho ds. W e therefore considered the R pac k age Mclust (Banfield and Raftery, 1993; F raley, 1999) whic h is one of the most widely used and arguably most adv anced program to p erform clustering. This program implemen ts inference for Gaussian mixtures and as suc h deals solely with con tinuous quan titative data. It implements a non-spatial algorithm and in its default setting p erforms inference b y lik eliho o d maximization via the Exp ectation Maximization (EM) algorithm. It implements a wide class of sub-mo dels regarding the co v ariance structure of the data. In its default option (which we used) it p erforms mo del selection (cov ariance structure and num b er of clusters) by optimizing a Ba yesian Information Criterion (BIC). W e set the maxim um n umber clusters to the K max = 10 , i.e. to the same v alue as in analyzes with our metho d. W e stress here that the goal of this exp eriment is not to rank our method and Mclust as the t wo methods/programs differ in man y imp ortan t resp ects. They differ regarding the t yp e of data handled ( Mclust is not aimed at genetic data and do es not implement any spatial model) and the breadth of cov ariance structure considered (our approac h assumes conditional indep endence while Mclust considers in excess of ten t yp es of co v ariance structures). It would b e therefore difficult to design an efficient and fair comparison. Results are mostly giv en here to supp ort the claim that our metho d compares with state-of-the-art metho ds and to assess the magnitude of impro vemen t brough t b y the use of a spatial mo del in a b est-case scenario when data are spatially structured (see also discussion). Most of the numerical results are summarized in figure 3. T o understand better ho w the metho d b ehav es as a function of the pairwise phenot ypic 11 differen tiation betw een clusters, w e also rep ort the classification error as a function of the T 2 statistic in a Hotelling T test (Anderson, 1984) on figure 4. See also supporting material for further details. Inference from Phenot ypic and Genetic Data jointly W e illustrate here ho w combining phenot ypic and genetic data can improv e the accuracy of inferences compared to inferences carried out from one t yp e of data only . T o do so, w e sim u- lated 500 data-sets consisting of t w o clusters each. There were five phenot ypic v ariables and ten co-dominan t genetic markers. W e inv estigated a broad range of phenot ypic and genetic differen- tiation and it app ears that on a v erage com bining the t w o types of data increases the accuracy of inferences. See figure 5. Anal ysis of D a t a from the Litera ture Analysis of Iris Morphometric Data Fisher’s iris data-set (Anderson, 1935; Fisher, 1936) giv es the measuremen ts in centimeters of the v ariables sepal length and width and p etal length and width, resp ectiv ely , for 50 flo w ers from each of 3 sp ecies of iris. The sp ecies are Iris setosa , versic olor , and vir ginic a . W e applied our metho d to the data transformed into log shap e ratios (see Claude, 2008, and references therein). Since the data are not georeferenced, we used the non-spatial prior. W e launched ten indep enden t MCMC runs. Seven of them return correctly ˆ K = 3 , the other three runs return ˆ K = 4, 5 and 6 respectively . Ranking the runs according to the a v erage posterior densit y , the b est run corresp onds to one of the seven runs that estimate K correctly (according to the num b er of actual sp ecies in the data set). This run ac hiev es a classification error of 6 % (see Fig. 6). Mclust returns an estimate of K equal to 2 (ra w data or log shap e ratio data) and 50 out of 150 individuals are misclassified, thus failing to identify the three sp ecies of the data set. AFLP Data of Calop o gon from Eastern North America and the Northern Caribb ean The w ay our mo del deals with genetic data and the accuracy resulting from this metho d based on genetic data only has been in v estigated by Guillot et al. (2005, 2008); Guillot (2008); Guillot 12 and San tos (2010); Safner et al. (2011); Guillot and Carp en tier-Sk andalis (2011) and further discussion can b e found in Guillot et al. (2009), how ev er, to further illustrate the accuracy of our metho d when used with genetic data only , w e study here a dataset pro duced and first analyzed b y Goldman et al. (2004). This dataset consists of sixt y Calop o gon samples genot yp ed at 468 AFLP mark ers. Goldman et al. (2004) iden tified the presence of fiv e species ( C. b arb atus, C. oklahomensis, C. tub er osus, C. p al lidus, C. multiflorus ) and t wo hybrids sp ecimens ( C. tub er osus × C. p al lidus and C. p al lidus × C. multiflorus ). According to Goldman et al. (2004), C. tub er osus has b een widely considered to hav e three v arieties: v ar. tub er osus , v ar. latifolius and v ar. simpsonii . In addition, the dataset con tains samples from tw o outgroups so that one could consider that the dataset con tains up to elev en distinct sp ecies. W e analysed this dataset under the same setting as the previous dataset. Under the UFM, the estimated K ranges b etw een 2 and 3 . The b est run (in terms of a verage p osterior density) corresp onds to ˆ K = 3 . In this clustering, one cluster con tains the samples of the C. tub er osus sp ecies, a second cluster merges the samples of the C. b arb atus, C. oklahomensis, C. p al lidus, C. multiflorus species and the h ybrids. The last cluster con tains the samples from the t w o outgroups. Under the CFM, the estimated K ranges b etw een 7 and 8 . The b est run (in terms of av erage p osterior density) corresponds to ˆ K = 8 . It clusters the individuals of the v arious sp ecies as follo ws: C. oklahomensis / C. multiflorus / C. b arb atus / C. p al lidus , C. tub er osus × C. p al lidus and C. p al lidus × C. multiflorus / C. tub er osus tub er osus except three samples / the three C. tub er osus tub er osus previous samples / t w o extra clusters for the outgroups. Anal ysis of Myo des V ole D a t a Data and statistical analysis W e no w study an original dataset of geo-referenced genetic and phenotypic mark ers of the v oles of the gen us Myo des in Sw eden. This dataset has sev eral interests to inv estigate the efficiency of our metho d on a complex real case. (i) F ennoscandia has b een recognised as a zone where the mito c hondrial DNA of the northern red-bac k ed v ole Myo des rutilus in trogressed its southern relativ e, the bank vole M. glar e olus (T egelstr¨ om, 1987). This mak es the identification of these t wo sp ecies impossible based on common mitochondrial markers. (ii) The bank v ole is further c haracterized b y in tra-sp ecific lineages (Deffon taine et al., 2009). Two of them are documented in 13 Sw eden (Razzauti et al., 2009), pro viding a complex case for disen tangling intra- and inter-specific structure. (iii) Both genetic and morphological data are a v ailable on this model to confron t the structure provided b y the t w o kinds of markers, and test for their combination. The dataset consists of 182 individuals. These individuals w ere genot yp ed at 14 microsatel- lite lo ci (Lehanse, 2010). The phenotypic dataset corresp onds to a subsample of 69 individuals (Ledevin, 2010). W e used measurements of the third upp er molar shap e, for which a pheno- t ypic differentiation has b een evidenced at the phylogeographic scale (Deffontaine et al., 2009; Ledevin et al., 2010a). The t wo-dimensional outline w as man ually registered from n umerical pictures, starting from a comparable starting point among teeth (Ledevin et al., 2010a). F or eac h molar, the outline is describ ed b y the Cartesian co ordinates of 64 p oin ts sampled at equally spaced interv als along the outline. These 64 landmarks are strongly correlated and therefore carry redundant information. T o summarize this information into a low er n umber of v ariables and decrease the intensit y of correlation b et ween v ariables, w e first performed an elliptic F ourier transform (EFT Kuhl and Giardina, 1982). The EFT pro vides shap e v ariables standardized by size, the F ourier coefficients that w eigh t the successive functions of the EFT, namely the h ar- monics. A study of the successive con tribution of each harmonic to the description of the original outline sho w ed that considering the first ten harmonics offered a go o d compromise b etw ee n the n umber of v ariables and the efficien t description of the outline (Ledevin et al., 2010a). Then w e p erformed a principal comp onent analysis of the F ourier co efficients and retained the scores on the first fiv e principal comp onents, whic h contained more than 80% of the v ariance (PC1=26.6%, PC2=21.6%, PC3=15.2%, PC4=7.4%, PC5=6.5%). These scores w ere used as phenot ypic data input (the y data matrix) to our clustering metho d. W e analysed this dataset with our mo del first under the UFM allele frequency prior then under the CFM prior. F or eac h allele frequency prior, we fed the mo del with fiv e types of data com bination: using the georeferenced phenotypic data under the spatial mo del (PS), using the phenot ypic data under the non-spatial mo del (PnS), using the georeferenced genetic data under the spatial mo del (GS), using the genetic data under the non-spatial mo del (GnS), using the georeferenced phenotypic and genetic data under the spatial mo del (PGS). In eac h case, w e p erformed 10 indep enden t MCMC runs of 100000 iterations discarding the first 10000 iterations as burnin. 14 Results F or eac h t ype of analysis, we observed an excellen t congruence across the ten independent MCMC runs. The UFM and the CFM mo del provide qualitatively similar results with a tendency of the CFM mo del to return sligh tly larger estimates of K . While the CFM option has pro ven to detect finer differen tiation than the UFM option (see analysis of AFLP data abov e), a detailed analysis and in terpretation of the fine scale structure inferred b y the CFM mo del w ould require extended data analysis, including some extra data still under pro duction. W e therefore fo cus on the results obtained under the UFM option. In the analysis based on georeferenced phenotypic data (PS), we inferred t wo clusters with one cluster in the top North of Sweden (Fig. 7 top panel), all remaining samples b elonging to the other cluster. These clusters corresp ond to the inter-specific differentiation b etw een the red-bac ked v ole to the North and the bank v ole to the South. Analysing these data without spatial information (PnS), w e also inferred also t w o clusters (Fig. 7 middle panel). The areas o ccupied b y the t w o clusters under the PS and the PnS analyzes match in the sense that they b oth correspond to a top North vs. South dic hotom y with a region of marked transition estimated to b e along the same line in Swedish Lapland with a SW-NE orientation. In the PnS analysis, the clusters displa y a large amoun t of spatial ov erlap with a regular North to South cline. In the analysis based on georeferenced genetic data (GS), w e inferred the presence of four clusters. The most northern cluster corresp onds to the samples iden tified as b elonging to the top North cluster in the phenotypic clustering, and hence to the Northern red-bac ked vole (Fig. 7 bottom panel). The three other clusters corresp ond to the intra-specific structure within the bank vole. This hierarchical pattern of in ter- and intra-specific differences is confirmed by estimates of inter- p opulation differen tiation provided by Fst v alues. The top North p opulation attributed to the red-bac ked v ole appears as strongly differen tiated from all other p opulations (N Sweden vs. NE Sw eden: F ST = 0.15; N vs. Central Sweden: F ST = 0.19; N Sw eden vs. South Sw eden: F ST = 0.17). In comparison, the differentiation is of smaller magnitude among bank vole p opulations (NE vs. C: F ST = 0.07; NE vs. S: F ST = 0.07; C vs. S: F ST = 0.06). Analysing these data without spatial information (GnS), we inferred four clusters whose lo cations match tigh tly those obtained under analysis GS (results not sho wn). In the joint analysis of georeferenced phenotypic and genot ypic data (PGS), we obtained results similar to those obtained with georeferenced genetic data (results not sho wn). allele frequencies allele frequencies in "ancestral population" location Scale population memberships number of clusters parameters parameters drifts Phenotypic data Spatial data Genetic data m K λ K max z f d p c u α β µ ξ κ g h ˜ f σ λ max δ y s Figure 2: Graph of prop osed mo del. Con tinuous black lines represen t sto chastic dep endencies, dashed black lines represen t deterministic dep endencies. Boxes enclose data or fixed hyper- parameters, circles enclose inferred parameters. Bold symbols refer to v ector parameters. The red, green and blue dashed lines enclose parameters relative to the phenotypic, geographic and genetic parts of the mo del respectively . The parameters of interest to biologists are the n um b er of clusters K , the vector p which enco de the cluster memberships, and p ossibly allele frequencies f , mean phenot ypic v alues µ , phenotypic v ariance σ 2 whic h quan tify the genetic and phenot ypic div ergence b etw een and within clusters. Other parameters can be view ed mostly as nuisance parameters. 16 (a) Our metho d (b) Mclust Figure 3: Classification error from simulated data. The v ariable plotted on the y-axis is the prop ortion of misclassified individuals (after correction for p oten tial lab el switching issues). Each bar is obtained as an av erage ov er 500 data-sets consisting of n = 200 individuals. Both metho ds are excellent at a voiding false p ositiv es (i.e. rep orting ˆ K = 1 when K =1) and ha ve a clear ability to reduce the error rate when the num ber of v ariables increases. They seem to lose accuracy in the same fashion when they are given an increasingly difficult problem (i.e. when the true K increases) and ha ve difficult y fully exploiting all of the av ailable information when the num ber of v ariables is large (cf. loss of accuracy for 50 v ariables compared to 20 v ariables). In the ov erall, under this type of simulated data, our metho d is t ypically t wice as accurate as the competing metho d. 17 q = 5 q = 10 q = 20 q = 50 Figure 4: Classification error for simulated data-sets consisting of K = 2 clusters as a function of the phenotypic differentiation b et ween the clusters. The v ariable plotted on the y-axis is the prop ortion of misclassified individuals (after correction for p oten tial lab el switching issues). The v ariable plotted on the x-axis is the Hotelling T statistic and assesses the magnitude of the phenot ypic differentiation. Our metho d: red triangles ( 4 ), Mclust : blac k circles ( ◦ ). 18 Av erage error: 4 5.2%, + 8.7%, × 2.4% Figure 5: Classification error for 500 sim ulated data-sets consisting of 200 individuals belonging to K = 2 clusters and recognized by q = 5 quantitativ e v ariables and l = 10 co-dominan t lo ci. The v ariable plotted on the y-axis is the prop ortion of misclassified individuals using our metho d (after correction for p otential label switc hing issues). 19 Figure 6: P airs plots of Fisher’s Iris data (transformed in to log shape ratios). Colors indicate individual species estimated b y our metho d. The true n um b er of species (three) is correctly estimated. Only 9 out of 150 individuals are misclassified. 20 Phenot ypic & Spatial - ˆ K = 2 Phenot ypic non Spatial - ˆ K = 2 Genetic & Spatial - ˆ K = 4 Figure 7: P opulation structure inferred on the bank v ole data. 21 Discussion Summary of Approac h Proposed Main features W e hav e prop osed the first metho d to date for analyzing georeferenced phenotypic and genetic data within a unified inferential framework, op ening the wa y to com bined analyses and robust comparison betw een markers. Our method tak es as input an y combination of phenot ypic and genetic individual data and these data can b e optionally georeferenced. Analyses can be run on phenotypic and genetic data separately or join tly . The main outputs of the method are estimates of the num ber of homogeneous clusters and of cluster mem b erships of each individual. If analyses are made on georeferenced data, the method also provides an estimate of the spatial lo cation of eac h cluster which can b e display ed graphically in form of contin uous maps (see program do cumentation for details on suc h graphic represen tation). Our approac h is based on an explicit statistical mo del. This contrasts with mo del-free metho ds suc h as P AM which roughly speaking attempts to cluster individuals in order to maximize some homogeneit y criterion. While such metho ds are fast and presumably robust to departure from sp ecific mo del assumptions they are exp ected to b eha ve p o orly compared to metho ds based on an explict model that fits the data to a reasonable exten t. This claim is supp orted by the recent study of Safner et al. (2011) in the case of spatial genetic clustering metho ds. In addition, because mo del-free metho ds do not rely on an explicit mo del, their output migh t b e difficult to interpret or relate to biological pro cesses. Main results from simulation study and analysis of classic data-sets Inference from Phenot ypic Data Only: All numerical results obtained here demonstrate the go o d accuracy of our metho d and its efficiency for iden tifying sp ecies and/or p opulations b oundaries. It is excellen t at a voiding false p ositives (i.e. at rep orting ˆ K = 1 when K =1) and has a clear ability to reduce the error rate when the num ber of v ariables increases. The metho d loses accuracy when it is giv en a difficult problem (i.e. when the true K is large). F or a fixed n umber of iterations, it also has increasing difficulty to exploit fully all of the a v ailable infor- mation when the n um b er of v ariables is large (cf. loss of accuracy for 50 v ariables compared to 20 v ariables), presumably due to loss of n umerical efficie ncy in the MCMC algorithm. W e also noted that Mclust is sub ject to similar difficulties for large num ber of clusters and/or large 22 n umber of v ariables presumably due to the existence of m ultiple maxima of the lik eliho o d. In our metho d, this problem can be resolved to a certain extent by longer MCMC runs, an asp ect not inv estigated in detail here. Ov erall, our metho d offers a notable impro vemen t ov er the non- spatial penalized maximum likelihoo d metho d of Mclust used under its default set of options. One factor resp onsible for this improv emen t could b e that our metho d exploits spatial informa- tion while Mclust does not. Results from section “Analysis of classic data of the clustering literature”, where our metho d still pro vides b etter results than Mclust even though the data are non-spatial, suggests this is not the sole factor. This might relate to mo del selection which is the second ma jor difference b etw een the t wo metho ds considered (Bay es vs. p enalized maxim um lik eliho o d) more so b earing that Mclust considers a broad family of co v ariance structure while our metho d assumes conditional indep endence. W e also stress that the n umerical v alues characterizing the accuracy of our metho d ha ve to b e taken with a grain of salt since the mo del used to analyze the data matc hes exactly the mo del that generated them. This situation is a b est case scenario and is unlik ely to be strictly met in real-life cases. Ho wev er, our results are informativ e about the p otential of the metho d and ev aluations of the iris data suggest a certain robustness of these results (see also analysis of crab morphometric data in supplemen tary material). As a final note, w e warn the reader unfamiliar with clustering metho ds against ov erly p es- simistic interpretation of figure 4. F rom this figure, it seems that the methods lose accuracy v ery quic kly as the the “phenotypic differentiation” decreas es and are in general not so efficient. This is b ecause detecting a hidden structure is a m uch harder statistical problem than testing the significance of a differentiation b etw een tw o known clusters (the former in v olving man y more pa- rameters and hence uncertain ty than the latter). More details are giv en in section ”P ow er to test the significance of a kno wn structure v ersus p o wer to detect a hidden structure” of Supp orting Material. Inference from Phenotypic and Genetic Data Genetic and phenotypic data can trace differen t ev olutionary histories, for instance phylogenetic divergence for neutral genetic mark ers and adaptation for a morphological structure (Renaud et al., 2007; Adams et al., 2009). Note that this is also true for any genetic marker that only traces its own ev olutionary history in a ph ylogenetic dynamics (T urmelle et al., 2011). Confronting the structure provided by different 23 mark ers emerges more and more as a w ay to get a comprehensive view of the dynamics and pro cesses of differen tiation among and within sp ecies. Our metho d, b y providing a unified in- feren tial framework for analysing differen t kind of data, including phenot ypic ones, app ears as a significan t improv emen t for v alid confrontation b et ween data sets. F urthermore, in situations when genetic and phenotypic patterns are susp ected to coincide, making inference from genetic and phenotypic data join tly has the p oten tial to increase the pow er to detect b oundaries b etw een ev olutionary units at different lev els (p opulations, sp ecies). Analysis of the Calop o gon AFLP data-set The abilit y of our mo del under the CFM prior to detect and classify sp ecies is excellent. This dataset has b een re-analyzed by Hausdorf and Hennig (2010) who carried out a comparison of Structure , Structurama , a metho d known as “field of recom bination” (Do yle, 1995) and a h ybrid metho d mixing sequentially multidimensional scaling and mo del-based Gaussian cluster- ing. The Str ucture program and the “field of recom bination”metho d w ere not able to detect an y structure. Structurama identified only three clusters and misclassifies 44% of the samples. The h ybrid metho d of Hausdorf and Hennig (2010) identifies 5 clusters but misclassifies 15% of the samples. Our metho d under the CFM prior also iden tifies 5 clusters but misclassifies only 5% of the samples. Under the UFM mo del, the results we obtain are higly consisten t with those obtained with the CFM. W e also refer the reader to the Supplemen tary Material where w e analyze AFLP data of V er onic a (p entasep alae) from the Iberian Peninsula and Moro cco pro duced and first analysed b y Mart ´ ınez- Ortega et al. (2004). The results w e rep ort there confirm the excellent p erformance of our method compared to the four metho ds inv estigated by Hausdorf and Hennig (2010). Finally , all the anal- ysis carried out in the presen t article show that concerns of Hausdorf and Hennig (2010) against metho ds for dominant mark ers based on Hardy-W ein b erg equilibrium were not grounded, pro- vided the dominan t nature of AFLP markers is taken in to accoun t at the likelihoo d level as w e did. W e susp ect that the p o or p erformances of Structure observed b y Hausdorf and Hennig (2010) relate to the pro cedure used to estimate K (Ev anno et al., 2005), as noted earlier by W aples and Gaggiotti (2006). 24 The Myo des data-set W e confronted clustering hypotheses using v arious data subsets with or without spatial data and with or without genetic mark ers or morphometric v ariables. This shed new ligh ts on the p opulation structure of Myo des . The pattern of phenotypic and genetic differentiation can find an in terpretation in a complex pattern of con tact b etw een sp ecies and p opulations. The north- ernmost area corresp onds to the narro w zone of p ossible o verlap betw een Myo des glar e olus and its close northern relative Myo des rutilus . Both sp ecies are difficult to recognise based on exter- nal phenotypic characters, and imp ossible to iden tify based on common mito chondrial mark ers b ecause of the in trogression of M. rutilus m tDNA in to the northern fringe of M. glar e olus distribu- tion. The northern cluster detected by our metho d corresponds most probably to the o ccurrence of the northern red-bac ked v ole M. rutilus , that tends to differ in molar shap e from its relativ e M. glar e olus (Ledevin et al., 2010b). The tw o analyses based on phenotypic data with and without spatial information lead to sligh tly differen t results, the former suggesting the presence of an abrupt phenotypic discontin uit y in the North while the latter suggests clinal v ariation (Fig. 7 upper and middle panel). In absence of mo del fit criteria to assess the v alue of these tw o maps, we are reduced to sp eculate. W e note ho wev er that these maps are congruen t concerning the lo cation of the main area of transition b et ween the clusters and that the analysis based on spatial information is graphically more efficien t at displa ying the location of this transition. The bank v ole molar shap e has been shown to display a large v ariation ev en within populations, due to w ear and developmen tal factors (Gu ´ er ´ ec heau et al., 2010; Ledevin et al., 2010b). This may render even clear cut inter-specific b oundaries difficult to detect. Our georeferenced metho d may greatly help to make suc h signal emerge despite the in trinsic v ariability in the phenotypic mark ers. This suggests that our metho d could b e view ed as an efficient generalisation of the methods aimed at detecting abrupt changes of W om ble (1951) and Bo cquet-App el and Bacro (1994). Regarding the additional clusters detected based on genetic data, the location of tw o of them suggests that they correspond to bank v ole lineages already kno wn in this region based on mito c hondrial DNA data. Indeed, after the last ice age, Sweden has b een recolonized by differen t p opulations separated sev eral h undreds of thousand years ago coming from the South and from the North of F ennoscandia (Jaarola et al., 1999; Razzauti et al., 2009). Our new data therefore confirm the existence of tw o differen t bank v ole lineages in Sweden based on mito chondrial and 25 no w nuclear DNA markers. The existence of a fourth cluster lo cated in Central Sweden strongly suggests that the contact zone b etw een these tw o main lineages is situated in this latter region. Its origin may b e attributed to hybridization betw een animals of the tw o genetic lineages. The disco very of this last cluster is new and it w as never detected previously using only mitochondrial DNA marker. Com bining phenot ypic and genetic data in a join t analysis (PGS) did not allow us to detect an y extra structure (map not sho wn), p ossibly because b ey ond the inter-specific phenot ypic difference corresponding to the differen tiation betw een top North and the rest of Sweden, a cline in molar shape exists through Sw eden that is roughly congruen t with the genetic clusters (data not shown). It shows that the confron tation b etw een data sets ma y b e as informative as a join t analysis, by providing clues about the hierarchical pattern of differentiation. Morphometric clusters evidenced here inter-specific differences b etw een red-back ed and bank voles whereas based on microsatellite data, b oth inter- and in tra-sp ecific levels of differen tiation emerged as separate clusters. The structure of genetic differen tiation corrob orates this in terpretation. The inter- sp ecific differen tiation of the top North cluster from the rest of Sw eden is indeed m uc h stronger than the in tra-sp ecific differentiation among the bank vole p opulations from North-East, Cen tral and South Sw eden. Com bining together b oth data t yp es allows us to in terpret the complex ph ylogeographic structure of this sp ecies and helps to distinguish differences betw een true species and p opulations within a sp ecies. F uture Extensions Our metho d is based on an assumption of indep endence of the phenotypic v ariables within eac h cluster. This does not amoun t to independence b etw een these v ariables globally . Indeed, the fact that phenotypic v ariables are sampled with cluster-sp ecific parameters do es include a cor- relation (similarly to the dep endence structure assumed in a linear mixed mo del). How ever our metho d does not deal with residual dependence not accoun ted for at the cluster level suc h as that generated by allometry . Results from sim ulations and classic datasets suggest that this can b e partially dealt with b y pre-processing the data (e.g. transforming raw data into log-shap e ra- tio). Sev eral other procedures ma y b e applied for a voiding or reducing problems with co v ariation among phenot ypic v ariable. F or example, working on principal comp onents rather than on ra w data ma y help in this task. Procedures such as the Burnaby approach (Burnaby, 1966) may also 26 allo w to remo ve cov ariance structures due only to gro wth or other confounding factors that the user ma y wish to filter out. A more rigorous approach w ould consist in allowing the v ariables to co v ary within clusters whic h would also allow one to quantify these cov ariations. P oten tial Applications Ev olutionary biology has b een flo o ded b y molecular data in the recen t y ears. How ev er, efficien t metho ds to deal with phenotypic data alone are still needed when this t yp e of data is the only a v ailable. This includes the imp ortant case of fossil data. W e note that in sys tematic paleon tology , the metho ds used are often simpler than those discussed in the presen t pap er and chosen as a matter of tradition in the field rather than on ob jectiv e basis. Implemen ting our method in a free and user-friendly program should help pro vide more ob jective methods in this context. Our method was specifically tailored for biometric/morphometric measuremen ts whic h are t ypically obtained at a few tens of phenotypic v ariables. The metho d proposed is therefore computer in tensive and not expected to b e w ell suited for large datasets such as expression data pro duced in functional genomics. Ho w ever, in the situations where the scien tist is able to select some v ariables of particular interest and reduce the dimensionalit y of the mo del (as w e did for our analysis of the Myo des molar shap e data), our method could b e used and play a role in the emerging field of landscap e genomics (Sc h wartz et al., 2010). The sub-mo del for genetic data used here w as presen ted and discussed in detail by Guillot et al. (2005) and Guillot (2008). It has b een used mostly to analyse v ariation and structure in neutral n uclear markers (Guillot et al., 2009) and pro v ed useful to detect and quantify fine-scale structure t ypical of landscape genetics studies. The nov el possibility brough t here to com bine it with morphometrics data might p opularize this genetic mo del among scientists in terested in larger spatial and temp oral scale typical of ph ylogeography . In the latter field, the use of mtDNA is common. As noted earlier, the analysis of suc h non-recom bining DNA sequence data using our metho d is technically p ossible and meaningful by reco ding the v arious observ ed haplotypes as differen t alleles of the same lo cus. W e stress that this approach is an exp edient which incurs a considerable loss of information and that our approac h should not b e viewed as a substitute to those that mo del the genealogy of genes (including the mutational pro cess) explicitly . Extending our mo del to deal with non recom bining DNA in a more rigorous w a y is a natural direction for future work. 27 Our method for the combined analysis of phenot ypic and genetic data can b e used to assess the relative imp ortance of random genetic drift and directional natural selection as causes of p opulation differen tiation in quan titative traits, and to assess whether the degree of divergence in neutral mark er lo ci predicts the degree of divergence in quan titative traits (Meril¨ a and Crnokrak, 2001). F urthermore, our metho d should b e useful in the study of hybrid zones where, as noted b y Ga y et al. (2008), comparing clines of neutral genetic mark ers with clines of traits known to b e under selection also indicates the extent to whic h the o verall genome is under selection. Lastly , because phenot ypic and genetic mark ers ma y reflect differen t ev olutionary or demo- graphic history , combined analyses can help to understand the hierarch y b etw een ev olutionary units (sp ecies and p opulations) as sho wn in the Myo des example. 28 Computer pr ogram a v ailability: The mo del presented here will b e a v ailable so on as part of a new version of the R pack age Geneland (version ≥ 4.0.0). Information will b e found on the program homepage http://www2.imm.dtu.dk/ ~ gigu/Geneland/ . A cknowledgments: The first author is most grateful to Cino Pertoldi for discussions that prompted him to develop the mo del for morphometric data. Our w ork benefitted from discussions with Jean-Marie Cornuet and comments of Andrew J. Crawford. P art of the original data of the Myo des analysis b elong to Bernard Lehanse’s Master thesis (genetic data). W e thank him for sharing these data with us. W e are also grateful to Mon tse Mart ´ ınez-Ortega and Doug Goldman for making there data a v ailable to us. This w ork has b een supported b y the F renc h National Researc h Agency (pro ject EMILE, gran t ANR-09-BLAN-0145-01) and the Danish Centre for Scien tific Computing (gran t 2010-06-04). 29 References Adams, D. C., C. M. Berns, K. H. Kozak, and J. J. Wiens. 2009. Are rates of sp ecies diversification correlated with rates of morphological evolution? Proceedings of the Roy al So ciet y of London, Biological Sciences (serie B) 276:2729–2738. Anderson, E. 1935. The irises of the Gasp e p eninsula. Bulletin of the American Iris So ciet y 59:2–5. Anderson, T. 1984. An in tro duction to multiv ariate statistical analysis. Probabilit y and mathe- matical statistics second ed. Wiley , New Y ork. Balding, D. and R. Nichols. 1995. A metho d for quan tifying differentiation b etw een p opulations at m ulti-allelic loci and its implications for in v estigating iden tit y and paternity . Genetica 96:3–12. Balding, D. and R. Nichols. 1997. Significant genetic correlation among Caucasians at forensic DNA lo ci. Heredit y 78:583–589. Banfield, J. D. and A. E. Raftery . 1993. Mo del-based gaussian and non-gaussian clustering. Biometrics 49:803–821. Beadell, J. S., C. Hyseni, P . P . Abila, R. Azab o, J. C. K. Eny aru, J. O. Ouma, Y. O. Mohammed, L. M. Ok edi, S. Akso y , and A. Caccone. 2010. Ph ylogeography and population structure of Glossina fuscip es fuscip es in Uganda: Implications for con trol of tsetse. PLoS Neglected T rop- ical Diseases 4. Biernac ki, C., G. Celeux, G. Gov aert, and F. Langrognet. 2006. Model-based cluster and dis- criminan t analysis with the MIXMOD soft ware. Computational Statistics and Data Analys 51:587–600. Bo cquet-App el, J. and J. Bacro. 1994. Generalized Wom bling. Systematic Biology 43:442–448. Burnab y , T. P . 1966. Growth-in v arian t discriminant functions and generalized distances. Biomet- rics 22:96–110. Claude, J. 2008. Morphometrics with R. Springer. 30 Corander, J., P . W aldmann, P . Martinen, and M. Sillanp¨ a¨ a. 2004. Baps2: Enhanced possibilities for the analysis of genetic population structure. Bioinformatics 20:2363–2369. Corander, J., P . W aldmann, and M. Sillanp¨ a¨ a. 2003. Bay esian analysis of genetic differentiation b et ween p opulations. Genetics 163:367–374. Coulon, A., G. Guillot, J. Cosson, J. Angibault, S. Aulagnier, B. Cargnelutti, M. Galan, and A. Hewison. 2006. Genetics structure is influenced by lansdcap e features. Empirical evidence from a roe deer p opulation. Molecular Ecology 15:1669–1679. Deffon taine, V., R. Ledevin, M. C. F ontaine, J.-P . Qu´ er ´ e, S. Renaud, R. Lib ois, and J. R.Michaux. 2009. A relict bank vole lineage highlights the biogeographic history of the pyrenean region in europ e. Molecular Ecology 18:2489–2502. Do yle, J. 1995. The irrelev ance of allele tree top ologies for sp ecies delimitation and a non- top ological aletrnative. Systematic Biology 20. Ev anno, G., S. Regnault, and J. Goudet. 2005. Detecting the num b er of clusters of individuals using the soft w are structure: a simulation study . Molecular Ecology 14:2611–2620. Excoffier, L. and G. Heck el. 2006. Computer programs for p opulation genetics data analysis: a surviv al guide. Nature Review Genetics 7:745–758. F alush, D., M. Stephens, and J. Pritc hard. 2003. Inference of p opulation structure using m ulti- lo cus genotype data: Linked loci and correlated allele frequencies. Genetics 164:1567–1587. Fisher, R. A. 1936. The use of m ultiple measuremen ts in taxonomic problems. Annals of Eugenics 7:179–188. F ontaine, M., S. Baird, S. Piry , N. Ra y , K. T olley , S. Duke, A. Birkun, M. F erreira, T. Jauniaux, A. Lla vona, B. ¨ Ost ¨ urk, A. ¨ Ost ¨ urk, V. Ridoux, E. Rogan, M. Sequeira, U. Siebert, G. Vikingson, J. Bouquegneau, and J. Mic haux. 2007. Rise of o ceanographic barriers in contin uous p opula- tions of a cetacean: the genetic structure of harb our p orp oises in old world w aters. BMC Biology 5. F raley , C. 1999. Mclust:softw are for mo del-based cluster analysis. Journal of classification 12:297– 306. 31 F r ¨ uh wirth-Sc hnatter, S. 2006. Finite Mixture and Marko v Switching Mo del. Series in Statistics Springer. Galarza, J., J. Carreras-Carbonell, E. Macpherson, M. Pascual, S. Roques, G. T urner, and C. Ri- co d. 2009. The influence of oceanographic fron ts and early-life-history traits on connectivity among littoral fish sp ecies. Pro ceedings of the National Academy of Sciences 106:1473–1478. Ga y , L., P . Cro chet, D. Bell, and T. Lenormand. 2008. Comparing genetic and phenotypic clines in hybrid zones: a window on tension zone mo dels. Ev olution 62:2789–2806. Gelman, A., J. Carlin, H. Stern, and D. Rubin. 2004. Bay esian data analysis. Chapman and Hall. Gilks, W., S. Richardson, and D. Spiegelhalter, eds. 1996. Mark o v Chain Monte Carlo in Practice. In terdisciplinary Statistics Chapman and Hall. Girard, C. and S. Renaud. 2011. The species concept in a long-extinct fossil group, the conodonts. Comptes Rendus P alev ol 10:107–115. Go dsill, S. 2001. On the relationship b et ween Mark ov c hain Monte Carlo metho ds for mo del uncertain ty . Journal of Computational and Graphical Statistics 10:230–248. Goldman, D. H., R. K. Jansen, C. V an Den Berg, I. J. Leitch, M. F. F a y , and M. W. Chase. 2004. Molecular and cytological examination of calop ogon (Or chidac e ae, Epidendr oide ae) : Circumscription, phylogen y , polyploidy , and p ossible hybrid sp eciation. American Journal of Botan y 91:707–723. Gu ´ er ´ ec heau, A., R. Ledevin, H. Hen ttonen, V. Deffon taine, J. R. Mic haux, P . Chevret, , and S. Renaud. 2010. Seasonal v ariation in molar outline of bank voles: an effect of wear? Mam- malian Biology 75:311–319. Guillot, G. 2008. Inference of structure in sub divided populations at lo w lev els of genetic differ- en tiation. The correlated allele frequencies mo del revisited. Bioinformatics 24:2222–2228. Guillot, G. and A. Carpentier-Sk andalis. 2011. On the informativ eness of dominant and co- dominan t genetic markers for Ba y esian sup ervised clustering. The Open Statistics and Proba- bilit y Journal 3:7–12. 32 Guillot, G., A. Estoup, F. Mortier, and J. Cosson. 2005. A spatial statistical mo del for landscap e genetics. Genetics 170:1261–1280. Guillot, G., R. Leblois, A. Coulon, and A. F rantz. 2009. Statistical metho ds in spatial genetics. Molecular Ecology 18:4734–4756. Guillot, G. and F. San tos. 2010. Using AFLP mark ers and the Geneland program for the inference of p opulation genetic structure. Molecular Ecology Resources 10:1082–1084. Guillot, G., F. Santos, and A. Estoup. 2008. Analysing georeferenced p opulation ge netics data with Geneland: a new algorithm to deal with n ull alleles and a friendly graphical user in terface. Bioinformatics 24:1406–1407. Hannelius, U., E. Salmela, T. Lappalainen, G. Guillot, C. Lindgren, U. von D¨ ob eln, P . Lahermo, and J. Kere. 2008. P opulation substructure in Finland and Sweden rev ealed by a small num ber of unlinked autosomal SNPs. BMC Genetics 9. Hausdorf, B. and C. Hennig. 2003. Biotic element analysis in biogeography . Systematic Biology 52:717–723. Hausdorf, B. and C. Hennig. 2010. Sp ecies delimitation using dominant and codominant m ulti- lo cus markers. Systematic Biology 59:491–503. Hubisz, M., D. F alush, M. Stephens, and J. K. Pritchard. 2009. Inferring weak p opulation struc- ture with the assistance of sample group information. Molecular Ecology Resources 9:1322– 1332. Jaarola, M., H. T egelstr¨ om, and K. F redga. 1999. Colonization history in Fennoscandian ro den ts. Biological Journal of the Linnean Society 68:113–127. Joseph, L., G. Dolman, S. Donnellan, K. Saint, M. Berg, and A. Bennett. 2008. Where and when do es a ring start and end? testing the ring-sp ecies h yp othesis in a sp ecies complex of australian parrots. Pro ceedings of the Roy al So ciety of London, series B 275:2431–2440. Klingen b erg, C. P ., L. J. Leam y , E. J. Routman, and J. M. Cheverud. 2001. Genetic architecture of mandible shape in mice: effects of quantitativ e trait lo ci analyzed b y geometric morphometrics. Genetics 157:785–802. 33 Kuhl, F. P . and C. R. Giardina. 1982. Elliptic Fourier features of a closed contour. Computer Graphics and Image Pro cessing 18:236–258. Ledevin, R. 2010. La dynamique ´ ev olutive du campagnol roussˆ atre (Myo des glar e olus) : structure spatiale des v ariations morphom´ etriques. Ph.D. thesis Universit ´ e Ly on 1. Ledevin, R., J. R. Mic haux, V. Deffon taine, H. Henttonen, and S. Renaud. 2010a. Evolutionary history of the bank v ole Myo des glar e olus : a morphometric p ersp ective. Biological Journal of the Linnean Society 100:681–694. Ledevin, R., J.-P . Qu´ er´ e, and S. Renaud. 2010b. Morphometrics as an insigh t in to pro cesses b ey ond tooth shape v ariation in a bank v ole p opulation. PLoS One 5:e15470. Lehanse, B. 2010. ´ Etude g ´ en ´ etique d’une zone de contact en Su` ede entre deux lign´ ees de cam- pagnols roussˆ atres Myo des glar e olus . Master’s thesis Univ ersit´ e de Li ` ege. Luo, A., A. Zhang, S. Y. Ho, W. Xu, W. Shi, C. S.L., and C. Zh u. 2011. P otential efficacy of mito c hondrial genes for animal DNA barco ding: a case study using eutherian mamals. BMC Genomics 12. Marin, J., K. Mengersen, and C. Rob ert. 2005. Handb o ok of Statistics v ol. 25 chap. Bay esian mo delling and inference on mixtures of distributions. Elsevier-Sciences. Mart ´ ınez-Ortega, M. M., L. Delgado, D. C. Albac h, J. A. Elena-Rossello, and E. Rico. 2004. Sp ecies Boundaries and Ph ylogeographic Patterns in Cryptic T axa Inferred from AFLP Mark- ers: V er onic a sub gen. Pentasep alae (Scr ophulariac e ae) in the W estern Mediterranean. Sys- tematic Botany 29:965–986. McLac hlan, G. J., D. P eel, and P . Basford, K. E. Adams. 1999. The EMMIX soft ware for the fitting of mixtures of normal and t-comp onents. Journal of Statistical Soft w are 4:1–4. Meril¨ a, J. and P . Crnokrak. 2001. Comparison of genetic differen tiation at mark er loci and quan- titativ e traits. Journal of Evolutionary Biology 14:892–903. Møller, J. and D. Stoy an. 2009. T essellations in the Sciences: Virtues, T ec hniques and Applica- tions of Geometric Tilings chap. Sto c hastic geometry and random tessellations. Springer. 34 N ´ eraudeau. 2011. The sp ecies concept in palaeon tology: Ontogen y , v ariabilit y , evolution. Comptes Rendus Palev ol 10:71–75. Nesi, N., E. Nak oum ´ e, C. Cruaud, and A. Hassanin. 2011. DNA barco ding of African fruit bats ( Mammalia, Pter op o didae ). the mito ch ondrial genome do es not pro vide a reliable discrimination b et ween Ep omophorus gambianus and Micr opter opus pusil lus . Comptes Rendus Biologies 334:544–554. Pritc hard, J., M. Stephens, and P . Donnelly . 2000. Inference of p opulation structure using m ul- tilo cus genotype data. Genetics 155:945–959. Razzauti, M., A. Plyusnina, T. Sironen, H. Henttonen, and A. Plyusnin. 2009. Analysis of pu- umala han tavirus in a bank vole p opulation in northern Finland: evidence for co-circulation of t wo genetic lineages and frequen t reassortmen t b etw een strain. Journal of General Virology 90:1923–1931. Renaud, S., P . Chevret, and J. Mic haux. 2007. Morphological vs. molecular evolution: ecology and phylogen y b oth shap e the mandible of ro den ts. Zo ologica Scripta 36:525–535. Ric hardson, S. and P . Green. 1997. On Ba yesian analysis of mixtures with an unkno wn n um b er of comp onents. Journal of the Ro yal Statistical So ciety , series B 59:731–792. Rob ert, C. and G. Casella. 2004. Mon te Carlo statistical metho ds. second ed. Springer-V erlag, New York. Ro dr ´ ıguez, F., T. P´ erez, S. E. Hammer, J. Albornoz, and A. Dom ´ ınguez. 2010. In tegrating ph ylogeographic patterns of microsatellite and mtDNA div ergence to infer the ev olutionary history of c hamois (genus Rupic apr a) . BMC Evolutionary Biology 10:222. Sac ks, B., D. L. Bannasc h, B. B. Chomel, and H. Ernst. 2008. Co yotes demonstrate how habitat sp ecialization b y individuals of a generalist sp ecies can diversify p opulations in a heterogeneous ecoregion. Molecular Biology and Evolution 25:1354–1395. Safner, T., M. Miller, B. McRae, M. F ortin, and S. Manel. 2011. Comparison of Ba yesian cluster- ing and edge detection metho ds for inferring b oundaries in landscape genetics. International Journal of Molecular Sciences 12:865–889. 35 Sc hw artz, M. K., G. Luik art, K. S. McKelvey , and S. A. Cushman. 2010. Spatial complexit y , in- formatics, and wildlife conserv ation c hap. Landscape genomics: A brief p ersp ective, P ages 165– 174. Springer. Sir ´ en, J., P . Marttinen, and J.Corander. 2011. Reconstructing population histories from single n ucleotide p olymorphism data. Molecular Biology and Ev olution 28:673–683. Stephens, M. 1997. Discussion of the pap er by Richardson and Green “On Ba yesian analysis of mixtures with an unknown num ber of comp onen ts”. Journal of the Ro y al Statistical So ciety , series B 59:768–769. T egelstr¨ om, H. 1987. T ransfer of mitochondrial DNA from the northern red-back ed v ole ( Clethri- onomys rutilus ) to the bank v ole ( C. glar e olus ). Journal of Molecular Ev olution 24:218–227. T urmelle, A. S., T. H. Kunz, and M. D. Sorenson. 2011. A tale of tw o genomes: contrasting patterns of ph ylogeographic structure in a widely distributed bat. Molecular Ecology 20:357– 375. W aples, R. and O. Gaggiotti. 2006. What is a p opulation? An empirical ev aluation of some genetic methods for indentifying the n umber of ge ne po ols and their degree of connectivity . Molecular Ecology 15:1419–1439. W asser, S., C. Mailand, R. Bo oth, B. Mutay oba, E. Kisamo, and M. Stephens. 2007. Using DNA to trac k the origin of the largest iv ory seizure since the 1989 trade ban. Pro ceedings of the National Academy of Sciences 104:4228–4233. W omble, W. 1951. Differen tial systematics. Science 28:315–322. Y ang, Z. and B. Rannala. 2010. Ba y esian species delimitation using m ultilocus sequence data. Pro ceedings of the National Academy of Sciences 107:9264–9269. 36 Appendix: Det ail of MCMC Inference Algorithm Ov erview The v ector of unknown parameters is θ = ( K , λ , m , u , c , f , ˜ f , d , µ , σ , β ) which can b e decomp osed in to θ S = ( λ , m , u , c ) , θ G = ( f , ˜ f , d ) and θ M = ( µ , σ , β ) blo cks of parameters of the spatial, genetic and phenotypic data resp ectiv ely . W e alternate blo c k up dates of Metrop olis-Hastings or Gibbs t yp e and also trans-dimensional up dates inv olving c hanges of K and of parts of other parameters. The up dates of blocks of parameters that do not in v olve phenot ypic data are described in Guillot et al. (2005) and Guillot (2008). W e describ e b elo w updates in v olving phenotypic data. Join t Updates of ( c , µ , σ ) W e up date join tly c , µ and σ as follows. W e propose a new v ector c ∗ b y pic king tw o clusters at random and re-assigning some individuals of one of those tw o clusters to the other one at random. Then w e prop ose µ and σ b y sampling from the full conditional distribution π ( µ , 1 / σ 2 | y , c ∗ ) . The Metrop olis-Hastings ratio is R = π ( θ ∗ | y ) π ( θ | y ) q ( θ | θ ∗ ) q ( θ ∗ | θ ) = π ( µ ∗ , 1 / σ 2 ∗ , c ∗ | y ) π ( µ , 1 / σ 2 , c | y ) q ( µ , 1 / σ 2 | c ) q ( µ ∗ , 1 / σ 2 ∗ | c ∗ ) q ( c | c ∗ ) q ( c ∗ | c ) = π ( c ∗ | y ) π ( c | y ) π ( µ ∗ , 1 / σ 2 ∗ | c ∗ , y ) π ( µ , 1 / σ 2 | c , y ) π ( µ , 1 / σ 2 | c , y ) π ( µ ∗ , 1 / σ 2 ∗ | c ∗ , y ) q ( c | c ∗ ) q ( c ∗ | c ) = π ( c ∗ | y ) π ( c | y ) q ( c | c ∗ ) q ( c ∗ | c ) (5) In terestingly , the latter expression does not depend on ( µ ∗ , σ 2 ∗ ) , whic h in principle w ould allo w us to decide whet her a new state θ ∗ is accepted prior to proposing ( µ ∗ , σ 2 ∗ ) . Unfortunately , expression (5) can not b e used as π ( c | y ) is not known analytically under the presen t model. The ratio in equation (5) has therefore to b e written as R = π ( y | µ ∗ , 1 / σ 2 ∗ , c ∗ ) π ( y | µ , 1 / σ 2 , c ) π ( c ∗ ) π ( c ) π ( µ ∗ , 1 / σ 2 ∗ ) π ( µ , 1 / σ 2 ) π ( µ , 1 / σ 2 | y , c ) π ( µ ∗ , 1 / σ 2 ∗ | y , c ) q ( c | c ∗ ) q ( c ∗ | c ) (6) whic h inv olv es only analytically known expressions. Join t Updates of ( K , c , µ , σ ) W e tak e the same strategy as Guillot et al. (2005). The algorithm follows ideas of Richardson and Green (1997). It consists in up dating K by prop osing to split a cluster in to t w o clusters or 37 merge t wo clusters, in a wa y that complies with the spatial constrain ts and multiv ariate nature of the mo del. Since w e use the natural prior conjugate family for parameters µ ∗ and σ ∗ the full conditional π ( µ , 1 / σ 2 ∗ | y , K ∗ , c ∗ ) is a v ailable and can b e used as prop osal distribution as adv o cated for example by Godsill (2001). The acceptance ratio takes essentially the same form as in equation 6 although it is now a genuine transdimensionnal mov e. Detail on Hyp er-Parameters Although we do not use exactly the same prior structure as Ric hardson and Green (1997), w e follo w largely these authors. W e take ξ j = P i y ij , h j = κ j = 2 / R 2 j where R j is the range of observ ed v alues of the j -th phenot ypic v ariable. β j | g j , h j , ∼ G ( g j , h j ) . W e also set α j = 2 and g j = 1 / 2 . Since E [1 /σ 2 ] = α/β , β represents 2 / E [1 /σ 2 ] . Also 1 / 2 h represents the prior mean of b eta. 38
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment