ProDiGe: PRioritization Of Disease Genes with multitask machine learning from positive and unlabeled examples
Elucidating the genetic basis of human diseases is a central goal of genetics and molecular biology. While traditional linkage analysis and modern high-throughput techniques often provide long lists of tens or hundreds of disease gene candidates, the…
Authors: Fantine Mordelet (CBIO, CREST), Jean-Philippe Vert (CBIO)
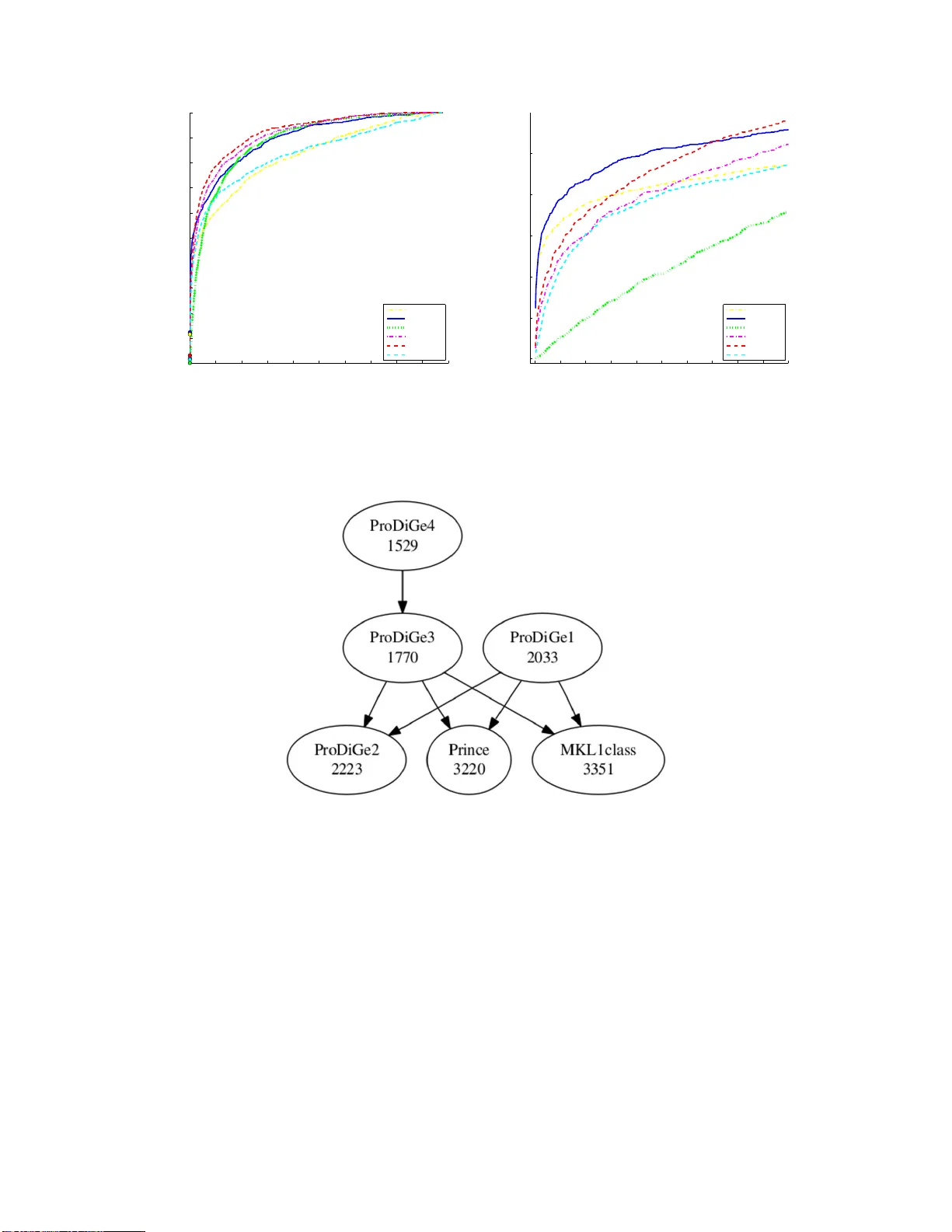
ProDiGe: PRioritizat ion Of Dis ease Genes with m ultitask mac hine learning from p ositive and unlab eled examples F an tine Mordelet 1 , 2 , 3 , 4 and Jean-Philip p e V ert 1 , 2 , 3 1 Mines P arisT ec h, C entre for Computational Biology , F on tainebleau, F-77 300, F rance 2 Institut Curie, P aris, F -75248 F r ance 3 INSERM, U900, P aris, F-75 248 F rance 4 CREST, INSEE, Malak off, F- 92240 F rance fantinemord elet@gmail.com jean-philip pe.vert@mines.org Octob er 1, 2018 Abstract Elucidating the genetic basis of human diseas es is a central goal o f ge ne tics and molecular biolo gy . While traditional link a g e analysis and mo dern high-thro ughput techniques often provide long lists of tens or h undreds of disease gene candidates, the ident ificatio n o f disea se ge ne s amo ng the candidates remains time-consuming and expens ive. Efficient computational metho ds ar e therefore needed to prioritize genes within the list of candidates, by exploiting the wealth of information av ailable ab out the genes in v ario us databases. Here w e prop ose Pr o DiGe, a nov el algorithm for Prioritizatio n of Disease G enes. ProDiGe implemen ts a novel machine learning strategy based on lea r ning fro m p os itive and unlabeled examples , which allows to integrate v ario us sources of information ab o ut the genes, to share info r mation ab out known disea se genes a cross diseases , a nd to p erfo rm geno me- wide sear ches for new dise a se genes. Experiments o n real da ta show that Pro DiGe outp er fo rms state- of-the-art metho ds for the prior itization of genes in h uman diseas es. In tro duc tion During the last decades, considerable efforts hav e b ee n made to elucidate th e genetic basis of rare and common human d iseases. The disco v ery of so-called dise ase genes , whose disr uption causes congenital or acquired disease, is in d eed imp ortan t b oth to wards diagnosis and to wa rd s new therapies, through the elucidation of the biologica l bases of diseases. T raditional approac hes to disco ver disease genes first iden tify c h romosomal regions lik ely to con tain the gene of interest, e.g., by link age analysis or s tudy of c hromosomal ab errations in DNA samples from large case-con trol p opulations. The regions ident ified, ho we ver, often conta in tens to hundreds of ca n d idate genes. Finding the causal gene(s) among these candidates is then an exp ensive and time-consuming p ro cess, wh ich requires extensiv e lab orato ry exp eri- men ts. Pr ogresses in sequencing, microarra y or proteomics tec hn ologies h a v e also facilitat ed the disco v ery of genes whose structure or activit y are mo difi ed in disease samples, on a full genome scale. Ho w ev er, again, these approac hes routinely iden tify long lists of candidate d isease genes among whic h only one or a few are truly the causativ e agen ts of th e disease pro cess, and further b iologica l inv estigatio ns are required to iden tify them. In b oth cases, it is therefore imp ortan t to select the most promising genes to b e further studied among th e candidates, i.e., to prioritize them from the most likely to b e a disease gene to the less lik ely . Gene p rioritization is t ypically based on prior information we ha v e about the genes, e.g., their bi- ologic al functions, patterns of expression in differen t conditions, or interac tions with other genes, and follo w s a “guilt-b y-asso ciation” strategy: the most promising candidates genes are those wh ic h share 1 similarit y with the disease of interest, or with other genes kno wn to b e associated to the disease. The a v ailabilit y of complete genome sequences and the w ealth of large-scale biologica l data sets n o w p ro vide an unpr eceden ted opp ortun it y to sp eed up the gene hun ting pro cess [1]. Integ rating a v ariet y of h et- erogeneous inf orm ation stored in v arious d atabases and in the literature to obtain a go o d fi nal ranking of h und reds of candidate genes is, how eve r, a difficult task for h uman exp erts. Unsu r prisingly man y computational app roac hes hav e b ee n p rop osed to p erform this task automatically via statistical and d ata mining approac hes. While some previous wo rks atte mp t to iden tify p romising candidate genes without prior kno wledge of an y other disease gene, e.g., by matc hin g the functional annotatio ns of candidate genes to the disease or ph en ot yp e under inv estigation [2–4], many successful approac hes assume that some d isease genes are already kno wn and try to d etect candidate genes w hic h share sim ilarities with kno wn disease genes for th e p henot yp e un der inv estigation [5 – 10] or for related p henot yp es [5, 9, 11 – 14]. These metho ds v ary in the algorithm th ey implement and in the data they use to p erform gene pr ioritiza- tion. F or example, Endeav our and r elated work [6, 7, 1 0] use state-o f-the-art mac hine learning tec hniques to in tegrate heterogeneous inf orm ation and rank the candid ate genes by d ecreasing similarit y to kno wn disease genes, while PRINCE [14] uses lab el propagation o v er a protein-protein in teraction (PPI) net work and is able to b orr o w information fr om kn o wn disease genes of related d iseases to fi nd new disease genes. W e refer the reader to [15] for a recen t review of gene prioritizatio n to ols av ailable on the w eb. Here we prop ose Pr oDiGe, a new metho d for p rioritization of disease genes based on the guilt-b y- asso ciation concept. ProDiGe assumes th at a set of gene-disease asso ciations is already known to infer new ones, and brings three main n ov elties compared to existing metho ds. First, Pr oDiGe imp lemen ts a no v el mac hin e learning p aradigm to score candidate genes. While existing metho d s lik e those of [6, 7, 10] score indep enden tly the differen t candid ate genes in terms of similarit y to known disease genes, ProDiGe exploits the relativ e similarit y of b ot h kno wn and cand id ate disease genes to jointly score and rank all candidates. This is done by form ulating the disease gene prioritization p roblem as an ins tance of the problem k n o wn as le arning fr om p ositive and unlab ele d examples (PU learning) in the mac h ine learning comm unity , wh ic h is kno wn to b e a p o werful paradigm wh en a set of candidates has to b e ranked in terms of similarit y to a set of p ositiv e d ata [16–18]. Second, in order to rank candidate genes for a disease of in terest, ProDiGe b orr o ws inform ation not only from genes known to b e asso ciated to the disease, b ut also from genes kn o wn to pla y a role in d iseases or p henot yp es related to the disease of interest. This again d iffers fr om [6, 7, 10] whic h treat diseases indep end en tly f r om eac h other. It allo ws u s, in particular, to rank genes ev en for orphan dise ases , w ith no kn o wn gene, by r elying only on kno wn disease genes of related diseases. In the mac hine learning jargon, we implement a multi- task strategy to sh are information b et ween differen t diseases [19–21], and weigh t the sharing of inform ation b y the ph enot ypic similarit y of diseases. Third, ProDiGe p erforms heterogeneous data integ ration to combine a v ariet y of information ab out the genes in the scoring function, including sequence features, expression lev els in d ifferen t cond i- tions, PPI int eractions or p resence in the scien tific literature. W e use the p ow erfu l f ramew ork of kernel metho ds for data integrat ion [22–24], akin to the work of [6, 7, 10]. This differs from appr oac hes like that of [14], w h ic h are limited to scoring o v er a gene or protein net work. W e test ProDiGe on real data extracted from the O MIM d atabase [25]. It is able to rank the correct disease gene in the top 5% of the candidate genes f or 69% of the d iseases with at least one other kno wn causal gene, and f or 67% of the diseases when no other disease genes is kn o wn, outp erforming state-o f- the-art metho ds like Endeav our and PRINCE. 2 Results Gene priorit ization without sharing of information across diseases W e fir st assess the abilit y of ProDiGe to retriev e n ew disease genes for diseases with already a few kno wn disease genes, without sh aring information across different d iseases. As a gold standard we extracted all kno wn disease-gene asso ciations from th e OMIM database [25], and w e b orro wed from [7] nine sources of in formation ab out the genes, in cluding expression profiles in v arious exp erimen ts, functional anno- tations, kno wn protein-protein inte ractions (PPI), transcriptional motifs, pr otein domain activit y and literature data. Eac h sour ce of in formation w as enco ded in a k ernel functions, whic h assesses pairwise similarities b et we en eac h pair of genes according to eac h sour ce of information. W e compare t wo wa ys to p erform d ata in tegration: first b y simply a veraging the nine kernel fu nctions, and second by letting ProDiGe optimize itself the relativ e con tribu tion of eac h source of inform ation wh en the mo del is esti- mated, through a multiple k ernel learnin g (MKL) app roac h. W e compare b oth v arian ts with the b est mo del of [10], n amely , the MKL1Class mo d el which differs from ProDiGe in this case only in the mac hin e learning paradigm imp lemen ted: while ProDiGe learns a m o del from p ositiv e and unlab ele d examples, MKL1class learns it only from p ositiv e examples. W e tested these three algorithm in a leav e-one-out cross-v alidation (LOOCV) setting. In short, for eac h disease, eac h kn o wn disease gene w as remo v ed in turn, a mo d el was trained on u sing the r emaining d isease genes as p ositiv e examples, and all 19540 genes in our database were rank ed; w e then recorded the rank of the p ositiv e gene that was remov ed in this list. W e fo cused on the 285 diseases in our d ataset ha ving at least 2 known disease genes, b ec ause all three metho d s require at least one kno wn disease gene for tr aining, and for the pu rp ose of LOOCV w e need in addition one kn o wn d isease gene remo ved from the trainin g set. Figure 1 pr esen ts the cum ulative distribution fun ction (CDF) of the rank of th e left-out p ositive gene, i.e., the n umber of genes that w ere rank ed in the top k genes of the list as a function of k , for eac h metho d. Note that the rank is alwa ys b etw een 1 (b est prediction) and 19540 − | P | , where | P | is th e num- b er of genes kno wn to b e associated to the d isease of in terest. The r igh t panel zo oms on the b eginning of this cur ve whic h corresp onds to the distribu tion of s m all v alues of th e rank. W e see clearly that b oth ProDiGe v arian ts outp erform MKL1class in the sen s e that they consisten tly reco v er the hid den p ositi ve gene at a b etter r an k in the list. A Wilco xon signed rank test confirm s these visual conclusions at 5% lev el with P -v alues 6 . 1 e − 29 and 8 . 8 e − 28 , resp ect ive ly , for the a verag e and MKL v arian ts of ProDiGe. This illustrates the b enefits of form ulating the gene ranking prob lem as a PU learning problem, and not as a 1-class learning on e, since apart fr om this formulati on b oth MKL1Class and ProDiGe1 use v ery similar learning engines, b ased on SVM and MKL . Both ProDiGe1 v ariants reco ver roughly one third of correct gene-disease associations in the top 20 genes among almost 19540, i.e., in the top 0 . 1%. Ho w eve r, we found no significan t d ifference b et we en the mean and MKL v arian ts of ProDiGe in this setting (P-v alue=0.619 ). This means that in this case, assigning equal we ights to all data sources w orks as well as trying to optimize these w eigh ts by MKL. Supp orted b y this result and b y the fact that MKL is muc h more time-consuming than a SVM with the mean k ernel, w e decided to restrict our exp erimen ts to the mean kernel in the f ollo win g exp erimen ts. Gene priorit ization with information sharing across diseases In a second run of exp eriments, w e assess the p erforman ce of ProDiGe w h en it is allo w ed to share in- formations across diseases. W e tested three v ariants of ProDiGe, as explained in Material and Metho ds: ProDiGe2, which uniformly shares information across all diseases without using p articular informations ab out the diseases, P r oDiGe3, w hic h weigh ts the sharing of in formations across diseases by a phenotypic similarit y b et wee n the diseases, and ProDiGe4, a v ariant of ProDiGe3 which additionally controls the 3 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 x 10 4 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 Rank Cumulative distribution ProDiGe1−Mean ProDiGe1−MKL MKL1class 0 2 4 6 8 10 12 14 16 18 20 0.1 0.15 0.2 0.25 0.3 0.35 Rank Cumulative distribution ProDiGe1−Mean ProDiGe1−MKL MKL1class A B Figure 1: Cum ulative distribut ion function of the rank for lo cal metho ds, in the LOOCV exp eriment. ProDiGe1 -Mean and ProDiGe1-M KL refer to the Pr oDiGe1 v arian t com bined with the mean kernel or the MKL str ategy to fu se heterogeneous gene information. P anel (A) Global curve, Pa nel (B) Zo om on the b eginning of the curv e. sharing of information b et we en diseases that w ould ha ve ve ry similar phenot yp ic d escription bu t whic h remain differen t diseases. All v arian ts are based on the same m etho dological bac kb one, namely , the use of a multit ask learning strategy , and only differ in a function used to cont rol the sharing of inf ormation. W e limit ourselv es to the 1873 diseases in the disease-gene asso ciation dataset which were also in the phenot ypic similarit y matrix that we used . This corresp onds to a total of 2544 asso ciations b et we en these diseases and 1698 genes. W e compare these v arian ts to PRINCE [14], a metho d recen tly p rop osed to rank genes by s haring information across diseases through lab el propagation on a PPI n etw ork. Figure 2 sh o ws the CDF curv es for the four metho ds. Comparing areas under the global curve , i.e., the av erage rank of th e left-out disease gene in LOOCV, the four metho ds can b e rank ed in the fol- lo wing order: ProDiGe4 (1682 ) > ProDiGe3 (1817) > ProDiGe2 (2246) > P R I NCE (3065). The fact that ProDiGe3 and ProDiGe4 outp erform ProDiGe2 confirms the b enefits of exploiting prior kn o wledge w e ha ve ab out the disease p henot yp es to wei ght the sharing of information across diseases, instead of follo w in g a generic str ategy for multita sk learning. The fact that ProDiGe4 outp erf orms Pr oDiGe3 is not su rprisin g and illustrates the fact that the diseases are not fu lly c haracterized by the phenot ypic description we use. Zo oming to the b eginning of the curve s (righ t p icture), we see that the relativ e order b et ween the metho ds is conserv ed except for PRI NC E whic h outp erforms ProDiGe2 in that case. In fact, ProDiGe2 has a v ery lo w p erformance compared to all other metho d s for lo w ranks, confirming that the generic m ultitask strategy should n ot b e pursued in p ractice if ph enot ypic information is a v ailable. The fact that ProDiGe3 and Pr oDiGe4 outp erform PRINCE for all rank v alues confir m the comp et- itiv eness of our approac h. On the other hand, the comparison with PRINC E is not completely fair since ProDiGe exploits a v ariet y of data sources ab out the genes, while PRINCE only uses a PPI net wo rk. In order to clarify wh ether the impro v ement of ProDiGe o ve r P R I NCE is due to a larger amount of data used, to the learning algorithm, or to b oth, we ran ProDiGe3 with only the kernel d eriv ed from the PPI net wo rk w hic h we call ProDiGe-PPI in Figure 2. In that case, b oth ProDiGe and PRINCE u se exactl y the same inf orm ation to rank genes. W e see on th e left picture that this v arian t is o ve rall comparable to PRINCE (no significan t d ifference b et ween PRI NCE and Pr oDiGe-PPI w ith a Wilco xon p aired signed 4 rank test), confirmin g that the m ain b enefit of ProDiGe o v er PRINCE comes from data inte gration. In- terestingly though, at the b eginning of the cu rv e (right picture), ProDiGe-PPI is far ab ov e PRINCE, an d ev en b eha ves comparably to the b est metho d Pr oDiGe4. Since Pr oDiGe-PPI and PRINCE use exactly the same in put data, this means that the b etter p erformance of ProDiGe-PPI for lo w ranks comes from the learnin g metho d based on P U learning with SVM, as opp osed to label propagation o ver the PPI net wo rk. 0 0.5 1 1.5 2 x 10 4 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 Rank Cumulative distribution ProDiGe2 ProDiGe3 ProDiGe4 ProDiGe−PPI Prince 0 50 100 150 200 250 300 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5 Rank Cumulative distribution ProDiGe2 ProDiGe3 ProDiGe4 ProDiGe−PPI Prince A B Figure 2: Cumulativ e distribution function of ranks in the LOOC V exp eriments, for global approac hes . ProDiGe2, 3, 4 refer to the three v arian ts of ProDiGe whic h sh are information, while ProDiGe-PPI refers to ProDiGe3 trained only the PPI net w ork data. P anel (A) Global curve. P anel (B) Zo om on the b eginning of the cu rv e. T o b ett er visualize the d ifferences b et wee n the different v ariants of Pr oDiGe, the scatter plots in Figure 3 compare directly the r anks obtained by the differen t v arian ts for eac h of the 2544 left-out asso ciations. Note th at smaller ranks are b etter than large ones, s in ce the goal is to b e ranke d as close as p ossible to the top of the list. On the left panel, w e compare ProDiGe3 to ProDiGe4. W e s ee that man y p oints are b elo w the diagonal, meaning that add ing a Dirac k ernel to the Phenotype kernel (ProDiGe4) generally impro ves the p erformance as compared to using a Ph enot yp e k ern el (ProDiGe3) alone. On the righ t panel, the Pr oDiGe2 is compared to the ProDiGe3. W e see that th e p oin ts are more concen trated ab ov e the diagonal, bu t with large v ariabilit y on b oth sid es of the diagonal. Th is indicates a clear adv an tage in fa v or of the Phenot yp e k ern el compared to the generic Multitask k ernel, although the differences are quite flu ctuant. Is sharing information across diseases b eneficial? In order to c hec k whether sh arin g inf ormation across diseases is b eneficia l, we restrict ourselv es to d iseases with ph enot ypic informations and with at least t wo kno wn asso ciated genes in the OMIM database. This w a y , w e are able to share information across diseases and, at the same time, to r un m etho ds that do not share information b ecause w e en s ure that ther e is at least one training gene in the LOOCV p ro cedure. This lea v es us with 265 diseases, corresp on d ing to 936 asso ciations. Figure 4 sho ws the C DF curves of the rank f or th e v arious metho ds on these data, including the 5 0 0.5 1 1.5 2 x 10 4 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 x 10 4 ProDige3 ProDige4 0 0.5 1 1.5 2 x 10 4 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 x 10 4 ProDige3 ProDige2 B A Figure 3: Comparison of rank measures b etw een differen t v arian ts of ProDiGe. Eac h p oint represent a disease-gene asso ciation. W e plot the rank they obtain from the differen t metho ds when they are left out in the LOOCV pro cedu re. S mall rank are ther efore b etter than large r anks. t w o method s MKL1class and ProDiGe1 (with th e mean ke rn el for data inte gration), whic h do not share information across diseases, and Pr oDiGe 2, 3, 4 and PRINCE, wh ic h do sh are inf ormation. Int erestingly , w e observ e different retriev al b eha viors on these curves, dep en ding on the part of the curv e w e are in ter- ested in. On the one hand, if we look at the curve s globally , P roDiGe 4 and 3 p erform v ery w ell, having high area und er the CDF cur v e, i.e., a lo w a v erage rank (resp ectiv ely 1529 and 177 0). PRINCE an d MKL1class ha ve the w orse a v erage ranks (resp ectiv ely 322 0 and 3351). A s y s tematic test of differences b et ween the metho d s, using a Wilco xon paired signed rank test ov er the r anks for eac h pair of metho ds, is su mmarized in Figure 5 . I n this picture, an arro w indicates that a metho d is significantly b etter than another at lev el 5%. This confirms that ProDiGe 4 is the b est metho d, s ignifi can tly b etter than all other ones except Pr oDiGe 1. Thr ee v arian ts of Pr oDiGe are signifi can tly b etter than PRINCE and MKL1Class. On the other hand , in th e con text of gene prioritizatio n, it is u seful to fo cus on th e b eg inn ing of the curv e and n ot on the full CDF curves. Indeed, only the top of th e list is lik ely to deserv e an y serious biologica l inv estigation. Therefore we present a zo om of the CDF curv e in p anel (B) of Figure 4 . W e see there that the lo cal metho ds ProDiGe1 and MKL1class p resen t a sharp er increase at the b eg inn ing of the cur v e than the global metho ds, m eaning that they yield more often tru ly disease genes near the v ery top of the list than other metho ds. Additionally , we observ e th at ProDiGe1 is in fact the b est method when w e focu s on the prop ortion of disease genes correctly identified in up to the top 350 among 19540, i.e., in u p to the top 1.8% of the list. These results are further confirmed by the quantita tiv e v alues in T able 1, whic h s h o w the recall (i.e., CDF v alue) as a function of the rank. ProDiGe 1, wh ich do es not share inf ormation across diseases, is the b est when we only fo cus at the v ery top of the list (up to the top 1.8%), while ProDiGe 4, which shares information, is then the b est metho d when w e go deep er in the list. A t this p oin t it is in teresting to question wh at p osition in th e list we are interested in. In classical applications where w e start from a sh ort list of, sa y , 100 candidates, then b eing in the top 5% of the list means that the correct gene is r ank ed in the top 5 among the 100 cand idates, while the top 1% corresp onds to the firs t of the list (see the last 3 column s of table 1). If we on ly fo cus on the first 6 top 1 top 10 top 1% top 5% top 10% MKL1class 11.5 25.3 41.1 52.8 59.9 ProDiGe1 12.3 27.8 49.2 61.9 71.2 ProDiGe2 0.1 0.7 17.8 51.2 66.9 ProDiGe3 1.9 11.4 38.6 64.0 74.2 ProDiGe4 3.1 14.6 43.4 68.9 78.4 PRINCE 1.5 6.8 37.3 57.1 65.4 T able 1: Recall of different metho ds at differen t rank levels, for diseases with at least one kno wn disea se gene. T he recall at rank lev el k is the p ercen tage of disease genes that w ere correctly rank ed in the top k candidate genes in the LO OCV p ro cedure, wh ere the num b er of candidate genes is near 19540. T op 1 and top 10 (first t wo columns) corresp ond resp ectiv ely to the recall at the first and first ten genes among 19540, while top X% (last three columns ) refer to the recall at the fi rst X% genes among 19540. gene of a sh ort list of 100 candidates, then ProDiGe1 is the b est metho d, with almost half of the genes (49 . 2%) found in the first p osition, follo wed by ProDiGe4 (43 . 4 %) and MKL1class (41 . 1%). As so on as we accept to look further than the first place only , ProDiGe 4 is the b est metho d, with 68 . 9% of disease genes in th e top 5 of a list of 100 candidates, f or example. On the other han d , if we consider a scenario w here w e s tart from no short list of candidates, and directly wish to p redict disease genes among the 19540 h um an genes, then only the few top genes in the list are interesting (see the fi r st 2 columns of table 1). I n that case, the metho d s that do n ot share information are clearly preferable, with 27 . 8% (resp 25 . 3%) of genes correctly found in the top 10 among 19540 for Pr oDiGe 1 (resp. MKL1class). In su mmary , sharing in f ormation is not b eneficial if w e are intereste d only in the v ery top of the list, t ypically the top 10 among 19540 cand id ates. This setting is h o we ver v ery c hallenging, wh ere ev en the b est metho d ProDiGe1 only find s 12 . 3% of all disease genes. As soon as we are intereste d in more than the top 2% of the list, whic h is a reasonable lev el when w e start from a short list of a few tens or h un dreds of candidate genes, sharin g in formation across d iseases b eco mes in teresting. In all cases, some v arian t of Pr oDiGe outp erform s existing method s. In p articular Pr oDiGe4, which sh ares information using p henot ypic information across diseases while keeping different diseases distinct, is the b est wa y to share information. Predicting causal genes for orphan diseases Finally , we inv estigate the capacit y of the different gene p rioritization metho d s to id en tify disease genes for orphan d iseases, i.e., d iseases with no kno wn causativ e gene y et. ProDiGe1 and MKL1class, whic h treat d iseases indep enden tly from eac h other and requ ire kno wn disease genes to find new ones, can not b e used in this setting. Metho ds that sh are information across d iseases, i.e., ProDiGe2, 3, 4 and PRINCE, can b e teste d in this con text, since they ma y b e able to disco ver causativ e genes for a giv en orph an diseases by learnin g from causativ e genes of other diseases. In fact, Pr oDiGe3 and P roDiGe4 b oil down to the same metho d in this context , b ecause the con tribu tion of th e Dirac kernel in (6) v anishes when no kno wn disease gene for a d isease of interest is av ailable durin g training. W e s u mmarize them b y the acron ym P r oDiGe3-4 b elow. T o simulate this setting, we start fr om the 1608 diseases with only one kno wn disease gene in OMIM and p henot ypic information, resulting in 1608 disease-gene asso ciations inv olving 1182 genes. F or eac h disease in turn, the asso ciated gene is remo ved from the training set, a s coring function is learned from 7 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 x 10 4 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 Rank Cumulative distribution MKL1class ProDiGe1 ProDiGe2 ProDiGe3 ProDiGe4 Prince 0 50 100 150 200 250 300 350 400 450 500 0 0.1 0.2 0.3 0.4 0.5 Rank Cumulative distribution MKL1class ProDiGe1 ProDiGe2 ProDiGe3 ProDiGe4 Prince A B Figure 4: Cum ulative distribution function of ra nks for lo cal and multitask approac hes. (A) Global curve. (B) Z o om on the b eginning of the curv e. Figure 5: Wilco xon paired signed rank tests for significan t rank difference b etw een all metho ds. ProDiGe1 and MKL1class are th e only lo cal approac hes, whic h do not share information across d iseases. Th e num b er in eac h ellipse is the av erage rank obtained by the metho d in the LOOCV pro cedur e. An arr o w indicates that a metho d is signifi can tly b etter than another. the asso ciations inv olving other diseases, and the r emov ed causal gene is rank ed for the disease of int erest. W e compute the r an k of th e true disease gene, and rep eat this op eration for eac h d isease in turn. Figure 6 and T able 2 show the p erformance of the different global metho ds in this setting. I n terestingly , they are v ery similar to the resu lts obtained in the multita sk setting (Figure 2 and T able 1), b oth in r elativ e ord er of the metho d s and in their absolute p erformance. Overall , ProDiGe3-4 p erforms b est, retrieving the true causal gene in the top 10 genes of the list 13 . 1% of times, and in the top 5% of candidate genes 66 . 9% of times. This is only sligh tly w orse than the p erf ormance reac hed for diseases with kn o wn disease genes (resp ectiv ely 14 . 6% and 68 . 9%), highlight ing the promising ability of global app r oac h es to deorphan ize diseases. 8 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 x 10 4 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 Rank Cumulative distribution ProDiGe2 ProDiGe3−4 Prince 0 50 100 150 200 250 300 0 0.05 0.1 0.15 0.2 0.25 0.3 0.35 0.4 0.45 0.5 Rank Cumulative distribution ProDiGe2 ProDiGe3−4 Prince A B Figure 6: Cum ulative distribution function of ra nks for prioritization of causal genes for orphan diseases. Panel (A) Global cur v e. P anel (B) Zo om on the b eginning of the curve. top 1 top 10 top 1% top 5% top 10% ProDiGe2 0.1 1.4 16.8 50.4 68.1 ProDiGe3-4 1.9 13.1 42.7 66.9 76.1 PRINCE 0.5 4.8 36.9 52.9 60.6 T able 2: Recall of different metho ds at different rank lev els, for orphan diseases. In this case, since the disease has no known causal genes, only the causal genes of other diseases interv ene in the learning, meaning th at ProDiGe3 and 4 are equiv alen t approac hes. V alidation on selected diseases T o further v alidate ProDiGe, we used the whole tr aining set to pr ioritize the un lab eled genes for a few particular diseases with P r oDiGe4. W e completed the training set with a list of genes collected through the use of Ingen uity Pat hw a ys Analysis (IP A, Ingenuit y Systems). In T able 3, we rep ort the r esults of this v alidation for a first set of diseases ha ving a training set of p ositive genes of r easonable size (more than 11 genes). These diseases are in the same order: prostate cancer [MIM 176807], colorecta l cancer [MIM 1145 00], diab etes mellitus [MIM 125853], Alzheimer [MIM 104300], gastric cancer [MIM 137215], leuk emia acute m yelo id [MIM 601 626], b reast cancer [MIM 114480], sc hizophrenia [MIM 18150 0]. The columns rep ort successiv ely the disease name, th e MIM id of the disease, the size of the training set, the size of the intersectio n b et ween the training set and the Ingenuit y list, the estimated precision and r ecall of th e top 100 genes in the prioritized list and the p-v alue of a hypergeometric test. The p recision is estimated as the fraction of the top 100 genes that are also in the IP A list while recall is the fraction of the IP A list that intersects the top 100 genes of the prioritized list. Of course, the tru e precision v alue is unkno wn and the v alue we rep ort u n derestimates th e true v alue. The hyp ergeometric test allo ws to test f or the enric hment of th e top 100 genes of our prioritized list in genes kn o wn to b e asso cia ted to the disease, whic h w ere n ot in the training set (namely genes p r eviously extracted fr om IP A). W e can see that precision is goo d , except f or sc h izophrenia, gastric cancer and leuk emia. Recall on the other hand 9 is n ot v ery h igh b ut the v alues are limited by the large size of IP A lists. All tests are s ignifican t at 5% leve l. Disease name MIM Id T raining s et T rainin g T IP A Pr ecision (%) Recall (%) P-v alue Prostate cancer 17680 7 12 12 41 7.5 5 . 3 e − 40 Colorectal cancer 11450 0 17 17 51 5.7 7 . 3 e − 44 Diab etes mellitus 12585 3 26 22 21 1.4 2 . 1 e − 06 Alzheimer 10430 0 11 10 23 2.3 3 . 8 e − 11 Gastric cancer 13721 5 12 12 16 7.1 9 . 3 e − 16 Leuk emia acute m ye loid 60162 6 17 16 13 10.0 2 . 8 e − 15 Breast cancer 11448 0 19 16 33 3.7 6 . 4 e − 22 Sc hizophren ia 18150 0 17 11 6 3.2 4 . 5 e − 05 T able 3: Prioritization with ProDiGe4 for 8 diseases with a large training set of kno wn genes. The results were v alidated by comparin g our top 100 genes with a list of genes r elated to the d isease, extracted from I n gen uit y database. W e th en did the same for 8 diseases with only 2 k n o wn genes in our training set: glaucoma [MIM 60665 7], Creu tzfeld-Jacob [MIM 123400 ], hyp erparathyroidism [MIM 145000] , psoriasis [MIM 177900], glioblastoma [MIM 137800 ], cystic fi brosis [MIM 219700] , pancreatic carcinoma [MIM 260350], tha- lassemia [MIM 60413 1]. Results are giv en in T able 4. As exp ected, precision is m uch smaller for these diseases. Ho wev er, we s ee that sh aring information across diseases still allo ws to r etriev e n ew d isease genes for diseases where the training set is very small. Disease name MIM Id T raining T IP A Pr ecision (%) Recall (%) P-v alue Glaucoma 60665 7 2 8 12.5 2 . 0 e − 11 Creutzfeld-Jacob 12340 0 2 2 40.0 1 . 3 e − 06 Hyp erparathyroidism 14500 0 2 3 18.7 1 . 1 e − 06 Psoriasis 17790 0 2 4 6.0 1 . 8 e − 05 Glioblastoma 13780 0 2 16 10.7 8 . 4 e − 19 Cystic fib r osis 21970 0 2 5 10.6 9 . 3 e − 08 P ancreatic carcinoma 26035 0 1 8 9.6 2 . 3 e − 10 Thalassemia 60413 1 0 2 25.0 2 . 6 e − 06 T able 4: Prioritization with ProDiGe4 for 8 diseases with only 2 kno wn genes. The results w ere v alidated b y comparing our top 100 genes with a list of genes related to the disease, extracted from Ingen uity d atabase. F ur th er v alidation include T able 5 which rep orts th e top ten genes of the p rioritized list for prostate cancer, colorect al cancer, diab etes mellitus, Alzheimer, gastric cancer, leukemia acute m yel oid, breast cancer, schizo ph renia. These lists were analyzed with GeneV aloriza tion [26], a text-mining to ol f or automatic bibliography searc h . Discussion W e h a v e in tro duced ProDiGe, a new set of metho ds for disease gene prioritization. ProDiGe int egrates heterogeneous information ab out the genes in a un ified PU learning strategy , and is able to share infor- mation across different diseases if w an ted. W e h a v e prop osed in particular t w o flav ours for disease gene 10 ranking: ProDiGe1, which learns new causal genes for eac h disease separately , based on already known causal genes for eac h disease, and ProDiGe4, whic h additionally transf er s information ab out kno wn d is- ease genes across different diseases, w eigh ting information sharing by disease phenot ypic similarit y . W e ha v e d emonstrated th e efficiency of b ot h v arian ts on real data fr om th e O MIM database where they outp erform End ea v our and PRINCE, t w o state-of-the-a rt gene prioritization metho d s. A particularit y of ProDiGe is the p ossib ilit y to enco de p r ior kno wledge on disease r elatedness through the disease ke rn el. While a Dirac kernel pr even ts sharing of in f ormation across diseases, we tested dif- feren t v ariants to share information includ ing a generic m ultitask kernel and k ernels taking into account the p henot ypic similarit y b et wee n diseases. W e demonstr ated the relev ance of u sing the ph enot ypic sim- ilarit y , compared to th e generic multitask ke rn el, and h a v e enhanced it by the addition of a Dirac kernel. Giv en the infl uence of the d isease ke rn el on the fin al p erf orm ance of the metho d , we b eliev e that there is still m u ch ro om for imp ro ve ment in the d esign of the pr ior, using the general ProDiGe framewo rk. W e note in particular that if other d escriptors w ere a v ailable for phenot yp es, one could also integ rate th ese data and the pr ior they ind uce on task relatedness in the disease ke rn el. A imp ortant question in p r actice is to c ho ose b et wee n the tw o v ariants. W e ha ve seen that Pr oDiGe1 has higher recall in the top 1 or 2% of the list, w hile ProDiGe4 is b etter after. Hence a first criterion to c hose among them is the rank lev el that we are ready to in vesti gate. In add ition, one could th ink that Pr oDiGe1, whic h can not b e used for orphan disease, is more generally handicapp ed compared to ProDiGe4 when the num b er of kno wn d isease genes is small, while it is in a b etter situation when many genes are already known. In deed, if enough causal genes are kno wn f or a giv en d isease, there is intuitiv ely no need to b orr ow information from other diseases, wh ic h ma y m islead the prediction. T his dep end ency of the relativ e p er f ormance of a lo cal and a global approac h on the num b er of training samples has previously b ee n observ ed in other conte xts [21] where a global approac h w as sh o wn to bring tangible impro vemen ts o v er a lo cal one when th e num b er of p osit ive examp les w as lo w. W e ha v e ho wev er c hec ke d for the p resence of suc h an effect, and found that it could n ot b e brough t to ligh t, as illustrated in Figure 7 whic h plots the mean and stand ard deviation of the rank of the left-out gene in LO OCV as a f unction of th e num b er of kno w n genes of the disease d uring training. W e obs erv e no tr end in dicating that the p erforman ce increases with the n umb er of tr aining genes, and no different b ehaviour b etw een the lo cal and m ultitask app roac h es, as long as at least one d isease gene is kno wn. This sur prising finding, which is coheren t with the observ ation that th er e is no big difference in p erf orm ance for orphan and non-orphan diseases, suggests that the num b er of kno wn disease genes in not a relev an t criterion to c h o ose b et ween the local and multitask v ers ion of ProDiGe. Instead, w e suggest in practice to use the lo ca l version ProDiGe 1 if we are in terested only in genes ranke d in the v ery top of the candid ate gene lists (b elo w the top 1%), and ProDiGe 4 as so on as we can afford going deep er in the list. Finally , except for th e w ork of [27], the PU learning p oint of view on this long-studied gene prioriti- zation problem is nov el. Classical one-cla ss approac hes w hic h learn a scoring function to rank candidate genes u s ing kn o wn disease genes only are prone to o ver-fitting in large dimensions wh en the training set if small, w h ic h results in p oor p erform an ce. W e obs er ved that our PU learning strategy , augment ed b y a multita sk p oint of view to share inf orm ation across diseases, was us eful to ob tain b etter results in the disease gene identificati on task. In fact, learning from p o sitive and un lab eled examples is a common situation in bioinform atics, and PU learning m etho ds combined or not with m ultitask k ernels h a v e a go o d p oten tial for solving m an y p roblems suc h as pathw a y completion, prioritization of cancer patien ts with a higher risk of relapse, or prediction of p rotein-protein or pr otein-liga nd int eractions. 11 0 5 10 15 20 25 −2000 0 2000 4000 6000 8000 Rank Number of known causal genes ProDiGe4 ProDiGe3 ProDiGe1 Figure 7: Effect of the num b er of related genes on the p erformance. Material and Metho ds The gene prioritiz at ion problem Let u s first f orm ally defi ne the disease gene p rioritization p roblem we aim to solv e. W e start from a list of N human genes G = { G 1 , . . . , G N } , which typical ly can b e the full h uman genome or a subset of in terest where disease genes are susp ec ted. A multitude of d ata sources to c h aracterize these genes are giv en, includin g for instance expression profiles, fu nctional annotation, sequence p rop erties, r egulatory information, interact ions, literature d ata, etc... W e assu me that for eac h data source, eac h gene G ∈ G is represent ed by a fin ite- or infin ite-dimensional vec tor Φ( G ), which defin es an inner p ro duct K ( G, G ′ ) = Φ( G ) ⊤ Φ( G ′ ) b et we en an y t w o genes G and G ′ . K is called a kernel in the machine learning communit y [28]. Intuitiv ely , K ( G, G ′ ) may b e thought of as a measure of similarit y b et we en genes G and G ′ according to th e representat ion defi n ed by Φ. S ince several representati ons are a v ailable, w e assume that L feature v ector mappings Φ 1 , . . . , Φ L are av ailable, corresp onding to L k ernels for genes K 1 , K 2 , . . . , K L . Finally , w e supp ose giv en a collection of M d isorders or disease phenotypes D = { D 1 , . . . , D M } . F or eac h disord er D i , the learner is giv en a set of genes P i ⊂ G , which con tains kno wn causal genes for this p henot yp e, and a set of candid ate genes U i ⊂ G where we wan t to fin d new disease genes for D i . Typically U i can b e the complemen t set of P i in G if no further inform ation ab out the disease is av aila ble, or could b e a smaller subset if a short list of candidate genes is giv en for the disease D i . F or eac h d isease D i , our goal is to retriev e more causal genes for D i in U i . In p ractice, w e aim at ranking the elemen ts of U i from the most lik ely disease gene to th e less lik ely , and we assess the qualit y of a ranking b y its capacit y to rank the true disease genes at or near the top of the list. Gene priorit ization for a single disease and a single data source Let u s first describ e our gene pr ioritizatio n approac h ProDiGe for a single d isease ( M = 1) and a single data source ( L = 1). In that case, w e are giv en a s in gle list of disease genes P ⊂ G , and must rank the candidate genes in U ⊂ G using the k ernel K . As explained in the In tro duction, most existing approac hes 12 define a scoring f unction s : U → R , using only p ositiv e examples in P , to qu an tify ho w similar a gene G in U is to the kno wn disease genes in P . Here we prop ose to learn the scoring function s ( . ) b oth from P and U , b y form ulating the p roblem as an ins tance of PU learning. In tuitive ly , the motiv ation b ehin d PU learning is to exp loit the information provided by the distrib u- tion of u nlab eled examples to impro ve the scoring fu n ction, as illustrated in Figure 1. Here w e initially ha v e a set of p ositiv e examples (genes known to b e related to a giv en d isease for instance) which are represent ed on the graph b y b lu e crosses, and we wan t to retriev e m ore of them. T raditional app r oac h es whic h define a scoring function f rom P usually try to estimate the s u pp ort of the p ositiv e class d istribu- tion to defin e an area of “similar genes”, w h ic h could b e in that case delimited by th e dashed line. No w supp ose that we additionally observ e a set of un lab eled examples (candid ate genes), represent ed by U letters. Gr een Us are p ositive unlab ele d and red ones are negativ e u nlab eled, but this inform ation is not a v ailable. Then, we can hav e the feeling that the b oun dary sh ou ld rather b e set in the lo w d ensit y area as represent ed b y the solid line, whic h b etter captures realit y than the d ashed line. Consequently , u sing the distribution of U in addition to the p osit ive examples can help u s b ett er c haracterize the area of p ositiv e examples. This is particularly true in high d imension with few examples, w here densit y estimation f rom a few p ositiv e examples is known to b e very c hallenging. Figure 8: An in tuitive example of how the unlab eled examples could b e helpful. In pr actice, a simple and efficien t strategy to solv e a PU learning pr oblem is to assign n egativ e lab els to elemen ts in U , and train a binary classifier to discriminate P from U , allo wing errors in the training lab els. Assum ing that the binary classifier assigns a score to eac h p oint d uring training (which is the case of, e.g., logistic regression or SVM), the score of an element in U is then ju st the scored assigned to it by the classifier after training. T h is approac h is easy to implemen t and it has b een sho wn that b uilding a classifier that discrimin ates the p ositiv e from th e un lab eled set is a goo d pr o xy to buildin g a classifier that discriminates the p ositiv e f r om the negativ e set. When the b inary classifier used is a SVM, th is ap p roac h leads to th e biased SVM of [16], whic h wa s recen tly com bin ed with b agging to reac h faster trainin g time and equal p er f ormance [18]. In p ractice, the biased SVM o ver-w eights p ositiv e examples during training to accoun t for the f act that they represent high-confiden ce examples wh ereas the “negativ e” examples are kno wn to con tain false negativ es, namely , th ose w e hop e to disco v er. Here we use the v arian t of [18], whic h adds a b ootstrap pro cedur e to biased SVM. T he additional bagging-lik e feature tak es adv antag e of the con taminated nature of the u nlab eled set, allo win g to reac h the same p erformances while increasing 13 b oth sp eed and scalabilit y to large datasets. The algorithm tak es as input a p ositiv e and an u nlab eled set of examples, and a parameter B sp ec ifying the num b er of b o otstrap iterations. It discrim in ates the p ositiv e set from random subs amp les of the unlab el ed set and aggreg ates the successiv e classifiers into a single one (b o otstrap aggregating) . The output is a score function s s u c h that f or an y example G , s ( G ) r efl ects our confidence that G is a p ositiv e example. W e then rank elements in U by decreasing score. F or m ore details on the m etho d, w e refer the reader to [18]. In practice, w e implement the SVM with the libsvm implementat ion [29]. After observing in preliminary exp erimen ts that the regulariza- tion parameter C of the SVM did not dramatically affect the fi nal p erformance, we set it constan t to the default v alue C = 1 for all results s h o wn b elo w. The num b er of b o otst rap iterations w as set to B = 30. Gene priorit ization for a single disease and m ultiple data sources When seve ral data sources are a v ailable to charact erize genes, e.g., gene expr ession profiles and sequence features, we extend our PU learning metho d to learn s im ultaneously from m ultiple heterogeneous sour ces of data through kernel data fusion [24]. F ormally , eac h data source is encod ed in a k ern el function, resulting in L ≥ 1 k ernels K 1 , . . . , K L . W e inv estigate the f ollo win g t wo strategies to fus e the L data sources. First, w e simply d efine a new k ernel whic h in tegrates the information con tained in all k ernels as the mean of the L k ernels, i.e., we define: K int = 1 L L X i =1 K i . (1) In other w ords, the k ern el similarity K int ( G, G ′ ) b et ween t wo genes is defined as the mean similarit y b et ween the t wo genes o v er the different data sources. This simple app roac h is w idely used and often leads to ve ry go o d p erform an ce with S VM to learn classification mo dels fr om h eterogeneous inf orm ation [22, 30, 31]. In our setting, we simply use the integ rated k ernel (1) eac h time a SVM is trained in the PU learning algorithm d escrib ed in S ection , to estimate a prioritization score from m ultiple data sources. Alternativ ely , we test a metho d for multiple ke rnel le arning (MKL) p r op osed b y [24, 32], whic h amoun ts to building a we ighte d conv ex com bination of kernels of the form K M K L = 1 L L X i =1 β i K i , (2) where the non-negativ e weigh ts β i are automatic ally optimized d uring the learning phase of a SVM. By w eigh ting d ifferen tly the v arious inform ation sources, the MKL formulatio n can p oten tially d iscard irrelev an t sources or giv e more imp orta nce to gene descriptors with m ore predictive p ow er. Again, com bining MKL with our PU learning strategy d escrib ed in Section is straigh tforw ard: we simply us e the MKL formulation of SVM instead of the classical SVM eac h time a S VM is trained. Gene priorit ization for m ultiple diseases and m ultiple data sources When multiple d iseases are considered, a fi rst op tion is to treat the diseases indep enden tly from eac h other, and app ly the gene pr ioritizatio n strategy presen ted in Sections and to eac h disease in turn . Ho w ev er, it is kno wn that disease genes sh are some common c haracteristics [27, 33, 34 ], and that similar diseases are often caused by similar genes [5, 9, 11 – 14]. This su ggests that, in stead of treating eac h disease separately , it may b e b eneficia l to consider them join tly and sh are in formation of kno wn disease genes across d iseases. By m utu alizing in formation across diseases, one ma y in p articular atte mp t to p rioritize genes for orphan diseases, with no kno wn causal gene. This is an imp orta nt p rop erty since these diseases are obvi ously those for wh ich p redictions are the most n eeded. 14 W e p rop ose to joint ly solve the gene prioritization problem f or different diseases by f orm ulating it as a multitask learning pr ob lem, and we adapt the multit ask learning strategy of [19] to our PU learning framew ork. In this setting, instead of just learning a scoring f unction o v er ind ividual genes G ∈ G to rank candidates for a disease, w e learn a scoring fun ction o ve r dise ase-ge ne p airs of the f orm ( D , G ) ∈ D × G . In order to learn this scoring function, instead of starting from a s et of p o sitive examples P ⊂ G made of kno wn disease genes for a p articular disease, we start from a s et of p ositiv e pairs D d ( i ) , G g ( i ) i =1 ,...,T ⊂ D × G obtained by com bin ing the pairs where gene G g ( i ) is kno wn to b e a disease gene f or disease D d ( i ) . T is then the total num b er of kno wn disease-gene pairs. Giv en the training set of d isease-gene pairs, we then learn a scoring function s o ver D × G u sing our general PU learnin g algorithm describ ed in Section , where the ke rn el f unction b etw een tw o d isease-gene pairs ( D , G ) and ( D ′ , G ′ ) is of the form: K pair ( D , G ) , ( D ′ , G ′ ) = K disease ( D , D ′ ) × K g ene ( G, G ′ ) . (3) In this equation, K g ene is a kernel b et we en genes, typical ly equal to one of the k ernels d escrib ed in Sections and in the cont ext of gene pr ioritizatio n for a single disease. K disease is a k ernel b et we en diseases, wh ic h allo ws sharing of information across diseases, as in classical multitask learning with k ernels [19 – 21]. More pr ecisely , we consider the follo wing v ariants for K pair , wh ic h give rise to v arious gene pr ioritizatio n metho ds: • T he Dir ac kernel is defined as K D ir ac ( D , D ′ ) = ( 1 if D = D ′ , 0 otherwise. (4) Plugging the Dirac kernel int o (3), we see that th e p airwise k ernel b et we en tw o d isease-gene pairs for differen t diseases is 0. One can then sh o w that there is no sharing of information across diseases, and that learning o v er pairs in this con text b oils do wn to treating eac h disease indep enden tly f rom the others [19 – 21]. This is thus our baseline strategy , which treats eac h disease in turn, and do es not provide a solution for orphan d iseases. W e call th is metho d Pr oDiGe1 b elo w. • T he multitask kernel is defined by K multitask ( D , D ′ ) = 1 + K D ir ac ( D , D ′ ) . (5) This kernel, which w as p rop osed by [19], allo ws a b asic sharing of information across diseases. In addition to the genes known to b e causal for a disease of in terest through the Dirac k ern el, the addition of a constan t in (5) allo ws all other kn o wn d isease genes for other d iseases to play the role of p ositive training examples, although to a lesser exten t than the d isease genes for the disease of inte rest. Here w e do not use any sp ec ific knowledge ab out the differen t diseases and th eir similarit y , an d simp ly try to capture prop erties that ma y b e shared by disease genes in general. This corresp onds to a low information p rior b ecause a disease equally exp loits kn o wledge ab out all other diseases. W e call this v arian t P r oDiGe2 b elo w. • T he phenotyp e kernel is an attempt to capture ph enot ypic similarities b et ween diseases to con trol the sharing of in formation across diseases. Indeed, many pr evious works hav e used as prior kno wledge the fact that s im ilar ph enot yp es are lik ely to b e caused by similar genes [5, 9, 11–14, 35]. This suggests that, instead of sharing information u niformly across diseases as the m ultitask ke rn el (5) do es, it ma y b e b eneficial to do it in a more principled wa y . In p articular, the more similar t wo diseases are, the more they should share in formation. In pr actice, this is obtained by defining a kernel K phenoty pe b et ween diseases that measures their phenot ypic similarit y , and plugging it into the general pairwise k ernel (3). Here we prop ose to use the phenotypic similarit y measur e for diseases based on text 15 mining prop osed by [36]. Since this m easure is derive d as a correlation measur e, the matrix whose en tries con tain the pairwise similarit y measures is eligible for kernel learning. W e call the resulting gene prioritization metho d P r oDiGe3 b elo w. • T he phenotyp e+Dir ac k ernel. Finally , we prop ose a sligh t v arian t to th e ph enot yp e k ernel by adding to it the Dirac k ernel: K P + D ( D , D ′ ) = K phenoty pe ( D , D ′ ) + K D ir ac ( D , D ′ ) . (6) The motiv ation for this kernel is that, since the d escription of disease phenot yp es we us e to build K phenoty pe is incomplete and do es not fu lly c haracterize the d isease, it ma y o ccur that t w o differen t diseases, with different disease genes, hav e similar or ev en id entical phenot ypic descrip tion. In this case, the addition of the Dirac k ernel in (6) allo ws to still distinguish different diseases, and giv e more imp ortance to th e genes asso ciated to the d isease of in terest than to the genes asso ciated to different diseases w ith similar phenot yp es. W e call ProDiGe4 the resulting gene prioritization metho d. In s u mmary , eac h of the four kernels for diseases present ed ab ov e can b e plugged into (3) to define a k ernel for disease-gene pairs. Then , th e PU learning strategy presen ted in Section can b e applied to learn a scoring function ov er D × G . Finally , the rankin g of cand idate genes in U i for a particular d isease D i is obtained by decreasing score s ( D i , G ) f or G ∈ U i . W e thus obtain four v arian ts su mmarized in T able 6. When heterogeneous sour ces of information for genes are av ailable, the t wo str ategies prop osed in Section can b e easily combined with eac h of the four ProDiGe v ariants, since eac h particular gene k ernel translates int o a p articular d isease-gene k ernel through (3). In th e exp eriments b elo w, we only imple- men t the MKL approac h f or ProDiGe1 to compare it to the mean k ernel strategy . F or other v ariant s of ProDiGe, w e restrict ourselve s to the simplest strategy where the differen t information sources are fused through ke rn el a ve raging. Exp erimen tal setting W e assess the p erformance of v arious gene prioritization metho d s b y lea v e-one-out cross-v alidation (LOOCV) on the d ataset of kn o wn disease-gene asso ciation extracted f rom th e OMIM database. Given the list of all disease-gene asso ci ations ( D d ( i ) , G g ( i ) ) i =1 ,...,T in OMIM, we remo v e eac h pair ( D d ( i ) , G g ( i ) ) in turn f rom the training set, train the scoring function from the T − 1 remainin g p ositiv e pairs, rank all genes G not asso ciated to D d ( i ) in the training set b y decreasing s core s ( D d ( i ) , G ), and c hec k h o w we ll G g ( i ) is r ank ed in the list. Note that in this setting, we imp licitly assu me that the candidate genes for a disease are all genes not known to b e asso ciated to the disease, i.e., U i = G \ P i . In the LO OCV setting, eac h time a pair ( D d ( i ) , G g ( i ) ) is remo v ed from the training set, the ranking is then p erform ed on U d ( i ) ∪ { G g ( i ) } . W e monitor the success of the prioritization by the rank of G g ( i ) among candidate genes in U d ( i ) . Since we are doing a LOOCV pro cedur e, the rank of the left-out sample is directly related to the classical area un d er the Receiv er Op erating Ch aracteristics curve (A UC), via the f orm ula r an k = ( | U | + 1) × (1 − AU C ). Therefore, an easy w a y to visualize the p erformance of a gene prioritization metho d is to plot th e emp ir i- cal cum ulativ e d istribution function (CDF) of the ranks obtained for all asso ciations in the training set in the LOOC V pro ce du re. F or a giv en v alue of the r ank k , the CDF at leve l k is defined as the prop ortion of asso ciations D d ( i ) , G g ( i ) for w hic h gene G g ( i ) rank ed among the top k in the pr ioritization list f or d isease D d ( i ) , whic h can also b e called the r e c al l as a function of k . 16 Other gene prioritization metho ds W e compare ProDiGe to tw o state- of-the-art gene pr ioritization metho d s. First we consider the 1-SVM L2-MKL f rom [10], whic h extends and outp erforms the Endea vour metho d [10], and wh ic h we denote MKL1class b elo w. This metho d p erf orms one-class SVM [37] while optimizing the linear com b ination of gene k ernels with a MKL app roac h in the same time. W e do wn loaded a Matlab implementa tion of all functions f rom the sup p lemen tary in formation w ebsite of [10]. W e used as input the s ame 9 k ernels as for ProDiGe, and w e set the r egularization parameter of the algorithm ν = 0 . 5, as d one by [10]. Second, w e compare ProDiGe to the PRINCE m etho d introdu ced by [14], whic h is designed to share inform ation across the d iseases. Prior inf ormation consists in gene lab els that are a function of their relatedness to the query disease. They are h igher for genes kno wn to b e directly r elated to the query disease, high but at a lesser exten t for genes related to a disease which is v ery similar to the query , smaller for genes related to a d isease that b ea rs little similarity to the qu ery and zero for genes not related to any disease. PRINCE propagates these lab els on a PPI net work and pro du ces gene scores that v ary smo othly o ve r the netw ork. W e used the same PPI net w ork for PRINCE as th e one used by ProDiGe. Data The first type of data r equired b y ProDiGe is the d escrip tion of the set G of h um an genes. W e b orro wed the dataset of [7], based on Ensem bl v39 and whic h conta ins multiple data sources. W e remo v ed genes whose ID had a “retired” status in Ensembl v59, lea ving us with 19540 genes. Th ese genes are describ ed b y microarra y expression profi les from [38] and [39] (MA1, MA2), exp ressed s equence tag d ata (EST), functional annotation (GO) , path w ay mem b ership (KEGG), p rotein-protein inte ractions from th e Hu- man Pr otein Reference Data base (PPI), transcriptional motifs (MOTIF), protein domain activit y from In terPro (IPR) and literature data (TEXT). F or PPI data which consists in a graph of in teractions, a diffusion k ernel with p arameter 1 was compu ted to obtain a k ernel for genes [40]. All other data sources pro vide a v ectorial representa tion of a gene. The inn er pro duct b et ween these vect ors defin es the ke rn el w e create from eac h data s ou r ce. All kernels are normalized to un it d iagonal to ensu re that kernel v alues are comparable b et ween differen t d ata sources, using the form ula: ˜ K ( G, G ′ ) ← K ( G, G ′ ) p K ( G, G ) × K ( G ′ , G ′ ) . (7) Second, to define the phen otype kernel b et wee n d iseases w e b orro w the phenot ypic similarit y measure of [36]. The measure th ey prop ose is obtained by automatic text mining. A d isease is describ ed in the OMIM database by a text record [25]. In particular, its description con tains terms from the Mesh (medical sub ject h eadings) v o cabulary . [36] assess the similarit y b et wee n tw o diseases by comparing the Mesh terms con tent of their resp ectiv e r ecord in OMIM. W e do wnloaded the similarit y matrix for 5080 diseases from the MimMiner w ebpage. Finally , w e collected d isease-gene asso ciations from the O MIM d atabase [25], downloa ded on August 8th, 2010. W e obtained 3222 disease-gene asso ciations in vo lving 2606 disorders and 2182 genes. Ac kno wledgmen ts W e are grateful to L ´ eon-Charles T ranchev ent, Sh i Y u and Yve s Moreau for p ro viding the gene datasets, and to Ro ded Sharan and Od ed Magger for making their Matlab implemen tation of PRI NC E a v ailable to us. This wo rk was supp orted b y ANR grants ANR-07-BLAN -0311-03 and ANR-09- BLAN-0051-04 . 17 References [1] Giallourakis C , Henson C, Reich M, Xie X, Mo otha VK (2005) Disease gene d isco very through in tegrativ e genomics. Ann u Rev Genomics Hum Genet 6: 381–4 06. [2] P erez-Iratxeta C, Bork P , Andr ade MA (2002) Asso ciation of genes to genetical ly inherited diseases using data minin g. Nat Genet 31: 316 –319. [3] T urner FS, Clu tterbuc k DR, Semple CAM (2003) P o cus : min in g genomic sequence annotation to predict disease genes. Genome Biol 4: R75. [4] Tiffin N, Kelso JF, Po we ll AR, Pan H, Ba jic VB, et al. (2005) In tegration of text- and d ata-mining using ont ologies successfully selects disease gene candidates. Nucleic Acids Res 33: 1544–15 52. [5] F reu d en b erg J, Pr opping P (2002) A similarit y-based m etho d f or genome-wide pred iction of disease- relev an t h um an genes. Bioinformatics 18 Suppl 2: S110– S115. [6] Aerts S, L ambrec hts D, Mait y S, V an Lo o P , Co essens B, et al. (2006 ) Gene prioritizatio n through genomic data fusion. Nat Biotec hnol 24: 537 –544. [7] De Bie T, T ranc heven t LC, v an Oeffelen LMM, Moreau Y (2007) Kernel-based data fusion for gene prioritization. Bioinformatics 23: i125–i 132. [8] Linghu B, Snitkin E, Hu Z, Xia Y, Delisi C (2009) Genome-wide pr ioritizati on of disease genes and iden tification of disease-disease asso ciations from an integrat ed human functional link age net work. Genome Biol 10: R91. [9] Hw ang T , Kuang R (2010) A heteroge neous lab el p r opagation algorithm for d isease gene disco very . In: Pro ceedings of the S IAM International Conference on Data Minin g, SDM 2010, April 29 - Ma y 1, 2010, Columbus, Ohio, USA. pp . 583–594. [10] Y u S, F alc k T, Daemen A, T ranc hev ent LC , S u yk ens Y, et al. (2010) L2-norm multiple k ern el learning and its application to biomedical data fu sion. BMC Bioinformatics 11: 30 9. [11] Ala U, Piro R, Grassi E, Damasco C, Silengo L, et al. (2008) Prediction of human disease genes by h uman -mou s e conserv ed co exp ression analysis. PLoS Comput Biol 4: e100004 3. [12] W u X, Jiang R, Zhang M, Li S (200 8) Net wo rk-based global inferen ce of h uman disease genes. Mol Syst Biol 4: 189. [13] K¨ ohler S, Bauer S , Horn D, R ob in son P (2008) W alking the interacto me f or prioritization of candidate disease genes. Am J Hum Genet 82: 949–958. [14] V anun u O, Magger O, Rup pin E, S hlomi T, S haran R (2010) Ass o ciating genes and protein complexes with disease via n et w ork propagation. PLoS Comput Biol 6: e10006 41. [15] T r an chev ent LC, Cap devila FB, Nitsc h D, De Moor B, De Causm aec ker P , et al. (2010) A guide to w eb to ols to prioritize candidate genes. Brief Bioinform 11. [16] Liu B, Lee WS , Y u PS , Li X (2002 ) P artially sup ervised classification of text d o cuments. In: IC ML ’02: Pro ceedings of the Nineteen th Inte rn ational Conference on Mac h in e Learning. San F rancisco, CA, USA: Morgan K aufmann Publish ers Inc., pp. 387–394 . [17] Denis F, Gilleron R, Letouzey F (2005) Learning from p ositiv e and u nlab eled examples. Th eor Comput Sci 348: 70–83. 18 [18] Mordelet F, V ert JP (2010 ) A bagging SVM to learn from p ositiv e and unlab el ed examples. T ec h nical Rep ort HAL-005233 36. [19] Evgeniou T , Micc helli C, Po ntil M (2005) Learning multiple tasks with kernel metho ds. J Mac h Learn Res 6: 615-637 . [20] Jacob L, V ert JP (2008) Efficien t p eptide-MHC-I binding prediction for alleles with few kno wn binders. Bioinformatic s 24: 358–36 6. [21] Jacob L, V ert J P (2008) Protein-ligand interacti on prediction: an impro ved c hemogenomics ap- proac h. Bioinformatics 24: 2149– 2156. [22] P a vlidis P , W eston J, Cai J , Noble W (2002 ) Learning gene functional classificatio ns from multiple data t yp es. J C omput Biol 9: 401–4 11. [23] Sc h¨ olk opf B, Tsu da K, V ert JP (2004) Kernel Metho d s in Computational Biology . The MIT P r ess, Cam brid ge, Massac hussetts: MIT Press. [24] Lanc kriet GR G, De Bie T, C ristianini N, Jordan MI, Noble WS (2004 ) A statistic al framew ork for genomic data fusion. Bioinformatics 20: 2626-26 35. [25] McKusic k V (2007) Mendelian inheritance in man and its onlin e version, omim. Am J Hum Genet 80: 588–604. [26] Brancotte B, Biton A, Bernard -Pierrot I , Radv anyi F, Rey al F, Cohen-Boulakia S (2011) Gene list significance at-a-gla nce with GeneV alorization. Bioinformatics 27: 118 7–1189. [27] Calv o B, L´ op ez- Bigas N, F u rney S, Larr a ˜ naga P , Lozano J (200 7) A partially sup ervised classification approac h to dominan t and recessive human disease gene prediction. Compu t Metho d s Programs Biomed 85: 229–2 37. [28] Sc h¨ olk opf B, S mola AJ (200 2) Learning with Kernels: S upp ort Vector Mac hines, Regularization, Optimization, and Bey ond. Cam brid ge, MA: MIT Press. [29] Chang CC, Lin CJ (2001) LIBSVM: a library for su p p ort vect or mac h in es. S oftw are a v ailable at http://w ww.csie. ntu.edu.tw/ ~ cjlin/li bsvm . [30] Y amanishi Y, V ert JP , Kanehisa M (2004) Protein net work inference fr om multi ple genomic data: a sup ervised approac h. Bioinformatics 20: i363-i37 0. [31] Bleakley K, Biau G, V ert JP (2007) S up ervised r econstruction of b iologic al net w orks with lo cal mo dels. Bioinformatic s 23: i57–i65 . [32] Lanc kriet G, Cristianini N, Bartlett P , El Ghaoui L, Jordan M (2004) Learning the k ernel matrix with semidefinite pr ogramming. J Mac h Learn Res 5: 27-72. [33] L´ op ez-Bigas N, Ouzoun is CA (2004) Genome- wid e identi fication of genes likely to b e in volv ed in h uman genetic d isease. Nucleic Acids Res 32: 3108–31 14. [34] Adie EA, Adams RR, Ev ans KL, P orteous DJ, Pick ard BS (2005) Sp eeding d isease gene disco v ery b y sequen ce based candidate prioritization. BMC Bioinformatics 6: 55. [35] Lage K, Karlb erg E, Strling Z, Olason P , P edersen A, et al. (2007) A human ph enome-in teractome net wo rk of protein complexes imp licated in genetic disorders. Nat Biotec hnol 25: 309–3 16. [36] v an Driel M, Bruggeman J, V riend G, Brunner H, Leuniss en J (2006) A text-mining an alysis of the h uman ph enome. Eu r J Hum Genet 14: 535–542. 19 [37] Sc h¨ olk opf B, Platt JC, Sh aw e-T a ylor J, S mola AJ, Williamson R C (2001) Estimating the supp ort of a high-himensional distr ib utions. Neural Comput 13: 1443 –1471. [38] Son C, Bilk e S, Da vis S, Greer B, W ei J, et al. (2005) Database of mRNA gene expression profiles of m ultiple human organs. Genome Res 15: 443–4 50. [39] Su A, C o ok e M, Ch ing K, Hak ak Y, W alke r J, et al. (2002 ) Large-scale analysis of the human and mouse transcriptomes. Pro c Natl Acad Sci U S A 99: 4465–4 470. [40] Kondor RI, Laffert y J (2002) Diffusion kernels on graph s and other discrete inp ut. In: Pro ceedings of the Nineteen th In ternational Conference on Mac h ine Learning. San F rancisco, CA, USA: Morgan Kaufmann Pub lish ers Inc., pp. 315–322 . 20 Prostate cancer Gastric cancer CDKN2A(102 9) 210 1 EGFR(1956) 853 1 AKT1(207) 1058 1 AKT1(207) 272 0 IGF1R(348 0) 152 1 EXT1(2131) 4 0 MSX1(4487 ) 5 0 F AS(355) 180 0 P AX3(5077) 2 0 LRP5(4041) 8 0 CCND1(595) 3 72 1 MSX1(4487) 3 0 BRAF(673 ) 22 1 CCND1(595) 2 50 1 TP53(7157 ) 1378 1 BRAF(673) 32 1 WFS1(746 6) 0 0 TP53(7157 ) 1593 1 WT1(749 0) 37 1 WFS1(746 6) 0 0 Colorectal cancer Leuk emia acute my eloid CDKN2A(102 9) 415 1 AKT1(207) 233 0 EXT1(2131 ) 14 0 F AS(355) 136 0 IGF1R(348 0) 86 1 KRAS(3845) 457 1 SMAD4(408 9) 211 1 L YN(406 7) 26 0 MLH1(429 2) 406 4 1 MYC(4609) 381 0 PDGFRA(515 6) 19 1 RAF1(589 4) 30 1 PDGFRB(51 59) 45 1 ST A T3(6774 ) 95 0 BRAF(673 ) 430 1 STK11(6794 ) 2 0 WFS1(746 6) 0 1 BTK(695) 6 0 WT1(749 0) 15 0 TP53(7157 ) 474 1 Diab etes mellitus Breast cancer COL1A1(1277 ) 4 0 CDKN2A(1029) 572 1 COL2A1(1280 ) 6 0 CO L2A1(128 0) 9 0 CYP3A5(157 7) 5 0 COL3A1(128 1) 1 0 EXT1(2131 ) 20 1 EXT1(2131) 22 0 GHR(2690 ) 4 9 0 LR P 5(4041 ) 51 0 ABCC6(368) 43 0 MSX1(4487 ) 10 0 LEP(3952) 754 1 P AX3(5 077) 6 0 LRP5(4041 ) 5 8 0 PITX2(530 8) 310 1 CA CNA1S(779) 4 0 BRAF(673) 37 1 ADIPOQ(9370) 1635 1 WFS1(74 66) 4 0 Alzheimer Sc hizophren ia COL2A1(1280 ) 0 0 CO L1A1(127 7) 0 0 CYP1B1(15 45) 0 0 COL2A1(1280 ) 0 0 EXT1(2131 ) 4 1 A TN1(1822) 40 0 ALDH3A2(2 24) 4 0 EXT1(2131) 20 0 APOE(348) 4143 1 FGFR3 (2261) 78 0 ABCC6(368) 10 0 GJB1(270 5) 0 0 LRP5(4041 ) 3 0 ABCC6(368 ) 7 0 MA O A(4128) 5 1 LRP5(4041) 4 0 PSEN2(5664) 63 5 1 P ARK2(50 71) 1 0 WFS1(746 6) 1 0 WFS1(7466 ) 5 0 T able 5: The top t en genes for 8 diseases with a reasonable training set are scanned. Th ese diseases are in order: prostate cancer [MIM 176807], colorectal cancer [MIM 114500], diab et es mellitus [MIM 125 853], Alzheimer [MIM 104 300], gastric cancer [MIM 137 215], leukemia acute my eloid [MIM 60162 6], b reast cancer [MIM 114480], sc h izophrenia [MIM 1815 00]. Using GeneV alorization, we counte d the num b er of p ublication hits in NCBI whic h are found to b e relev an t to a query disease and a query gene. A t last, the third column indicates whether the gene b elongs to the list extracted from the In gen uit y P ath wa ys Analysis to ol. 21 Name Disea se k ernel Sharin g of disease gene inform ation across diseases Pro dige1 K D ir ac No sharing. Pro dige2 1 + K D ir ac Uniform sharing. Pro dige3 K phenoty pe Sharing weig hted b y phenot ypic similarit y . Pro dige4 K D ir ac + K phenoty pe Sharing weig hted b y phenot ypic similarit y and disease id en tit y . T able 6: Summary of ProDiGe v arian ts. W e prop ose four v ariants, which differ in th e wa y they share inf ormation across d iseases, as sum marized in th e third column of the table. T h e second column sho ws the k ern el for diseases u sed b y eac h v ariant to ac hieve the sharing of information. Apart from the c hoice of disease k ernel, the four v ariants f ollo w exactly th e same pro ce du re describ ed in Section . 22
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment