Probabilistic analysis of the human transcriptome with side information
Understanding functional organization of genetic information is a major challenge in modern biology. Following the initial publication of the human genome sequence in 2001, advances in high-throughput measurement technologies and efficient sharing of…
Authors: Leo Lahti
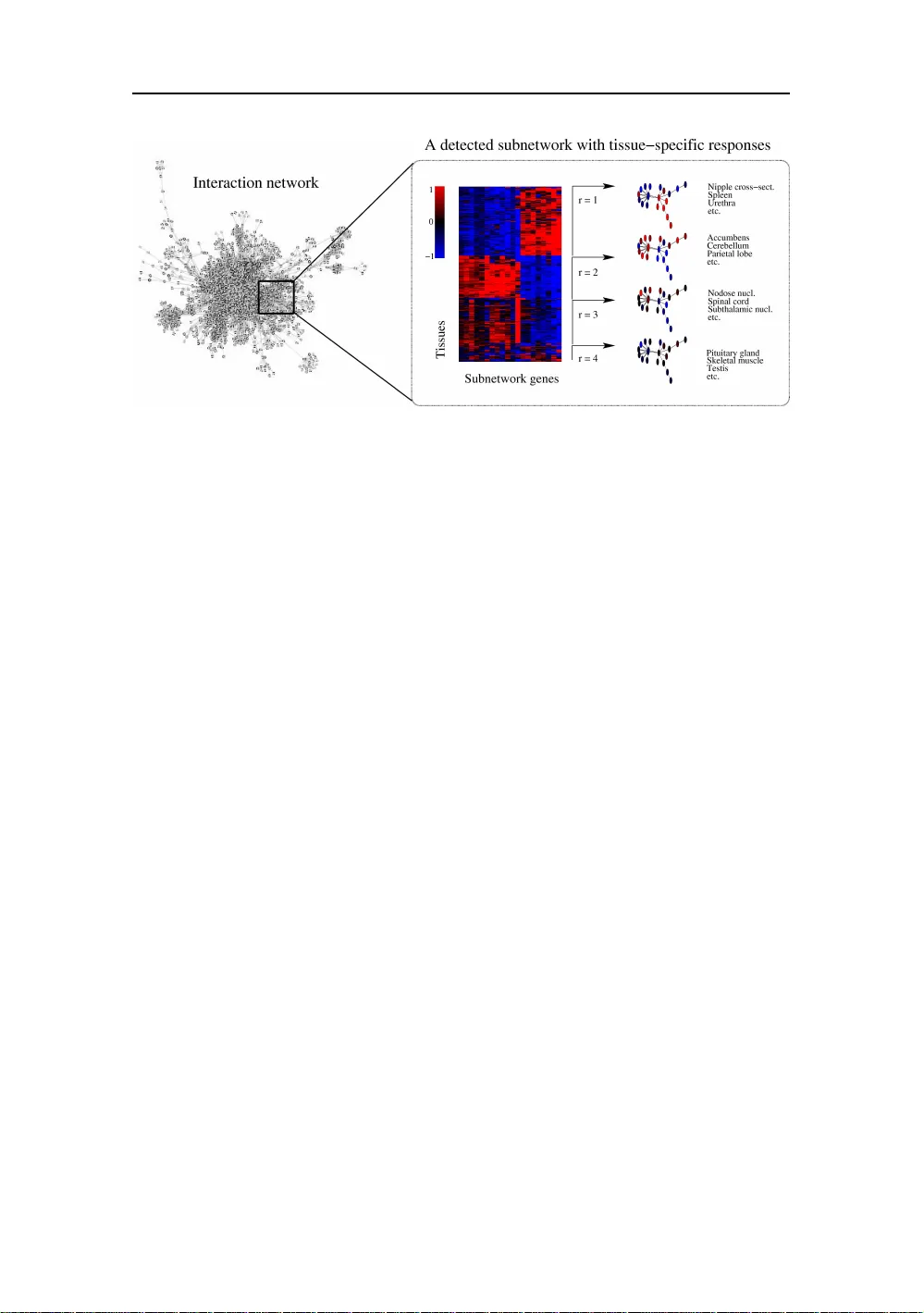
TKK Dissertations in In formation and Computer S cience Espo o 2010 TKK-ICS-D19 PR OBABILISTIC ANAL YSIS OF THE HUMAN TRANSCRIP TOME WITH SIDE INF ORMA TION Leo Lahti Dissertation for t he degree of Do ctor of S cience in T echnology to b e presen ted with d ue p ermission of the F aculty of Information and Natural Sciences f or public examination and debate in Auditorium AS1 at the Aalto Universit y Sc hool of Science and T ec hnology (Esp o o, Finland) on the 17th of December 2010 at 13 o’clock. Aalto Univ ersity School of Science and T echnology F aculty of Infor mation and Natur al Sciences Department of Information and Computer Science Aalto-yliopiston teknillinen korkeak oulu Informaatio- ja luonnontieteiden tiedekunta Tieto jenk¨ asittelytieteen laitos Distribution: Aalto Univ ersity School of Science and T echnology F aculty of Infor mation and Natur al Sciences Department of Information and Computer Science P .O.B ox 1 5400 FI-0007 6 Aalto FINLAND T el. +3 58-9-4 70 23 272 F ax +35 8-9-47 0 232 77 Email: series @ics.t kk.fi Copyrigh t c 2010 Leo Lahti First Edition. Some Rights Reser ved. ht tp://www.iki.fi/Leo.L aht i (leo.lahti@iki.fi) This thesis is licensed und er the t erms o f Cr e ative Co mmons A t tribution 3.0 Un- p orte d license av aila ble from http://www.creativecommons.org/ . Accordingly , you are free to co p y , distribute, display , perform, remix, tw eak, and build upon this work e ven for commercial purposes, assuming that you give the original autho r credit. See the licensing terms for details . F or Appendices and Figur e s, consult the separate copyrigh t notices. ISBN 978-9 52-60 -3367-9 (Print) ISBN 978-9 52-60 -3368-6 (Online) ISSN 1797- 5 050 (Print) ISSN 1797- 5 069 (Online) URL: http:/ /lib.t kk.fi/Diss/2010/isbn9789526033686/ Multiprint Oy Esp o o 2010 ABSTRAC T Lahti, L. (20 10): Probabilistic analysi s of the human transcriptome with side info rm ation Do ctoral thesis, Aalto Universit y School of Scie nc e and T ech- nology , Dissertations in Information and Computer Science, TKK-ICS- D19, Espo o , Finland. Keyw ords: data int egration, explor atory data analysis, functional genomics, probabilistic mo deling, transcripto mics Recent adv ances in hig h-throughput measurement technologies and efficient sharing of biomedica l data thr ough communit y databa s es hav e made it p ossible to inv es tig ate the complete collection of genetic material, the genome, which enco des the heritable genetic progra m of an o rganism. This has op ened up new views to the study of living org anisms with a profound impact on biolo g ical r e s earch. F unctional geno mics is a sub discipline of molec ula r biology that inv estigates the functional organiza tion of genetic information. This thesis dev elops c o mputational strategies to inv estigate a key functional lay er of the g e no me, the trans c r iptome. The time- and context-sp e cific transcr iptional activity of the g enes regulates the function of living cells thr o ugh protein syn thesis. Efficient co mputational tec h- niques are needed in order to extract useful information from high-dimensional genomic observ ations that ar e associa ted with hig h le vels of complex v a r iation. Statistical lear ning and pro babilistic mo dels provide the theoretical framew ork for combining statistical evidence acros s m ultiple observ a tions and the wealth o f background informatio n in genomic data rep ositor ies. This thesis addr esses three key challenges in transcr iptome analy sis. First, new prepro cess ing tec hniques that utilize side information in genomic sequence databases and microarr ay collections ar e dev elop ed to improve the accura cy of high-throughput microar ray measur emen ts. Second, a novel e xplorator y approach is prop os ed in or der to construct a global view of cell-biolog ical netw ork a cti- v ation patterns and functional rela tednes s b etw een tissues acr oss normal human bo dy . Information in g enomic int eraction databases is used to derive constraints that help to foc us the mo deling in those parts of the da ta that ar e suppo rted by k nown or po tential interactions b etw e e n the genes, and to scale up the analy- sis. The third co n tribution is to develop nov el approaches to mo del dep endency betw een co- o ccurring mea surement sour ces. The methods are used to s tudy can- cer mec hanisms and tra nscriptome ev olution; in tegrative analys is of the human transcriptome a nd other lay ers o f geno mic information a llows the identification of functional mechanisms and interactions that could not b e detected based on the individual measurement sources. Open source implemen tations of the key metho d- ologica l contributions hav e b een released to facilitate their further a doption b y the resear ch communit y . TI I V ISTELM ¨ A Lahti, L. (2010): Ihmi s en gee nien ilment ymis en ja taustatiedon tilasto lli- nen mallitus V¨ ait¨ o skirja, Aalto-ylio piston teknillinen korkeak oulu, Disser tations in Information and Computer Science, TKK-IC S- D19, Es p o o, Suomi. Av ainsanat: aineisto jen yhdistely , data-a nalyysi, toiminnallinen geno miikk a, ti- lastollinen mallitus, geenien ilment y minen Mittausmenetelmien kehit ys ja tutkim ustiedon laa jen tun ut saatavuus ov a t mah- dollistaneet ihmisen per im¨ a n eli genomin kokonaisv altaisen tark as telun. T¨ am¨ a on av a nn ut uusia n¨ ak¨ okulmia biolo giseen tutkimukseen ja auttanut ymm¨ art¨ am¨ a¨ an el¨ am¨ an synt y¨ a ja ra kennetta uusin tav oin. T oiminnallinen genomiikk a on molek yyli- biologian os a -alue, jok a tutkii p erim¨ an toiminnallisia ominaisuuksia. Perim¨ an toimint aan liitt yv¨ a¨ a mittausaineistoa on runsaa sti saata villa, mutt a k orkeaulot- teisiin mittauksiin liittyy monimutk ais ia ja tuntemattomia taustatekij¨ oit¨ a, joiden hu omiointi mallituksessa on haas teellista. T ehokk aat laskennalliset menetelm¨ at ov a t av ainasemas sa pyritt¨ aess¨ a jalostamaa n uusista hav ainnoista k¨ aytt¨ okelpo ista tietoa. T¨ ass¨ a v¨ ait¨ os k irjassa on kehitett y yleis k¨ aytt¨ oisi¨ a laskennallisia menetelmi¨ a, joilla voidaan tutkia ihmisen geenien ilmen t ymist¨ a k oko p erim¨ an tasolla . Geenien il- men t yminen viittaa l¨ ahetti-RNA-mole kyylien tuotto on solussa p er im¨ an sis¨ alt¨ a m¨ an informaation no jalla. T¨ am¨ a o n kesk einen p erinn¨ o llisen informaa tion s¨ a¨ atelyta so, jonk a avulla solu s¨ a ¨ atelee pro teiinien tuottoa ja solun toimintaa a jasta ja tilanteesta riippuen. Tilastollinen oppiminen ja todenn¨ a k¨ oisyyksin per ustuv a probabilis tinen mallitus tarjoav at teor eettisen keh yksen, jonk a avulla rinnakk a isiin mittauksiin ja taustatietoihin sis¨ altyv¨ a¨ a infor maatiota voidaan k¨ aytt¨ a¨ a k as v attamaan mallien tilastollista voimaa. Kehitetyt menetelm¨ at ov at yleisk¨ aytt¨ ois i¨ a laskennallisen tie- teen tutkimusv¨ alineit¨ a, jotk a tekev¨ at v¨ ah¨ a n, mutta selke¨ asti ilmaistuja mallitusole- tuksia ja siet¨ av¨ at korkeaulotteisiin toiminnallisen genomiik an hav aintoaineistoihin sis¨ altyvi¨ a ep¨ av a rmuuksia. V¨ ait¨ oskirjass a kehitet y t menetelm¨ at tarjoav at r atk aisuja kolmeen keskeiseen mallitusongelmaa n toiminnallisess a ge nomiik assa . Luotettavien esik¨ a sittelymene- telmien kehitt¨ aminen on ty¨ on ensimm¨ ainen p¨ a¨ atulos, jo s sa tietok an toihin sis¨ a lt y- v¨ a¨ a taustatietoa k¨ aytet¨ a¨ an p erim¨ anlaa juisten mittausaineisto jen ep¨ av a rmuuksien v¨ ahent¨ amiseksi. T oisena p¨ a¨ atuloksena v¨ ait¨ os kirjassa kehitet¨ a¨ an uusi alia v aruus- k asautuks een p erustuv a menetelm¨ a, jonk a avulla voidaan tutkia ja kuv ata solu- biologisen vuorov aikutusverkon k¨ aytt¨ aytymist¨ a kok onaisv altaises ti ihmiskehon eri osissa. T austatietoa geenien vuorov aikutuksista k¨ aytet¨ a¨ an ohjaamaan ja no peut- tamaan mallitusta . Menetelm¨ all¨ a saadaa n uutta tietoa geenien s¨ a¨ atelyst¨ a ja ku- dosten toiminnallis is ta yhteyksist¨ a. Kolmanneksi v ¨ ait¨ oskirjaty¨ oss¨ a kehitet¨ a¨ an uu- sia menetelmi¨ a p erim¨ a nlaa juisten mittausaineisto jen yhdistelyyn. Ihmisen geenien ilmen t ymisen ja m uiden a ineisto jen r iippuvuuksien mallitus mahdollistaa sellaisten toiminnallisten yht eyksien ja v uorov aikutusten hav aitsemisen, joiden tutkimiseks i yksitt¨ aiset hav aintoaineistot o v at riitt¨ am¨ att¨ omi¨ a. Aineisto jen yhdistelyyn kehitet- t yj¨ a menetelmi¨ a so v elletaan sy¨ o p¨ amek anismien ja la jien v¨ a listen er oav a isuuksien tutkimiseen. Julk a istuilla av oimen l¨ a hdekoodin toteutuksilla on pyritty v a rmista- maan k ehitett yjen menetelmien s a atavuus ja laa jempi k¨ aytt¨ o¨ ono tto laskennallisen biologian tutkim uksessa. Con ten ts Preface . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . v List of pu blications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . vii Summary of p ublications and the author’s con tribution . . . . . . . . . . . . . viii List of abbrev iations and symbols . . . . . . . . . . . . . . . . . . . . . . . . . . ix 1 Introduction 1 1.1 Con tributions and organization of the th esis . . . . . . . . . . . . . . . . . 2 2 F unctional ge nomics 4 2.1 Universal genetic co de . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4 2.1.1 Protein synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 2.1.2 La yers of regulation . . . . . . . . . . . . . . . . . . . . . . . . . . 6 2.2 Organization of genetic information . . . . . . . . . . . . . . . . . . . . . . 7 2.2.1 Genome structure . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 2.2.2 Genome function . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 2.3 Genomic data resources . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9 2.3.1 Comm unity databases and evol ving biological k n o wledge . . . . . . 9 2.3.2 Challenges in high-t hroughput data analysis . . . . . . . . . . . . . 12 2.4 Genomics and health . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13 3 Statistical le arning and exploratory data analysi s 14 3.1 Modeling tasks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14 3.1.1 Cen tral concepts in data analysis . . . . . . . . . . . . . . . . . . . 15 3.1.2 Exploratory data analysis . . . . . . . . . . . . . . . . . . . . . . . 17 3.1.3 Statistical learning . . . . . . . . . . . . . . . . . . . . . . . . . . . 18 3.2 Probabilistic mo d eling paradigm . . . . . . . . . . . . . . . . . . . . . . . 18 3.2.1 Generative mo deling . . . . . . . . . . . . . . . . . . . . . . . . . . 19 3.2.2 Nonparametric mo dels . . . . . . . . . . . . . . . . . . . . . . . . . 21 3.2.3 Ba ye sian analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 3.3 Learning and inference . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 3.3.1 Model fitting . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 3.3.2 Generalizabilit y and ove rlearning . . . . . . . . . . . . . . . . . . . 28 3.3.3 Regularization and mo del selection . . . . . . . . . . . . . . . . . . 29 3.3.4 V alidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 4 Reducing uncertaint y in hi gh-throug hput m icroarra y studies 31 4.1 Sources of uncertaint y . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 4.2 Preprocessing microarra y data with side information . . . . . . . . . . . . 32 4.3 Model-b ased noise reduct ion . . . . . . . . . . . . . . . . . . . . . . . . . . 36 4.4 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41 iii 5 Global analy sis of the human transcriptome 42 5.1 Standard approaches . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42 5.2 Global mo deling of transcriptional activity in interaction netw ork s . . . . 46 5.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50 6 Human transcriptome and other layers of genomic information 51 6.1 Standard approaches for genomic data integration . . . . . . . . . . . . . 52 6.1.1 Com bining statistical ev id ence . . . . . . . . . . . . . . . . . . . . 52 6.1.2 Role of side information . . . . . . . . . . . . . . . . . . . . . . . . 53 6.1.3 Modeling of mutual dep end ency . . . . . . . . . . . . . . . . . . . 54 6.2 Regularized dep endency detection . . . . . . . . . . . . . . . . . . . . . . 57 6.2.1 Cancer gene discov ery with dep endency detection . . . . . . . . . . 60 6.3 Associative clustering . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63 6.3.1 Exploratory analysis of tran scriptional divergence b etw een species 66 6.4 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 7 Summa ry and conclusions 69 References 71 iv Preface This w ork has b een carried out at the Neural Net w orks Resea rch Centre and Adap- tive Infor matics Resea rch Cent re of the Lab or atory of Computer and Infor mation Science (Department of Infor ma tion and Computer Scie nc e since 20 08), Helsinki Univ ersity of T echnology , i.e., as of 20 10 the Aalto University School o f Science and T echnology . Part of the work was done a t the Department of Computer Sci- ence, Univ er sity of Helsinki, when I was visiting ther e for a y ear in 20 0 5. I am also pleased to having had the opp ortunity to b e a part of the Helsinki Institute for Infor mation T echnology HIIT. The work has b een suppo r ted by the Gradua te School of Computer Science and Engineer ing, as well as by pro ject funding from the Academy of Finland through the SYSBIO progr am and from TEKES through the MultiBio research co nsortium. The Graduate Sc hoo l in Computational Bi- ology , Bioinformatics, and Biometr y (ComBi) has suppo rted my par ticipa tion to scientific conferences and workshops abr oad dur ing the thes is work. I wish to thank my sup erviso r , professo r Sa m uel Kaski for giving me the o p- po rtunit y to work in a truly interdisciplinary resea r ch field with the freedom a nd resp onsibilities o f scientific work, and with the necessa ry amount o f guida nce. These hav e b een essential parts of the lea rning pro cess. I would also like to express my gratitude to the re v iewers of this thesis, Professor Juho Rousu and Do ctor Simon Rogers for their exp ert feedback. Research on computational bio logy has given me the ex cellent o ppo r tunit y to work with and learn from exp erts in tw o traditionally distinct disciplines , co mpu- tational science and geno me biolo gy . I am par ticularly gr ateful to professo r Sak ari Knuutila for his en th usiasm, curiosity , and p ersonal example in collab ora tion and daily resea rch w ork. Resear chers in the Lab orator y of Cyto mo lecular Gene tics at the Haartman Institute hav e provided a friendly a nd inspiring environmen t f or active collab or ation during the las t years. My sincere compliments belong to a ll of my other co- authors, in particular to T ero Aittok allio , Laura Elo-Uhlgre n, Jaakko Hollm´ en, Juha Knuuttila, Sa m uel Myllyk ang a s and J anne Nikkil¨ a. It has b een a plea sure to w ork with y ou, and your contributions extend beyond what we wro te together. I would also like to tha nk the fo rmer and present members of the MI resear ch group for working bes ide me through these y ears, as well as for in tr iguing discussions abo ut science and life in general. I w o uld also like t o thank t he perso nnel of the ICS department, in particular profes s ors E rkki Oja and O lli Sim ula, who have help ed to pr ovide an excellent academic resear ch environmen t, as well as our secretarie s T arja Pihamaa and Leila Koiv is to, and Markku Ranta a nd Miki Sirola , who hav e given v aluable help in so many practica l matters during the years. Science is a communit y effort. O pen sha r ing of ideas, knowledge, publication v PREF A CE material, data, softw a re, code, exp er ie nces and emotions h as had a tremendous impact to this thesis. I will expr ess my s incere g ratitude to the communit y by contin ued participa tion a nd co ntributions. I w ould also lik e to thank m y earliest scient ific advisors; Reijo, who brought me writings ab out the chemistry of life and help ed me to grow bacteria and prepar e space dust in the 1980 ’s, Pekk a, who ha s demonstrated the p ow er of criticism and emphasized that natura l science has to be exact, T apio, for the attitude that maths can b e just fun, and Risto, for showing how rational thinking can b e applied also in real life. Thanks also g o to my science fr iends, Manu and Ville; we have shar ed the pas sion for natura l science, and I wan t to thank you for our co n tin uous and inspiring discussions along the wa y . I am grateful to m y grandfather Osmo, who shared with me the wonder towards life, science, and humanities, a nd was willing to discuss it a ll thr ough days and nights whe n I was a child, questioning himself the self-evident truths a gain and a gain, rema ining as puzzled as I was. And for Alli and Arja, m y grandmothers, for their understanding, and all suppor t a nd lo v e. My F riends. With you I hav e explored other facets of nature, science , and life... Thank you for staying with me throug h all these years and sharing so many asp ects of curiosity , explo ration a nd m utual under standing. Finally , I a m gr ateful to my parents and sister , Pips a , Kari, and T uuli. Y ou hav e acc epted me and lov ed me, supp orted me on the paths that I hav e chosen to follow, and understo o d that freedom can create the stro ngest ties. Cambridge, Nov em ber 23, 2010 Leo Lahti vi LIST OF PUBLICA TIONS LIST OF PUBLICA TIONS This thesis consists of an ov erview and of the following publications whic h are referred to in the text by their Roman numerals. 1. Laura L. Elo, Leo Lahti, Heli Skottman, Minna Ky l¨ a niemi, Riitta Lahesmaa, and T ero Aittok allio. Integrating prob e-level ex pression changes acr oss gener- ations of Affymetrix arrays. Nucleic A cids R ese ar ch , 33 (22):e193, 2 005. 2. Leo Lahti, La ur a L. Elo , T ero Aittok allio , and Sa m uel Ka ski. Probabilistic analysis of pr ob e re lia bilit y in differ e ntial g ene ex pr ession studies with sho r t oligonucleotide arr ays. IEEE/A CM T r ansactions on Computational Biolo gy and Bioinformatics , 8(1):217– 225, 2011 . 3. Leo La h ti, Juha E .A. K nuuttila, a nd Samuel Kaski. Globa l mo deling of tran- scriptional resp onses in in ter action net w orks. Bioinformatics , 26(2 1 ):2713– 2720, 2010 . 4. Leo Lahti, Samuel Mylly k angas, Sak ari Knuutila, a nd Samuel K aski. Dep en- dency detection with similar it y constraints. In T ¨ ulay Adali, Jo celyn Cha nus- sot, Chr istian Jutten, and Jan Larsen, editor s , Pr o c e e dings of the 2009 IEEE International Workshop on Machine L e arning for Signal Pr o c essing XIX , pages 89–94 . IEE E, Pis c a taw ay , NJ, 2 009. 5. Janne Sinkkonen, Janne Nikkil¨ a, Leo La ht i, and Samuel K aski. Assoc ia - tive clustering. In Boulicaut, E sp o sito, Giannotti, and P edreschi (editors), Machine L e arning: ECML200 4 (Pr o c e e dings of the ECML’0 4, 15th Eur op e an Confer enc e on Machine L e arning) , Lectur e No tes in Computer Science 3201 , 396–4 06. Springe r , B erlin, 2004. 6. Samuel Kaski, Janne Nikkil¨ a, Janne Sinkkonen, Leo Lahti, Juha E.A. K n uut- tila, and Cristo phe Roos. Asso ciative c lus tering fo r exploring dep endencies betw een functional genomics da ta s e ts. IEEE/A CM T r ansactions on Compu- tational Bi olo gy and Bioinformatics: S p e cial Issue on Machine L e arning for Bioinfo rmatics – Part 2 , 2(3):20 3–216 , 20 05. vii THE AUTHOR’S CON TRIBUTION SUMMAR Y OF PUBLICA TIONS AND THE A UT HOR’S CONTRIBUTION The publications in this thesis have been a joint effort of all authors; key contri- butions by the author of this thesis ar e summarized b elow. Publication 1 introduces a nov el a nalysis stra tegy to improv e the a ccuracy and r e pr o ducibilit y of the measurements in genome-wide tra nscriptional profiling studies. A cen tr al part of the approach is the utilization of side information in external genome sequence databas e s. The author participated in the design of the study , sugg ested the utilization of external s equence da ta , implemen ted this, as well as participated in preparing the manuscript. Publication 2 provides a pro babilistic framework fo r pr ob e-level g ene ex pres- sion analysis. The mo del combines statistica l pow er a cross multiple microarray exp eriments, a nd is shown to outperform widely-used preproces sing metho ds in differential gene expression analysis . The mo del pr ovides tools to a ssess prob e per formance, whic h can p otentially help to improv e pro be and microarr ay design. The author had a ma jor role in designing the study . The author derived the for- m ulation, implemented the mo del, p erfor med the prob e-level ex per imen ts, as w ell as co ordinated the manuscript preparation. The a uthor prepared an accompa - nied open source implemen tatio n which has b een published in BioCo nductor, a reviewed op en source rep osito r y for computationa l biolog y a lgorithms. Publication 3 introduces a nov el approach for or ganism-wide mo deling of tran- scriptional activity in genome-w ide interaction net works. The metho d provides to ols to analyze lar ge collections o f genome- wide transcriptiona l profiling data. The author had a ma jor ro le in designing the study . The author implemen ted the algo rithm, p erformed the exp e r iment s, as w ell as co or dinated the manuscript preparatio n. The a uthor par ticipated in and sup ervised the prepara tion of an accompanied op en source implementation in BioCo nductor. Publication 4 introduces a regularized dep endency modeling framework with particular applications in cancer genomics. The author had a ma jor role in for mu- lating the biomedical mo deling ta s k, and in des igning the study . The theoretica l mo del was jointly develop e d by the a uthor a nd S. Kaski. The autho r derived and implemented the mo del, ca rried out the exp eriments, a nd co ordina ted the manuscript prepara tio n. The author s up erv ised and par ticipated in the prepara- tion of an accompanied op en source implemen tation in BioConducto r. Publication 5 introduces the asso cia tive clustering pr inc iple , whic h is a nov el data in tegration framework fo r dep endency d etection with direct applications in functional g enomics. The author participated in implemen tation o f the metho d, had the main resp onsibility in designing and p erforming the functional g enomics exp eriments, as well as par ticipated in prepar ing the manuscript. Publication 6 contains the mo st extensive treatment of the as so ciative cluster- ing principle. In addition to presenting detailed theo retical c onsiderations, this work in tro duces new sensitivity analys is of the results, a nd pr ovides a comprehen- sive v alida tion in bioinformatics case s tudies. The a utho r participated in designing the expe r iment s, p erfor med the compara tive functional genomics ex p er iments and techn ical v alidation, as well a s participated in preparing the manuscript. viii THE AUTHOR’S CON TRIBUTION LIST OF ABBREVIA TIONS AND SYMBOLS In this thesis b oldface symbols are used to de no te matrices and vectors. Capital symbols ( X ) signify matrices a nd low ercase symbols ( x ) column vectors. Normal lower c ase symbols indicate scalar v aria bles. R Real domain X , Y Data matrices ( D × N ) [ X ; Y ] Concatenated data x , y Data samples, vectors in R D x , y Scalars in R X , Y Random v ariable s I Ident it y matrix Σ , Ψ Cov a riance matrices p ( x ) Probability or proba bilit y density o f X p ( X ) Likelihoo d E [ · ] Exp ectation k · k Norm of a matrix or vector T r Matrix trace I ( X ; Y ) Mutual information b etw een r andom v ariables X and Y Beta( α, β ) Beta distribution with parameter s α and β Dir( θ ) Dirichlet distribution with pa rameter vector θ IG( α, β ) Inv er se Gamma distribution with pa r ameters α and β Mult( N , θ ) Multinomial distribution with sample s ize N and pa rameter v ector θ N ( µ , Σ ) Normal distribution with mean µ and cov ariance matr ix Σ A C Asso ciative clustering aCGH Array Compara tiv e genomics hybridization CCA Canonical correlatio n analysis cDNA Complementary DNA DNA Deoxyribonucleic acid DP Dirichlet pro cess EM Exp ectation – Maximization algor ithm IB Information b ottleneck KL–divergence Kullback-Leibler divergence MAP Maximum a p osterior i MCMC Marko v chain Monte Car lo ML Maximum likelihoo d mRNA Messenger- RNA tRNA T rans fer-RNA PCA Principal comp onent analys is RNA Ribo n ucleic acid ix Chapter 1 In tro duction Revolutions in measurement technologies hav e led to rev olutions in science and so ciety . In troduction of the micr oscop e in the 17th century ope ne d a new view to the world of living organisms and enabled the study of life pr o cesses at ce llular level. Since then, new tec hniques hav e b een dev elop ed to in vestigate ever s maller ob jects. The dis c overy of the molecular str uc tur e of the DNA in 1 953 (W atso n and Crick, 1 953) led to the establishmen t of genes as fundamental units of ge- netic informa tion that is passed on betw e e n gener a tions. T he draft sequence of the hu man genome, cov ering three billion DNA base pairs, was published in 2001 (In ternational human genome s equencing consortium, 2 001; V enter et al., 2001 ). Mo dern meas ur ement technologies provide rese archers with large volumes of data concerning the struc tur e, function, a nd interactions of g enes and their pro ducts. Rapid accum ulation of genomic data in shared communit y databa ses has accel- erated biological r esearch (Co chrane a nd Galp erin, 2010 ), but the structural and functional organization of genetic infor mation is still po orly understoo d. While functional roles o f individual genes have b een characterized, little is known re- garding t he higher -level reg ularities and interactions fro m which the complex ity and diversit y of life emer ges. The quest for systems-level understanding of geno me function is a ma jor pa r adigm in mo der n biology (Collins et al., 2003 ). Computational science has a key ro le in tra nsforming the g enomic data co llec- tions into new biological knowledge (Cohen, 2004). New obser v ations a llow the formulation of new r esearch questions, but also bring new challenges (Barb our et al., 2005). The shee r size of high-thr o ughput data sets makes them incompre- hensible for human mind, and the co mplexity of biolo gical pheno mena a nd high levels of uncontrolled v ar iation set sp ecific c hallenges for computational analy sis (Tilstone, 2003; T r oy a nsk ay a, 20 0 5). Filtering relev an t information fro m s ta tisti- cally uncertain high-dimensional data is a challenging ta s k wher e new computa- tional metho ds a re needed to o rganize and summar ize the ov erwhelming volumes of o bserv atio nal data into a compr e hensible form to make new discoveries a bo ut the structure of life; computation is a new microscop e for studying mas s ive da ta sets. This thesis dev elops principled explorato ry methods to in vestigate the hum an tr anscriptome . It is a central functional lay er of the g enome and a significant source of phenotypic v ariation. The transcriptome refers to the co mplete colle c - tion of messenger -RNA tra nscripts of an orga nis m. The essentially static genome sequence regulates t he t ime- and c o nt ext-sp ecific patterns of transcriptiona l ac- 1 CHAPTER 1. INTRODUCTION tivit y o f the genes, and subsequently the function o f living cells throug h protein synthesis. An average c e ll contains ov er 300,0 0 0 mRNA molecules and the expres- sion levels of individual genes spa n 4-5 orders of magnitude (Ca rninci, 2 009). A wealth of a s so ciated ge no mic information resour ces ar e av aila ble in public rep osi- tories (Co chrane and Ga lp er in, 2010). By com bining heterog eneous informa tion sources and utilizing the wealth of background informatio n in public r epo sitories, it is p ossible to s olve some o f the problems that are rela ted to the statistical uncer- tainties and small sample size o f individual data sets, as w ell a s to for m a holistic picture of the genome (Huttenho wer and Hofmann, 20 10). The obser v ational da ta can pro vide the s ta rting p oint to disco ver nov el resea rch hypotheses of po o rly c haracterize d large-s c ale systems; the ana lysis pr o ceeds from general observ a tions of the data tow a rd more detailed inv estigations and hypothe- ses. This differs from traditional h ypo thesis tes ting wher e the investigation pro- ceeds from hypothese s to measurements that ta rget par ticular resea rch questions , in order to supp ort o r r eject a given h yp o thesis. Explor atory data analysis refers to the use of computational too ls to summar ize and visualize the da ta in orde r to ident ify p otentially interesting structure, and to fac ilitate the gener ation of new resear ch hypotheses when the search space would b e o ther wise exhaustively larg e (T ukey, 197 7). When the system is po o rly characterized, there is a need for meth- o ds that can adapt to the data and extract features in an a utomated wa y . This is useful since applica tion-oriented mo dels often re quire care ful prepro cessing of the data and a timely mo del fitting pro cess . They may also require prio r know- ledge o f the inv e s tigated system, which is often not av ailable. Statistic al le arning inv es tig ates so lutio ns to these pr oblems. 1.1 Con tributions and organization of the thesis This thesis introduces computational stra tegies for genome- and o r ganism-wide analysis of th e h uman transcr iptome. The thesis provides nov el to ols (i) to in- crease the reliability of hig h-throughput micro array measurements by co mb ining statistical ev idence from genome sequence databases and across multiple microar - ray exper imen ts, (ii) to mo del context-specific transcriptional activ ation patterns of geno me-scale interaction netw orks acros s normal h uman b o dy b y using back- ground information o f genetic interactions to guide the analysis, a nd (iii) to in te- grate measurements of the human transcripto me to other lay e rs of genomic infor- mation with novel dep endency mo deling techniques for co-o ccur ring data sources. The thre e strategies a ddress widely recog niz e d challenges in functional genomics (Collins et al., 2003 ; T royansk ay a , 20 05). Obtaining reliable measurements is the crucial starting point for an y da ta a nal- ysis task. The first contribution of this thesis is to dev elop computatio nal strategies that utilize side informatio n in genomic sequence and microa rray data collections in order to reduce noise and improv e the quality o f high-throug hput o bserv a tio ns. Publication 1 intro duces a pro be- level strategy for micr oarray prepro ce s sing, where upda ted ge no mic sequence data bases are used in orde r to remov e er roneously tar- geted prob es to reduce meas urement noise. The w ork is extended in Publica tion 2, which intro duces a principled proba bilistic framework for prob e-level analysis. A generative mo del for probe - level o bserv a tions co m bines evidence across multiple exp eriments, and allows the estimation of prob e p erformance directly from mi- croar ray measur ement s. The mo del det ects a lar ge n um ber of unrelia ble probes 2 1.1. CONTRIBUTIONS AN D ORGANIZA TION OF THE THESIS contaminated by kno wn prob e-level error sour ces, as well as many po or ly p er- forming pro be s where the s ource o f contamination is unknown a nd co uld not b e controlled based o n exis ting pro b e-le vel information. The mo del pr ovides a prin- cipled framework to incorp or ate prio r information of pr ob e per formance. The int ro duced alg orithms outp erform widely used alternatives in differential gene ex- pression studies. A nov el strategy for or ganism-wide a nalysis of transcriptio na l activity in geno me- scale interaction netw orks in Publica tion 3 forms the second main cont ribution of this thesis. The metho d sea rches for lo cal r egions in a netw ork exhibiting co ordi- nated trans c riptional resp onse in a s ubset of conditions. Constra in ts derived from genomic in tera ction database s are used to fo cus the mo deling o n those pa rts o f the data that are supp orted b y known or potential interactions b etw een the g enes. Nonparametric inference is used to detect a num ber of physiologically coheren t and repro ducible tr anscriptional resp onses, as w ell as context-specific r egulation of the genes . The findings provide a global view on tra nscriptional activity in cell-biologic a l netw orks and functional re la tedness b etw een tiss ues. The third contribution of the thesis is to integrate measurements of the human transcriptome to other lay ers of genomic information. No vel dep endency mo de ling techn iques for co -o ccurrence data are used to r eveal regula rities and interactions, which co uld not b e detected in individual observ ations. The r egularized dep en- dency mo deling framework o f Publication 4 is used to detect asso c iations betw een chromosomal m utations a nd transcriptional ac tivit y . Prior biological knowledge is used to constrain the latent v ar iable mo del and shown to improve cancer g ene detection performanc e . The ass o ciative cluster ing , in troduced in Publica tions 5 and 6, provides to ols to inv estigate evolutionary divergence of tra nscriptional ac- tivit y . Op en source implementations of the key metho dological contributions of this thesis hav e b e e n released in order to gua rantee wide access to the developed al- gorithmic to ols a nd to comply with the emerging standar ds of transpar ency and repro ducibility in computational science, where an increa sing prop or tion of re- search details ar e embedded in co de a nd da ta acco mpanying traditional publi- cations (Boulesteix, 2010 ; Carey and Sto dden, 2010; Ioa nnidis et al., 2 009) and transpare nt sharing of these resources can fo r m v aluable contributions to public knowledge (Sommer, 201 0; Sonnenburg et al., 20 07; Sto dden, 20 10). The thesis is org anized as follows: In C ha pter 2, there is a n ov erview of func- tional geno mics, related meas urement techniques, and genomic data resourc es. General methodo logical background, in par ticular of explor atory data a nalysis and the probabilis tic mo deling paradigm, is provided in Chapter 3. The metho dolo gical contributions of the thesis ar e presented in Chapter s 4-6 . In Chapter 4, str ategies to impro v e the reliabilit y of high-throughput micro array mea surements ar e pre- sented. In Chapter 5 metho ds for organism-wide a na lysis of the transc riptome a re considered. In Chapter 6, t w o general- purp ose algor ithms for depe ndency mo del- ing a r e intro duced and applied in in v estigating functional effects of chromosomal m utations and evolutionary div ergence of transcriptional activity . The conclusions of the thesis are summarized in Chapter 7. 3 Chapter 2 F unctional genomi cs F r om al l we have le arnt ab out the st ructur e of living matter, we must b e pr ep ar e d to find it working in a manner that c annot b e r e duc e d t o the or di- nary laws of physics - - b e c ause the c onstru ction is differ ent fr om anything we have yet teste d in the physic al lab or atory. E. Sc hr¨ odinger (1956) Living o r ganisms ar e co nt rolled no t only by na tural la ws but also b y inheritable genetic pr o gr ams (Mayr, 2 004; Schr¨ odinger, 19 44). Such double c ausation is a unique feature of life, and in fundamen tal contrast to purely physical pro cesse s of the inanimate world. Life ma y hav e emerg ed on earth mo re than 3.4 billion years ago (Schopf, 200 6; Tice a nd Low e, 2004 ). Genetic inf ormation evolves by means of natur al sele ction (Darwin, 18 59). Living o r ganisms maintain homeostasis, adapt to changing environments, resp ond to external stimuli, a nd co mm unicate. Peculiar fea tures of liv ing systems include metabolism, growth and hie r archical organiza tion, as well as the ability to r eplicate a nd r epro duce. All known life forms share fundamen tal mec hanisms a t molecular lev el, whic h suggests a co mmon evolutionary or ig in of the living or ganisms. The complete c ollection o f genetic mater ial, the genome , enco des the herita- ble genetic prog ram of an o rganism. Adv ances in measur emen t technology a nd computational sc ience hav e opened up new views to the large-sca le o rganizatio n o f the geno me (Car r oll, 2003; Lander, 19 96). F unctional genomics is a subdis c ipline of molecular biology inv estigating the funct ional org anization and proper ties of genetic information. In this thesis, new computatio nal approaches are developed for investigation of a cent ral f unctional layer o f the genome of our own sp ecies, the h uman tr anscriptome . This chapter gives an overview to the relev an t concepts in genome biolo gy in euk ar yotic organisms and asso ciated genomic data r esources. F or further background in molecular genome bio lo gy , see Alb erts et al. (2 002); Brown (2006). 2.1 Univ ersal gene tic c o de Cells a re fundamental building blocks of living organisms. All known life f orms maintain a carb on-based cellula r form that car r ies the genetic progr a m (Alberts et al., 20 02). E ach cell carrie s a co p y of the heritable g enetic co de, the genome . 4 2.1. UNIV ERSAL GENETIC CODE The h uman genome is divided in 23 pairs of chr omosomes , lo cated in the nu- cleus of the c ell, a s w ell as in additiona l mito chondrial geno me. Chromosomes are macrosco pic deoxyrib onucleic a cid (DNA) mo lecules in whic h the DNA is wra pped around histone molecules and pack ed into a p eculiar chr omatin structure that will ultimately constitute chromosomes. The genetic c o de in the DNA consists of four nucle otides : adenosine (A), th ymine (T), g uanine (G), and cytosine (C). In rib onu- cleic acid (RNA), the th ymine is replaced by uracil (U). Or de r ing of the n ucleotides carries genetic inf ormation. Nucleic acid sequences have a p eculiar base pairing prop erty , where only A-T/U a nd G-C pairs can h ybridize with ea ch other. This leads to the well-kno wn double-stranded structure of the DNA, and forms the basis for cellular informatio n pro c e ssing. The c entr al do gma of m ole cular biolo gy (Crick, 1970) sta tes that DNA enco des the information to construct pro teins throug h the irreversible pr o cess of pr otein synthesis . This is a central par adigm in mo le cular biology , describing the functional organiza tio n of life at the cellular level. 2.1.1 Protein syn thesis Genes ar e basic units of genetic info r mation. The gene is a sequence of DNA that contains th e informa tion to manufacture a protein or a set of related proteins. Genetic v ar iation a nd regulatio n of gene activity ha s therefore ma jor phenotypic consequences. The r e gulatory r e gion and c o ding se quenc e are tw o k ey elements of a gene. The r egulatory regio n regulates gene activity , while the co ding sequence carries the instructio ns for protein synthesis (Alb erts et al., 2002 ). Interestingly , the concept o f a ge ne remains contro v ersial despite c omprehensive identification of the protein- co ding genes in the human geno me and detailed knowledge of their structure and function (Pearson, 200 6). Proteins, e nco ded by the genes , are k e y functional en tities in the cell. They form cellular structures, a nd par ticipate in cell signaling and functional regulation. Pr otein synthesis refers to the cell- biological pro cess that conv erts ge ne tic infor ma - tion int o final functional protein pro ducts (Figure ?? A). Key steps in pro tein sy n- thesis include tra nscription, pre- mRNA splicing, a nd translation. In tr anscription , the do uble-stranded DNA is ope ned in a proximity of the g ene sequence and the pro cess is initiated on the reg ulatory regio n of the ge ne . The DNA sequence of the gene is then conv erted into a co mplemen tary pre- mRNA by a p olymerase enzyme. The pre-mRNA se q uence contains b oth pr otein co ding and no n-co ding segments. These ar e called exons and intro ns , respectively . In pr e-mRNA splicing , the introns are re moved and the exons are joined together to form mature messenger-RNA (mRNA) . A g ene ca n enco de multip le splice v ar iants, corr esp onding to differe n t exon definitions and their co mbinations; this is called alternative splici ng . The mature mRNA is exp or ted from nucleus to the cell cytopla sm. In tr anslation the mRNA is conv erted in to a corr esp onding a mino acid sequence in r ibo somes based on the universal genetic c o de that defines a mapping b etw een nucleic a cid triplets, so-called c o dons , and amino acids. The co de is common for a ll known life forms. Each cons ecutive co don on the mRN A sequence corresp onds to a n amino acid, and the co rresp onding sequence o f amino acids co nstitutes a protein. In the final stage of protein synthesis, the a mino ac id sequence folds into a three-dimensio nal structure and undergo es p ost-tr anslational mo dific ations . The structural charac- teristics of a protein molecule w ill ult imately determine its functional prop erties (Alber ts et al., 2002). 5 CHAPTER 2. FUNCTIONAL GENOMICS chromatin cell chromosome centromere nucleus histones DNA double helix pairs base nucleosomes Cell membrane Transport to cytoplasm Nucleus tRNA Translation mRNA A B Figure 2.1: A Key steps of protein synthesis. The t wo k ey pro cesses in protein synth esis are called tr anscription and tr anslation , resp ectively . In transcription, the DNA sequence of the gene is transcribed into pre-m RNA based on the base pairing property of n ucleic acid sequences. The pre-mRNA is modified to pr oduce mature messenger-RNA (mRNA), which is then transp orted to cytoplasm. T r ansfer-RNA (tRNA) carr ies the mRNA to rib osomes, where it is translated int o an amino acid sequence based on the universal genetic co de where each nucleotide triplet of the mRNA sequence, so-called co don , corresponds to a particular amino acid. The amino acid sequence is subsequent ly modified to form the final functional protein product. B Or- ganization of the genetic material in an euk aryotic cell. The nuc leotide base pairs form the double helix structure of DNA. Thi s is wrapped around histone m olecules to form nucleosomes, and the chromatin sequence. The chromatin is tight ly pack ed to form chromosomes that carry the genetic material and are located in the cell nu cleus. The image has b een mo dified from h ttp://commons.wikimedia.org/wiki/File:Chromosome en.svg. 2.1.2 La yers of regulation Phenotypic changes can rar e ly b e a ttributed to changes in individua l ge ne s ; c ell function is ultimately determined b y co or dinated activ ation of genes a nd o ther bio- molecular entit ies in resp ons e to changes in cell-biolog ical environmen t (Hartw ell et al., 199 9). Gene activit y is reg ulated a t all levels o f protein synthesis and cellu- lar pro cesses. A ma jor p ortio n o f functiona l genome sequence and pro tein co ding- genes themselves participate in the regula tory sys tem itself (Lauffenburger, 20 00). Epigenetic r e gulation refer s to c hemica l and structural mo difica tio ns o f chro- mosomal DNA, the chr omatin , for instance through methylation, acetylation, a nd other histone-binding molecules . Such mo difica tions a ffect the packing of the DNA molecule around histones in the cell nu cleus. The combinatorial regulation of such mo difications r egulates access to the gene sequences (Gibney and Nola n, 201 0). Epigenetic changes are b elieved to b e her itable and they constitute a ma jor source of v aria tion at individual and p opulation level (Jo hns on a nd T r ick er , 2010 ). T r an- scriptional r e gulation is the nex t ma jor regula tory lay er in pr otein synthesis. So- called tr anscription factor proteins can regulate the trans cription ra te by binding to cont rol elemen ts in gene regulatory region in a combinatorial fashio n. Post- tr anscriptional mo dific ations will then regulate pre-mRNA splicing. Up to 9 5% of human m ulti-exon genes a re estimated to hav e alterna tive splice v ariants (Pan et al., 2008). Cons equent ly , a v ar iety of related proteins can be enco ded by a single gene. This contributes to the str uctural and functional diversit y of cell function 6 2.2. O RGANIZA TION OF GENETIC IN F ORMA TION (Stetefeld and R uegg, 20 05). Several mechanisms will then affect mRN A de gr a- dation ra tes. F or instance , micr o-RNAs that are small, 21-2 5 basepair nucleotide sequences can inactiv ate sp ecific mRNA tra nscripts through complementary base pairing, leading to mRNA degrada tion, or preven tion of transla tion. Finally , p ost- tr anslational mo dific atio ns , pr otein de gr ada tion , and o ther mechanisms will a ffect the three-dimensio nal structure and life cycle of a pr otein. The proteins will pa r - ticipate in further c e ll-biological pr o cesses. The pro cesses ar e in contin uous inter- action and form complex functional netw orks, whic h regulate the life pro cess es o f an organism (Alberts et al., 2002 ). 2.2 Organization of genetic information The understanding of the str ucture and functional or ganization of the genome is rapidly a ccumu lating with the developing g e nome-scanning technologies and computational methods . This s e ction provides an ov erview to key structural and functional lay ers of the human genome. 2.2.1 Genome struct ure The genome is a dyna mic structure, or ganized a nd regulated at multiple levels of resolution from individual nu cleotide base pa irs to complete c hromosomes (Fig- ure ?? B; B rown (2006)). A ma jor po rtion o f heritable v ariation b etw een individu- als has been attributed to differences in the genomic DNA sequence. T raditionally , main genetic v a r iation was b elieved to arise fr om small po in t mutations, so -called single-nucle otide p olymo rphisms (S NPs) , in protein-co ding DNA. Recently , it ha s bee n increa singly recognized that st ructur al variation of the genome ma kes a re- mark a ble contribution to genetic v ar iation. Structural v aria tion is observed at all levels of organiza tion fro m single-nucleotide po lymorphisms to large chromosomal rearr angements, including deletions, inser tions, duplications, copy-n um ber v ari- ants, inv ersions a nd tr anslo cations o f g enomic r egions (F e uk et al., 200 6; Sharp et al., 200 6 ). Such mo difications ca n directly and indirectly influence transcr ipt- ional activity and contribute to hum an diversit y and health (Collins et al., 2003 ; Hurles et al., 200 8 ). The draft DNA sequence of the complete human genome w as published in 2001 (In ternational human genome s equencing consortium, 2 001; V enter et al., 2001 ). The human genome con tains three billio n base pairs and appro ximately 2 0,000- 25,000 protein-co ding genes (International Human Genome Sequencing Consor - tium, 2004 ). The pro tein-co ding exons comprise less than 1.5% of the human genome sequence. Appro ximately 5% of the human g enome sequence has been conserved in evolution for more than 200 million years, including the ma jority of protein-co ding genes ( The ENCODE Pr o ject Consortium, 200 7 ; Mo us e Genome Sequencing Consortium, 2 002). Half of the genome co nsists of highly rep etitive sequences. The genome sequence c o nt ains structural e le men ts such as cen tromeres and telomer es, r ep etitiv e and mobile elements, (Prak a nd K azazian Jr., 2000 ), retro elements (Banner t and Kur th, 2004), and non-co ding, non-rep etitive DN A (Collins et al., 2 0 03). The functional role of intergenic DNA, which forms 75 % of the genome, is to a large extent unknown (V enter et al., 2001). Recent evidence suggests that the three-dimens ional organiza tion of the chromosomes, which is to a large ex ten t regulated by the intergenic DNA is under a c tiv e selection, ca n hav e 7 CHAPTER 2. FUNCTIONAL GENOMICS a remark able r egulatory role (Lieb erman-Aiden et al., 200 9; Parker et al., 2 009). Compariso n o f the h uman genome with other org anisms, such as the mouse (Mouse Genome Sequencing Consortium, 2 0 02) can highlight impor tant evolutionary dif- ferences b e t w een sp ecies. F or a comprehensive revie w of the s tr uctural pro per ties of the human genome, see Brown (2006 ). 2.2.2 Genome function In protein synthesis, the gene sequence is transcrib e d in to pr e -mRNA, whic h is then further mo dified into matur e messenger -RNA and transp orted to cytoplasm. An av erage cell c o nt ains over 300,0 00 mRNA molecules, and the mRNA conc e n- tration, or expr ession levels of individual g enes, v ary according to Zipf ’s law, a power-law distribution where mos t genes are expr essed at low concentrations, pe r - haps only one or few copies of the mRNA p er cell o n average, and a small num b er of gene s are hig hly expre ssed, potentially with thousands of copies p er cell (see Carninci, 200 9; F urusaw a and Kaneko, 2 0 03). Cell-biologica l pro cess es ar e re- flected at the transcr iptional lev el. T ranscr iptional activit y v ar ies b y cell t ype, environmen tal conditions and time. Different collectio ns of gene s are active in different co n texts. Ge ne expr ession , or mRNA expr ession, refers to the expressio n level o f an mRNA transcript a t par ticular physiological condition and time p oint. In a ddition to pr otein-co ding mRNA mo lecules that are the main tar get of analy- sis in this thesis, the cell contains a v ar iet y of other functional and non-functional mRNA tr a nscripts, for instanc e micro-RNAs, r ibo s omal RNA and transfer -RNA molecules (Carninci, 2009 ; Johnso n et al., 200 5). The tr anscriptome refers to the complete colle c tion of mRNA s equences of an organis m. This is a central functional layer of the g enome that reg ulates pr o tein pro duction in the cells, with a s ignificant ro le in crea ting genetic v ariation (Jordan et al., 2005). According to cur rent estimates, up to 90 % of the euk aryotic g enome can b e transcr ibed (Consor tium, 2005; Gagneur et al., 20 09). The pro tein-co ding mRNA transcr ipts are transla ted into pr oteins at rib osomes during pro tein syn- thesis. The pr ote ome r efers to the co lle c tion o f protein pro ducts of an or ganism. The proteome is a main functional lay er of the genome. Since the final protein pr o ducts carry out a main p or tion of the a ctual cell functions, tec hniques for monitor ing the concentrations of all proteins and their mo dified forms in a cell simultane- ously would sig nificantly help to improve the understanding o f the cellular systems (Collins et al., 20 03). How ever, sens itiv e, r eliable and c o st-efficient g enome-wide screening techniques for measur ing pr otein expression are currently not av ailable. Therefore genome- w ide measur ement s of the mRNA expres s ion lev els are often used as an indirect estimate of protein activity . In addition to the DNA, RNA and pr o teins, the cell co ntains a v a riety of o ther small molecules. The ex treme functional diversity of living or ganisms emerges from the co mplex netw o rk of interactions betw een the bio molecular en tities (Bara b´ a si and Oltv ai, 2004; Hartw ell et al., 1999). Understanding of these netw orks a nd their functional pro p er ties is crucial in understanding ce ll function (Collins et al., 2003 ; Schadt, 2009). How ever, the systemic pro per ties of the inter actome are p o orly characterized and understo o d due to the co mplexity of bio logical phenomena and incomplete informatio n co ncerning the interactions. T he cell- biological pro cesses are inherently mo dular (Har t well et al., 199 9; Ihmels et al., 2002; Lauffenburger, 2000), and they ex hibit complex p athway cr oss-talk b etw een the cell-biolog ical 8 2.3. GENO MIC DA T A R ESOURCES pro cesses (Li et al., 2008). In mo dular sys tems, small changes can hav e significant regulator y effects (Espinosa- Soto and W a gner, 2010). 2.3 G enomic data resources Systematic observ a tions from the v arious functional and regulato r y layers of the genome are needed to understand cell-biolog ic al sys tems. Efficien t sharing and int egration of geno mic infor mation resources thro ugh digital media has enabled large-s cale inv estigations that no single institution could afford. The public human genome sequencing pro ject (International hu man genome se q uencing consor tium, 2001) is a prime example of suc h pro ject. Results from genome- wide transcriptio- nal profiling studies ar e ro utinely dep osited to public rep ositor ie s (Barrett et a l., 2009; Parkinson et al., 2009). Sharing o f orig inal data is inc r easingly accepted a s the scientific norm, often following explicit data release po licies. The e s tablishment of large-s cale databases and sta ndards for r epresenting biological information sup- po rt the efficient use of these reso urces (Bammler et al., 2 005; Br azma et al., 2006). A co n tin uously increasing array of genomic information is av ailable in these da ta - bases, concerning asp ects of genomic v ar iability a cross individuals, disease states, and sp ecies (Brent, 2008; Church, 2 005; Co chrane and Galp erin, 20 1 0; G1 0KCOS consortium, 200 9 ; The Cancer Genome At las Research Netw o r k, 2008). 2.3.1 Comm unity databa ses and ev olving biological kno w- ledge Genomic se quence databases During the human genome pro ject and preceding sequencing pro jects DNA se- quence reads were a mong the first so urces of biolog ic a l data that w ere colle c ted in large-scale public repo sitories, such as GenBank (Benson et al., 2010). Gen- Bank contains comprehensive sequence information of genomic DNA a nd RNA for a num ber o f orga nisms, a s w ell as a v ariet y of informatio n co ncerning the genes, non-co ding regio ns, disease asso ciatio ns , v ariation and o ther genomic fea- tures. Online ana lysis to ols, such as the E nsembl Geno me browser (Flicek et al., 2010), facilitate efficient use of these annotation resources. Next-genera tion se- quencing technologies provide rapidly increasing s e quencing capa cit y to in v estigate sequence v aria tion b etw een individuals, populations and disease s tates (Ledfor d, 2010; McP herson, 2 0 09). In par ticular, the human and mouse transcriptome se- quence collections a t the Entrez Nucleotide da tabase of GenBank are utilized in this thesis, in Publications 1 and 2. T ranscriptome databases Gene expression measure ment provides a snapshot of mRNA transc ript levels in a cell p opulation at a spe c ific time and co nditio n, reflecting the activ ation patterns of the v arious cell-biolo gical pr o cesses. While gene expr ession measurements pro- vide only a n indirect view to cellular pro cesses, their wide av ailability provides a unique reso urce for inv estigating ge ne co -regulatio n on a g enome- and or ganism- wide scale. V er satile collectio ns of microa r ray data in public re po s itories, such as the Gene Ex pression Omnibus (GEO; Bar rett e t a l. (2009)) and Arr ayExpre ss (Parkinson et al., 20 0 9) are av ailable for human and mo del organis ms, and they 9 CHAPTER 2. FUNCTIONAL GENOMICS contain v a lua ble information o f cell function (Consor tium, 2005 ; DeRisi et al., 1997; Russ and F utschik, 20 10; Zha ng et al., 2 004). Several techniques are av a ilable for qua ntitative and highly parallel measur e- men ts of mRNA or gene expr ession , a llowing the meas urement of the expressio n levels of tens of thousands of mRNA transcr ipts simultaneously (Br adford e t al., 2010). Micro array techniques are routinely use d to measure the ex pression levels of tens of thousands of mRNA transcr ipts in a given sa mple, and tra nscriptio- nal pro filing is curre ntly a ma in hig h-throughput tec hnique used to inv estigate gene function at genome- a nd o rganism-wide scale (Gershon, 2005; Y auk et al., 2004). Increas ing amoun ts of transcriptional profiling data are b eing pro duced by sequencing-based methods (Car ninci, 2009). A main difference b etw een the microarr ay- and sequencing -based techniques is that g ene expression arrays hav e bee n designed to measure predefined mRNA transcripts, whereas sequencing-bas ed metho ds do not r equire prior information of the measured sequences, and enable de novo discovery of ex pressed tra nscripts (Bra dford et al., 201 0; ’t Ho en et al., 2008). L a rge-sca le microarr ay rep ositories provide currently the most mature to o ls for data pro cessing and retr iev al, a nd form the main sour ce o f transcripto me data in this thesis. Microar r ay technology is ba sed on the base pairing prop erty of nucleic acid s e - quences where the DNA or RNA sequences in a sample bind to the complement ary nu cleotide sequences on the arr ay . This is c a lled hybridization . The measurement pro cess b egins b y the collectio n of c e ll samples and isolation of the sample mRNA. The isolated mRNA is conv erted to cDNA, lab ele d with sp ecific mar ker molecules, and hybridized on complementary prob e sequences o n the array . The array s ur- face may contain hundreds o f thousa nds of sp ots, eac h containing sp ecific prob e sequences designed to uniquely match with particula r mRNA seq uences. The hy- bridization level r e fle c ts the target mRNA concentration in the sample, a nd it is estimated by measuring the intensit y of lig h t emitted by the lab e l molecules with a laser sca nner. Short oligonucle otide arr ays (Lo ckhart et al., 1996) are among the most widely used microarray technologies, and they are t he main so urce of mRNA ex pression da ta in this thesis. Short oligonucleotide arrays utilize multi- ple, t ypically 10 -20, pr o be s for each transcr ipt target that bind to different regio ns of the same tr anscript s e quence. Use of sev eral 25-nucleotide prob es fo r each targe t leads to mo re robust estimates of tra nscript activity . E ach prob e is e x pected t o uniquely hybridize with its in tended targ et, and the detec ted hybridization level is used as a meas ure of the ac tivit y of the transc r ipt. A short olig o nu cleotide arr ay measures a bsolute express ion levels of th e mRNA sequences; relative differences betw een conditions ca n be in vestigated afterwards by comparing these mea s ure- men ts. A standard whole-g e no me array measures typically ∼ 20,000 -50,00 0 unique transcript seq uences. A single microarr ay exp eriment can therefore pro duce hun- dreds of thousands of raw obser v ations. Compariso n and in tegration of individual microar r ay exp eriments is often c hal- lenging due to r emark able exp erimental v ariation b etw een the exp eriments. Co m- mon standa rds hav e be e n develop ed to a dv ance the c o mparison a nd integration (Brazma et al., 20 01, 2006). Ca refully con trolled in teg r ative datasets, so - called gene expr ession atlases , contain thousands o f genome- wide measurements o f tran- scriptional activ ity acro ss div erse conditions in a dir ectly compar able format. Ex- amples of suc h data colle ctions include GeneSa piens (Kilpinen et al., 200 8 ), the hu man g ene expression a tla s o f the Euro pea n Bioinfor matics Institute (Lukk et al., 10 2.3. GENO MIC DA T A R ESOURCES 2010), as well as the NCI-60 cell line panel (Scherf et a l., 2000 ). In tegrative a nal- ysis of large a nd versatile transcriptome collections can provide a ho lis tic view of transcriptiona l a ctivit y of the v arious cell-biolog ical proce s ses, and op ens up pos s i- bilities to disc over previously unc haracterized cellula r mechanisms that contribute to h uman health and disease. Other types of microarra y da ta Microar r ay techniques can also b e used to s tudy other functional a s pects of the genome, including epigenetics and micro-RNA regulation, chromosoma l ab err a- tions and p olymo rphisms, alternative s plicing, a s well as transcr iption factor bind- ing (Butte, 2002 ; Hoheisel, 2006 ). F o r insta nce, chromosomal ab erratio ns can b e measured with the arr ay c omp ar ative genome hybridization metho d (aCGH; Pinkel and Alb ertson 2005), which is based on hybridization of DNA seq uences on the array surface. Copy num ber changes are a particula r type of chromosomal ab er- rations, which a re a ma jor mechanism for cancer dev elopment and progression. Copy num ber altera tions can cause changes in g ene- and micro - RNA expr ession, and ultimately cell-biolo g ical pro cesses (Bero ukhim et al., 2010). A public rep osi- tory of copy num ber measurement data is provided for instance by the Ca nGE M database (Sc heinin et al., 200 8). In Publication 4, microarray meas urements o f DNA copy num ber c hanges are integrated with transcriptional pr ofiling data to discov er p otential cancer genes for further biomedical ana ly sis. P ath wa y and intera ction databases Curated informa tion concerning cell-biolog ic al pro cesses is v aluable in b oth expe r - imen tal design and v alidation of computational studies (Blake, 2004). Represen- tation of dyna mic bio chemical rea ctions in their full richness is a challenging task beyond a mere listing of bio chemical events; a v arie ty of pr o teins and other com- po unds interact in a hierar chical manner throug h v ario us molecular mechanisms (Hartw ell et al., 199 9 ; Przytyc k a et al., 2010). Standa r dized data ba se formats such as the BioP AX (BioP AX workgr oup, 2005) a nd SBML (Str¨ omb¨ ac k and Lam- brix, 2005) adv ance the accum ulation of highly structure d biologica l knowledge and auto ma ted analys is of such data. A huge b o dy of information concerning cell-biologic a l pro ces ses is av a ilable in public rep ositories . The most widely used annotation r esources include the Gene Ontology (GO ) databas e (Ash burner et al., 2000) and the KEGG path w a y da tabase (K a nehisa et al., 2010). The GO databa se provides functiona l anno ta tions for genes and can b e used fo r instance to detect enrichmen t of cer tain functional categ ories a mong the key findings f rom compu- tational analysis, as in Publication 6, where enr ichm ent analysis is us e d for b oth v alidation a nd int erpretation purp oses. Pathw ays ar e mo r e s tr uctured repres e n- tations concerning cellular pro cesses and in teractions be t w een molecular en tities. Such pr ior infor mation can be use d to g uide computational mo deling, as in P ub- lication 3 , where pa th w ay informatio n derived fr om the KEGG pa thw ay databa se is used to g uide or g anism-wide discovery and analysis o f transc r iptional resp ons e patterns. Ev olving biol ogical kno wledge The collectiv e knowledge about genome or ganization a nd function is constantly upda ted and refined by improved measurement techniques a nd a ccumulation of 11 CHAPTER 2. FUNCTIONAL GENOMICS data (Sebat, 2007). This can alter the analysis and interpretation of res ults from large-s cale genomic scr eens. F or ins tance, evolving gene and tra nscript defini- tions a r e known to significantly affect micro a rray in terpr etation. Pr ob e design on microa rray technology r elies on sequence anno tations that may hav e changed significantly after the original array design. Reinterpretation of microarr ay data based on up dated prob e annotations has b een shown to improve the ac c ur acy and compar ability of micro array results (Dai et al., 2005 ; Hwang et al., 20 04; Mecham et al., 20 04b). B io informatics studies r o utinely take in to account updates in genome version, genome build , in new a nalyses. The constantly refined bio- logical data highlig h ts the need to a ccount for this uncertaint y in computational analyses. In Publications 1 and 2, explicit computational s trategies that are robust against evolving tra nscript definitions ar e de velop ed for micro array data analysis . 2.3.2 Challenges in high-throughput data analysis High-throughput genetic sc r eens a re inherently noisy . Con trolling all p otential sources o f v a r iation in the mea s urement pro cess is incre asingly difficult when a u- tomated measur emen t techniques can pro duce millions of da ta po in ts in a single exp eriment, concer ning extremely complex living systems that are to a larg e extent po o rly understo o d. Noise arises fr om b oth tech nical and biological so urces (Butte, 2002), and sys- tematic v aria tion betw een lab o ratories , measurement batches and measurement platforms has to b e ta ken into acc o un t when comb ining the results a c ross individ- ual studies (Heb er a nd Sick, 2 0 06; MA QC Consortium, 20 06). Moreov e r , genomic knowledge is constantly evolving, which can p otentially change the interpretation of previous exp eriments (see e.g. Dai et a l., 2005 ). The v arious sources of nois e and uncertaint y in micro array studies are discussed in more detail in Chapter 4. High dimensionality of the data and sma ll sample size form another challenge for the analys is of high-thro ughput functional genomics data. T ens of thousands of transcr ipts can b e mea sured simu ltaneously in a single micro array exp eriment, which greatly ex c eeds the num b er of av ailable sa mples in most bio medical studies. Small sample s izes leav e co nsiderable uncertaint y in the analyses; few observ a tions contain very limited information co nce rning the complex a nd hig h-dimensional phenomena and potential in teractions betw een differen t pa rts of the system. Over- fitting of the models and the problem of multiple tes ting forms co nsiderable chal- lenges in such situa tio ns. While a utomated analysis metho ds can gener a te thou- sands of hypotheses concer ning the system, prioritizing the fin dings and charac- terizing uncertaint y in the predictions b ecome central issues in the analysis. The curse of dimensionali ty , coupled with the high levels of noise in functional g enomics studies, is therefor e p osing particula r challenges for computational mo deling (Saeys et al., 2007). The challenges in controlling the v arious sour ces of uncertaint y hav e led to remark able problems in repro ducing microar ray results (Ioa nnidis et al., 200 9), but maturing technology and the development of common standards a nd a nalyti- cal pro c e dur es are cons ta n tly improving the reliability of hig h- throughput scr eens (Allison et al., 2006; Reimers, 2 010; MAQC Consortium, 2 006). The mo dels de- veloped in this thesis combine statistical evidence across re lated exp eriments to improv e the reliability of the analysis and to increa se modeling p ow er. Gener- ative probabilistic models provide a rigorous framework fo r handling noise a nd uncertaint y in the data and mo dels. 12 2.4. GENO MICS AND HEAL TH 2.4 G enomics and health Genomic v a riation betw een individuals ha s remark a ble and to a larg e ex ten t un- known contribution to hea lth and disease susceptibility . Lar ge-scale character iz a - tion of the v ariability b et ween indiv idua ls and p opulations is exp ected to elucidate genomic mechanisms asso cia ted with disea se, as well as to lead to t he discov ery of nov el medical treatments. High-throughput genomics can provide new tools to unders tand disease mechanisms (Brag a-Neto and Marques , 2006; Lag e et al., 2008), to ’hack the genome’ (Ev a nko, 20 06) to trea t diseases (V olinia et al., 20 10), and to g uide p ersonali ze d ther apies tha t take into a ccount the individual v ariabil- it y in sensitivity and res p ons es to tre a tmen ts (Ch urch, 2005; Down w ard, 2006; F o ekens et al., 2008; Ocana and Pandiella, 2010; v an ’t V eer and B ernards, 20 08). Disease signature s ar e potentially robust acro ss tissues a nd exp eriments (Dudley et al., 2009; Hu e t al., 2006). Genomic screens have revealed new disea s e subtypes (Bhattacharjee et al., 2001), and led to the discov ery of v a rious diagnostic (Lee et al., 2 008; Su et al., 2009; Tibshirani e t al., 20 02) and pr o gnostic (Beer et al., 2002) biomarkers. Diseases cause coordinated changes in gene activit y thro ugh biomolecular netw orks (Cabusor a et al., 2 005). Integration of chemical, genomic and pharmacolo gical functional geno mics data can als o help to pr edict new dr ug targets and resp ons es (Lamb et al., 2 006; Y amanishi et al., 2010 ). Genomic m u- tations can a lso affect geno me function and cause diseases (T aylor et a l., 2008 ). Cancer is an example of a prev alent g enomic disease. Bov eri (1914) discov er ed that cancer cells ha v e chromosoma l imbalances, and since then the understanding of ge- nomic c hanges a sso ciated with ca ncer has co ntin uously improv ed (Stratton et al., 2009; W underlich, 2007 ). F or instance, many human micro-RNA genes a r e lo cated at cancer -asso ciated genomic reg io ns and are functionally alter ed in cancer s (see Calin and Croce , 20 06). G enomic changes also affect transcriptional activity of the genes (Myllyk angas et al., 20 08). Publicatio n 4 introduces a nov el computationa l approach for s creening cancer-a s so ciated DNA mutations with functional implica- tions b y g enome-wide integration of ch romoso ma l ab err ations a nd tra nscriptional activity . This chapter has provided a n ov erview to central mo deling challenges and re- search topics in functiona l genomics. In the following chapters, particular metho d- ologica l approa ches ar e intro duced to solve res earch tasks in lar ge-scale analysis of the human tra nscriptome. In particular, methods are in tro duced t o increase the reliability of high- throughput measur ement s, to mo del lar ge-scale collections of transcr ipto me data and to integrate transcriptiona l profiling data to o ther lay- ers of genomic informatio n. The next chapter provides genera l methodolog ical background for these studies. 13 Chapter 3 Statistical learning a nd exploratory data analysis Essential ly, al l mo dels ar e wr ong, but some ar e useful. G.E.P . Box a nd N.R. Dra per (19 87) Mo dels ar e condens ed, simplified repres e n tations of obser ved phenomena. Mod- els ca n b e used to desc rib e obser v ations a nd to predict future ev en ts. Two key asp ects in mo deling are the construction and learning of formal representations of the observed da ta. Complex r eal-world obser v ations contain larg e a mounts of uncontrolled v ar iation, which is o ften ca lled n oise ; all asp ects of the da ta canno t be describ ed within a single mo del. Therefore, a mo deli ng c ompr omise is needed to decide what asp ects of data to describ e and what to ignor e. The seco nd s tep in mo deling is to fill in, to le arn , details of the formal repr esentation based on the actual e mpirical o bserv atio ns. V ar ious learning algo rithms are typically av a ilable that differ in efficiency and a ccuracy . F or instance, improv emen ts in computation time can often b e a ch ieved b y potential decrease in accuracy . An infer enc e c om- pr omise is needed to decide how to balance b etw een these and other p otentially conflicting ob jectives of the learning algorithm; the relative imp ortance of eac h factor dep ends on the particular application a nd av a ilable reso urces, and a ffects the choice of the lea rning pro cedure. The mo deling and infer ence compro mis e s are at the heart of data analysis. Ultimately , t he v alue of a mo del is determined by its ability to adv ance the solving of pra ctical pro blems. This chapter gives an overview of the key conc e pts in statistica l mo deling cen- tral to the topics of this thesis . The o b jectives of explorator y data ana lysis and statistical learning are cons ider ed in Section 3.1. The metho dolo gical framework is int ro duced in Section 3.2, which con tains an ov erview of central concepts in proba- bilistic mo deling and the Bayesian analysis paradigm. Key iss ues in implementing and v alidating the mo dels ar e discuss ed in Sectio n 3 .3 . 3.1 M o deling tasks Understanding requires generaliz ation b eyond par ticular obs e rv ations. While em- pirical observ a tions cont ain infor mation of the underlying pro cess that genera ted 14 3.1. MODELING T ASKS the data, a ma jor challenge in computationa l mo deling is that empirica l data is al- wa y s finite and cont ains only limited information of the system. T raditiona l sta tis- tical mo dels a re based o n car eful hypothesis formulation a nd systema tic collectio n of data to supp or t or reject a giv en hypothesis. How ev er, succes sful hypothesis formulation may require substantial pr ior knowledge. When minimal knowledge of the sy stem is av ailable, there is a need for explor atory m etho ds that can reco gnize complex patterns and ex tract features from empirical data in an automa ted way (Baldi and Bruna k, 199 9). This is a cen tral challenge in computational biolo g y , where the inv estigated s y stems a re extr emely complex and contain lar ge amounts of p o orly characterized and uncontrolled sources of v aria tion. Moreover, the data of genomic systems is often v ery limited and inco mplete. General-pur p os e algo- rithms that can lea r n relev ant fea tur es from the data with minimal ass umptions are therefore needed, and they provide v aluable to ols in functional genomics studies. Classical e x amples of suc h e xplorator y metho ds include clustering, classification and v isualization techniques. The extracted features can pr ovide hypo theses for more detaile d exp erimental testing and reveal new, unexp ected findings. In this work, genera l-purp ose explor atory to ols are developed for central mo deling tasks in functional genomics. 3.1.1 Cen tral concepts in data analysis Let us star t by defining so me of the ba sic co ncepts and terminology . Data set in this thesis refers to a finite collectio n of obs erv ations , or samples . In exp erimental studies, as in biology , a sample typically refer s to the particular ob ject of study , for instance a patient o r a tissue sample. In co mputational studies, sample r efers to a numerical observ atio n, or a subset of observ a tio ns, repres en ted by a numerical fe atur e ve ctor . Each element of the feature vector describ es a particular fe atur e of the o bserv a tion. Given D features and N sa mples, the data set is pre s en ted as a matrix X ∈ R D × N , wher e each column vector x ∈ R D represents a sample and each row corresp onds to a particular feature. The featur es ca n r e present for insta nce different exp erimental conditio ns, time po int s, or particular summaries ab out the observ ations. This is the g e neral str ucture of the observ a tio ns in vestigated in this work. The observ a tions are modeled in terms of proba bilit y densities; the s a mples are mo deled as independent instance s of a random v ariable. A central mo deling task is to characterize the underlying proba bilit y density of the observ a tio ns, p ( x ). This defines a top olog y in the sample space and pr ovides the bas is for gener alization beyond empirical observ a tions. As explained in more detail in Sec tion 3.2, the mo dels a re formulated in ter ms of observ ations X , mo del par ameters θ , and latent variables Z that ar e not directly observed, but characterize the underlying pro cess that generated the data. Ultimately , all mo dels describ e r elationships betw een ob jects. Similarity is therefore a key concept in data analy sis; the ba sis for characteriz ing the r elations, for summarizing the observ ations, and for predicting future even ts. Measures of similarity can be defined for diff erent classes of o b jects such a s feature vectors, data sets, or random v a riables. Similarity in general is a v ague concept. E uclide an distanc e , induced by the Euclidean metrics, is a co mmo n (dis- )similarity mea- sure for multiv a riate obser v ations. Corr elation is a sta ndard choice for univ a riate random v a riables. Mutual information is an information-theor etic measure of s ta - tistical dep endency b et w een tw o random v a riables, characterizing the decr ease in 15 CHAPTER 3. ST A TISTICAL LEAR NING AN D EXPLORA TOR Y DA T A ANA L Y SIS the uncer taint y concerning the realizatio n o f one v ariable, given the o ther one. The uncertaint y of a random v aria ble X is measure d in terms of entr opy 1 (Shannon, 1948). The m utual information bet ween tw o ra ndom v ariables is then given by I ( X , Y ) = H ( X ) − H ( X |Y ) (see e.g. Gelman et al., 2003). The Kullback-Leibler divergence, or KL–div er genc e , is a closely r elated non-s ymmetric dissimilar it y mea- sure for probability distributions p , q , defined a s d K L ( p, q ) = R x p ( x ) log p ( x ) q ( x ) d x (see e.g. Bis hop, 20 06). Mutual information betw een tw o rando m v aria bles can b e al- ternatively formulated as the KL –divergence b etw een their joint density p ( x , y ) and the product of their independent marginal dens ities, q ( x , y ) = p x ( x ) p y ( y ), which g ives the connection I ( X , Y ) = d K L ( p ( x , y ) , p x ( x ) p y ( y )). Mutual informa- tion and KL-divergence are central informatio n-theoretic measure s of dep endency employ ed in the mo dels of this thesis. It is impo rtant to notice that measures of similarity are inherently coupled to the statis tica l represen tation of data and to the goals of the analysis; differ- ent repr esentations can reveal differen t r elationships betw een observ a tions. F o r instance, the Euclidean distance is sensitive to scaling o f the features ; repr esen- tation in natura l or log arithmic scale, or with different units can p otentially lead to very different analysis results. No t a ll mea s ures ar e equally sensitive; mutual information can natura lly detect non-linear relatio nships, and it is inv a riant to the scale of the v ariables. On the other hand, estimating m utual information is computationally demanding. F e atur e sele ction refers to computational techniques for selecting, sca ling and transforming the da ta into a suitable form for further analysis. F eature selection has a central role in data analysis, and it is implicitly present in all analysis tas ks in selecting the inv e s tigated features for the ana lysis. There are no univ ersally optimal stand-alone fea ture selection techniques, since the problem is inher ent ly e ntangled with the analysis task and multiple equally op- timal featur e sets may be av ailable for instance in classification or predictio n tasks Guyon and Eliss eeff (2003); Saeys et al. (2007). S uccessful feature selection can re- duce the dimensionality of the data with minimal loss o f relev an t information, and fo cus the analysis on particular features. This can reduce model complexity , which is exp ected to y ield more efficient , ge ne r alizable and interpretable models . F eature selection is particularly imp orta nt in g enome-wide pro filing studies, where the di- mensionality of the data is larg e compared to the n um ber of av ailable samples, and only a small num b er o f features are relev a nt for the studied phenomenon. This is also known a s the lar ge p, smal l n pr oblem (W est, 2003). Adv anced feature selec- tion techniques can take in to account dep endencies b etw een the features, co nsider weigh ted combinations of them, and c a n be designed to in teract with the more general mo deling task, as for insta nc e in the near est shrunken centroids classifier of Tibshir ani et al. (20 0 2). The constrained subspace clus ter ing mo del of P ubli- cation 3 can b e viewed as a fea ture s election pr o cedure, where high-dimensional genomic obser v ations are deco mp os e d in to distinct feature subsets, ea c h o f which reveals different r elationships of the samples. In Publication 4 , iden tification o f maximally inf ormative features b etw een tw o data sets forms a cen tral par t of a regular iz ed dep endency modeling framew ork. In Publications 3-4 the pro cedure and representations are motiv a ted by biologica l reaso ning and analysis goals . 1 En trop y i s defined as H ( X ) = − R x p ( x ) log p ( x ) d x for a con tin uous v ariable. 16 3.1. MODELING T ASKS 3.1.2 Exploratory data analysis Explor atory data analysis refers to the use o f computational techniques to sum- marize and visualize data in order to facilitate the g eneration of new hypotheses for further study when the sear ch space w ould be otherwise exhaustively large (T ukey, 1977 ). The analys is strategy takes the obs e rv ations as the sta rting p oint for discov ering in ter e s ting regularities and nov el research hypotheses for p o orly characterized large- scale systems without prior knowledge. The analysis can then pro ceed from g eneral o bserv atio ns o f the data tow ard c onfirmatory data analysis , more detailed inv es tigations and hypo theses that ca n be tested in indep endent data sets with standar d statistica l pro ce dures. Ex ploratory da ta a nalysis differs from traditional hypothesis testing wher e the hyp o thesis is given. Light-w eight ex- plorator y to ols are par ticularly useful with larg e data sets when prior knowledge on the sys tem is minimal. Standard explora tory approaches in computational biolo g y include for instance c lus tering, class ification and visualization techniques (Ev anko, 2010; Polanski and Kimmel, 200 7). Cluster analysis r efers to a versatile fa mily o f methods that par tition data in to int ernally ho mogeneous groups of similar data po in ts, a nd o ften at the same time minimize the similar ity b e t w een distinct clusters . Clustering techniques enable class disc overy from the data. This differs from classifica tion where the target is to a ssign new obser v ations into known c lasses. The par titions pr ovided by clustering ca n be nested, pa r tially ov erlapping or mutually exclusive, and many clustering metho ds genera liz e the partitioning to cover previously unseen data po in ts (Jain and Dub es, 19 88). Clus tering can provide compressed representations of the da ta based on a s hared par ametric representation of the observ a tions within each cluster, as for instanc e in K-means or Gaussia n mixture mo deling (see e.g . Bishop, 20 06). Certain clustering approaches, such a s the hiera r chical clustering (see e.g. Hastie e t al., 2009 ), apply recursive schemes that partition the data into int ernally ho mogeneous groups without providing a para metric representation of the cluster s. Cluster structure can b e also discovered by linear algebraic op eratio ns on the distance matr ices, a s for instance in sp ectral clustering. The different approaches often hav e close theoretical connections. Clustering in general is an ill- defined concept that refers to a set of related but mutually inco mpatible ob jectives (Ben-David a nd Ack e rman, 2008 ; Kleinberg , 2002). Cluster ana lysis ha s b een tremendously popular in computational biology , and a comprehensive re v iew of the different applications ar e b eyond the sc op e of this thesis. It has b een observed, for instance, tha t g enes with related functions hav e often similar express ion profiles and are clus tered together, sugge s ting that clustering can be used to form ulate hypotheses concerning the f unction of previously unc haracterized genes (DeRisi et al., 1997 ; Eisen et al., 1 998), or to dis cov er nov el cancer subtypes with bio medical implications (Sørlie et al., 200 1). Visualization te chniques are another widely used explorator y a pproach in com- putational biolog y . Visualizations ca n provide c ompact a nd int uitiv e summar ies of complex, hig h-dimensional observ a tions on a low er-dimensional display , for in- stance by linear pr o jection methods such as princ ipa l comp onent a nalysis, o r by explicitly optimizing a low er-dimensional repres en tation as in the self-o rganizing map (Kohonen, 198 2 ). Visualization can provide the first step in inv estigating large data sets (Ev anko, 20 10). 17 CHAPTER 3. ST A TISTICAL LEAR NING AN D EXPLORA TOR Y DA T A ANA L Y SIS 3.1.3 Statistical learning Statistic al le arning r efers to computational mo dels that can learn to reco gnize structure and patterns from empirical data in an automated wa y . Unsup ervised and super vised mode ls form t w o ma in categor ie s of learning algorithms. Unsup ervise d le arning a pproaches seek compact descr iptions of the data with- out prior knowledge. In probabilistic mo deling, unsup ervised lear ning c a n b e for - m ulated as the task o f finding a proba bilit y distribution that describ es the o bs erved data and gener a lizes to new observ ations. This is also c a lled density estimatio n . The par ameter v alues of the model can be used to pr ovide compact represent a- tions o f the data. Examples of unsupervised a na lysis tasks include metho ds for clustering, visua lization and dimensionality reduction. In cluster analysis, gro ups of similar observ ations ar e so ught fro m the data. Dimensio nality reduction tech- niques provide compact lower-dimensional re presentations of the orig inal data, which is often useful for subsequen t mo deling steps. Not a ll observ ations of the data are equally v aluable, a nd assessing the relev ance o f the observed regular ities is problematic in fully unsup ervised ana ly sis. In su p ervise d le arning the task is to learn a function that ma ps the inputs x to the desired outputs y ba s ed on a set of t raining examples in a ge ne r alizable fashion, as in regres sion for contin uous outputs, and classific a tion for discr e te output v ariables. The sup e r vised learning tas k s ar e inherently asymmetric; the inference pro ceeds from inputs to outputs, and prior informatio n of the mo deling task is use d to sup ervise the a nalysis; the tra ining ex amples also include a desired output of the mo del. The mo dels develop ed in this thesis ca n be viewed as unsupe rvised explo ratory techn iques. Ho w ev er, the distinction b etw een sup ervised and unsup ervis ed mo dels is not strict, and the mo de ls in this thesis b orr ow ideas from b oth catego r ies. The mo dels in Publications 2-3 are unsupervised alg o rithms that utilize prior infor- mation derived fro m background databa ses to guide the mo deling by constra ining the solutions . How ever, since no desired outputs ar e av ailable for thes e models, the mo deling tas ks differ fr o m sup ervised ana ly sis. The dep endency mo deling a l- gorithms of Publications 4-6 have close theoretical connections to the s up er v ised learning task. In contrast to sup ervised learning, the learning task in these algo- rithms is symmetric; mo deling of the co- o ccurring data sets is unsup ervis e d, but coupled. Each da ta set affects the mo deling of the other data se t in a symmet- ric fa s hion, and, in analogy to super vised learning , p rediction can then pr o ceed to either directio n. Compar ed to supervised a nalysis tas ks, the emphasis in t he depe ndency detection algorithms introduced in this thesis is in the discovery and characterization of symmetric dep endencies , rather than in the constr uc tio n of asymmetric predictive mo dels. 3.2 Pr ob abilistic mo deling paradigm The main contributions of this thesis follow the g enerative probabilistic mo deling paradigm. Generative proba bilistic mo dels describ e th e observed data in terms of pr obability distributions . This a llows the calculation of exp ectations, v a riances and other standard summaries of the model par a meters, and at the same time allows to describ e the indep endence assumptions and relations b et ween v a riables, and uncertaint y in the modeling pro cess in an explicit manner . Measur e men ts 18 3.2. PROBABILISTIC MOD ELING P A RADIGM are regar ded as noisy observ ations of the genera l, underlying pro cesses; generative mo dels are used to ch aracteriz e the pro cess e s that generated the obser v ations. The first task in mo deling is the selection of a mo del family - a set of p otential formal representations o f the data. As discussed in Section 3 .2 .2, the repr esenta- tions can also to some e x ten t b e learned fro m the data. The second task is to define the obje ctive function , or cost function, which is used to meas ure the descriptive power of the mo dels. The third task is to iden tify the optimal mo del within the mo del family tha t b est de s crib es the obser ved data with r esp ect to the ob jective function. This is called le arning or mo del fitting . The details of the mo deling pro- cess are large ly determined by the exact mo deling tas k a nd particula r nature of the observ ations. The ob jectives of the mo deling task are enco ded in the selected mo del family , the ob jective function and to some extent to the mo de l fitting pro ce- dure. The mo del family determines the space of p ossible descriptions for the data and has ther efore a ma jor influence on the final s olution. The o b jectiv e function can b e used to prefer simple mo dels or o ther a spe c ts in the mo deling pro ce s s. The mo del fitting pro cedure affects the efficiency a nd accuracy of the learning pro cess. F or further infor mation of these and related co ncepts, see Bisho p (2006 ). A gene r al ov erview of the probabilistic mo deling framework is given in the r emainder of this section. 3.2.1 Generativ e mo deling Gener ative pr ob abi listic mo dels view the o bs erv ations as r andom samples fro m a n underlying proba bility distribution. The mo del defines a pro ba bilit y distribution p ( x ) ov er the feature spa ce. The mo del can b e para meterized by mode l pa r ame- ters θ that sp ecify a par ticular mo del within the mo de l family . F or conv enience, we assume that the mo del family is given, and leave it out fro m the notation. In this thesis, the a ppropriate mo del fa milies ar e selected ba sed on biologica l hy- po theses a nd a nalysis go als. Genera tiv e mo dels allo w efficient repr esentation of depe ndencie s b etw een v ariable s , indep endence assumptions and uncertaint y in the inference (Koller a nd F riedman, 2 009). Let us nex t consider central analysis tasks in generative mo deling. Finite m ixture mo del s Classical pr obability distributio ns provide well-justified and con v enien t to o ls for probabilistic mo deling, but in many pra c tical situations the o bserved regularities in the data ca nnot b e de s crib ed with a single standard distr ibution. How ev er, a sufficiently r ich mixture of standar d distributions can provide arbitra rily accurate approximations o f the o bserved data. In mixtur e mo dels , a set of distinct, latent pro cesses, or c omp onents , is used to des crib e the obser v ations. T he task is to ident ify and characterize the comp onents a nd their asso ciatio ns to the individual observ ations. The sta ndard formulation assumes independent and identically dis- tributed observ ations where each obse r v ation has b een generated b y ex a ctly one comp onent. In a standa rd mixture model the overall probability density of the data is mo deled as a weighted sum of comp onent distributions: p ( x ) = R X r =1 π r p r ( x | θ r ) , (3.1) 19 CHAPTER 3. ST A TISTICAL LEAR NING AN D EXPLORA TOR Y DA T A ANA L Y SIS where the comp onents are index ed by r , and R p ( x ) d x = 1. Ea c h mixture c o mpo - nent can hav e a different dis tributional form. The mixing prop or tion, or weigh t, and model par a meters o f each comp onent are denoted by π r and θ r , r esp ectively , with P r π r = 1. Many a pplications utilize conv enien t standa r d distributions, such as Gauss ians, or other distributions fr o m the exp onential family . Then the mixture model can b e learned for instance with the E M algo rithm descr ibed in Section 3.3.1. In practice, the mixing pr op ortions of the co mpo nen ts are often unknown. The mixing pr op o rtions can b e estimated from the data by conside r ing them a s stan- dard mo del parameters to b e fitted with a ML estimate. Howev er, the pro cedure is po ten tially prone to overfitting and lo cal optima, i.e., it may learn to descr ibe the tra ining data well, but fails to g eneralize to new obser v ations. An alter na tive, probabilistic wa y to determine the weigh ts is to treat the mixing prop or tions as latent v ariables with a prior distr ibutio n p ( π ). A standar d choice is a symmetric Dirichlet prior 2 π ∼ D ir ( α R ). This gives a n eq ual prio r weight for ea ch com- po nent and guarantees the standard exchangeability assumption of t he mixture comp onent lab e ls . A lab el deter mines cluster identit y . In tuitiv ely , exchangeability corres p onds to the assumption that the analysis is inv ar iant to the ordering of the data samples and mixture components. Compared to standard mixture models, probabilistic mixture mo dels hav e increas e d computationa l co mplexity . F urther prior knowledge can b e incorp orated in the model by defining prior distributions for the other para meter s of the mixture mo del. This ca n also be used to reg ularize the learning pr o cess to avoid overfitting. A typical prior distribution for the comp onents of a Gaus sian mixture mo del, parameterized by θ r = { µ r , Σ r } , is the normal-inv erse-Gamma prior (see e.g. Gelman et al., 200 3). Int erpreting the mixture comp onents as clusters provides an alternative, pr ob- abilistic formulation of the cluster ing task. This ha s made probabilis tic mixture mo dels a p opular choice in the a nalysis of functional genomics data sets that t ypically ha ve high dimensionality but small s ample size. Pro ba bilistic ana lysis takes the uncertain ties into acco un t in a rig orous ma nner, whic h is particular ly useful when the sample size is s ma ll. The n umber of mixture components is of- ten unknown in practica l mo deling task s , how ev er, and has to be inf erred ba s ed on the data. A straightforward so lution can be obtained by emplo ying a suffi- ciently la rge n um ber of comp onents in lea rning the mixture mo del, and selecting the compo nen ts ha ving non-zero weights as a p ost-pro ces sing step. An alternative, mo del-based treatment for lea rning the n um ber o f mixture comp onents from the data is provided by infinite mixture mo dels co ns idered in Section 3 .2.2. Laten t v ariables and marginalization The observed v aria bles are often affected b y latent variables that describ e relev ant structure in the mo del, but ar e not directly o bs erved. The latent v ariable v alues can b e, to some extent, infer red based on the obser ved v aria bles. Comb ination of latent a nd observed v ariables allows the des cription of complex probability spaces in terms of simple comp onent distributions a nd their r elations. Use of simple comp onent distr ibutions can provide a n intuitiv e and computationally tractable characterization of complex gener ative pro cesses underlying the observ a tions. 2 Dirichlet distribution is the pr obabilit y densit y D ir ( π | n ) ∼ Q r π n r − 1 r where the multiv ariate random v ariable π a nd the p ositive parameter v ector n hav e their element s indexed by r , 0 < π r < 1, and P r π r = 1. 20 3.2. PROBABILISTIC MOD ELING P A RADIGM A gener ative la ten t v aria ble mo del sp ecifies the distributional form and rela - tionships of the latent a nd observed v ariables. As a s imple example, co nsider the probabilistic in terpretation of probabilistic compo nent analysis (PCA), where the observ ations x are mo deled with a linear m o del x = Wz + ε w her e a normally distributed latent v ariable z ∼ N ( 0 , I ) is tr a nsformed w ith the parameter ma trix W and isotropic Gauss ian noise ( ε ) is assumed on the obser v ations. More co m- plex models can be constructed b y analog ous reaso ning. A c omplete-data likelih o o d p ( X , Z | θ ) defines a joint densit y for the observed a nd latent v aria bles. Only a sub- set of v ar iables in the model is t ypically of in terest for the actual analysis task. F or instance, the latent v aria bles may b e central for describing the genera tiv e pro cess of the da ta, but their actual v a lue s may b e ir r elev ant. Such v aria ble s are called nuisanc e variables . Their in tegr ation, o r mar ginalizatio n , pr ovides probabilistic av eraging o ver the p otential rea lizations. Marginalization ov er the la tent v a riables in the complete-data likelihoo d gives the likelihoo d p ( X | θ ) = Z Z p ( X , Z | θ ) d Z . (3.2) Marginaliza tion o v er the latent v a r iables collapses the mo deling task to finding optimal v alues for mo del par ameters θ , in a way that takes into acco un t the un- certaint y in latent v ariable v alues. This can r educe the num ber of v a riables in the learning phase, yield mo re stra ightf orward and robus t inferences, as well a s sp eed up computation. How ev er, marginalization ma y lead to ana lytically in tractable int egrals. As cer tain latent v ariables may b e direc tly relev an t, ma rginalizatio n de- pends on the ov er all goals of the ana lysis and may co ver only a subset of the latent v aria bles. In this thesis la ten t v ariables are utilized for instance in P ublication 3, which trea ts the sample-cluster as s ignments as discrete latent v aria bles, as well as in Publication 4 , where a regula rized la ten t v ariable mo del is int ro duced to mo del depe ndencie s b et w een co -o ccurring observ ations. 3.2.2 Nonparametric mo dels Finite mixture mo dels and latent v ar iable mo dels require the s pecific a tion of mo del structure prior to the analy sis. This can be pro blematic since for instance the nu m ber and distributional shap e of the gener ative pro cesses is unknown in many practical tasks. How ever, the mo del s tructure can a ls o to some extent b e learned from the data. Non-parametric mo dels provide principled a pproaches to lea r n the mo del structure from the data . In contrast to pa r ametric mo dels, the num ber and use of the parameters in nonparametric mo dels is flexible (see e.g. Hjort et al., 2010; M ¨ uller and Quintana, 2004 ). The infinite mixtur e of Gaussia ns, used as a part of the mo deling pro cess in Publicatio n 3, is an example of a non-par ametric mo del wher e b oth the num ber of comp onents, a s well as mixture prop or tions of the c o mpo nen t distr ibutions are inferred fro m the data . Lea rning o f Bayesian net work structure is another exa mple of no nparametric inference, where r elations betw een the mo del v aria bles a re lear ned from the data (see e.g. F riedman, 20 03). While mo re complex mo dels can describ e the training data mor e accurately , an increasing mo del complexity ne e ds to b e pena lized to avoid ov erfitting and to ensure generaliza bilit y of the mo del. Nonparametric mo dels provide flexible and theoretica lly principled appro aches for data-dr iven exploratory analysis. Howev er , the flexibility often comes with an increased computatio nal cost, and the models are po tentially more prone to 21 CHAPTER 3. ST A TISTICAL LEAR NING AN D EXPLORA TOR Y DA T A ANA L Y SIS ov erfitting than less flexible pa rametric mo dels. Moreover, co mplex mo dels can b e difficult to interpret. Many nonpar ametric probabilistic mo dels are defined by using t he theor y of sto chastic p ro cesses to impose prio r s o v er p otential model structure s . Stochas- tic pro cesses can b e used to define priors ov er function s paces. F or instance, the Dirichlet pr o c ess (DP) defines a probability density over the function space o f Dirichlet distributions 3 . The Chinese Re staur ant Pr o c ess (CRP) provides an in- tuitiv e description of the Dirichlet proc e ss in the cluster analysis context. T he CRP defines a prior distribution over the num ber o f cluster s and their size distri- bution. The CRP is a random pro cess in which n customers arrive in a res taurant, which has an infinite num ber of tables. The pro cess go es as follows: The fir s t customer c hoo ses the first table. E ach subsequent customer m will selec t a ta- ble based o n the state F m − 1 of the r estaurant tables after m − 1 customers hav e arrived. The new customer m will select a previously o ccupied table i with a probability which is pro p or tional to the num ber of customers s eated a t table i , i.e. p ( i | F m − i ) ∝ n i . Alternativ ely , the new customer will s elect an empty table with a probability whic h is prop or tional to a cons ta n t α . The mo del prefers tables with a la rger n um ber o f customers, and is analogo us to clustering, where the customer s and tables co rresp ond to sa mples and clusters, resp ectively . This provides an in- tuitiv e prior distribution for clustering tasks . The prio r pr efers compa c t m o dels with relatively few clusters, but the num ber of clusters is p otentially infinite, and ultimately determined based on the data. Infinite mixture mo dels Infinite mi xtur e mo dels are a general clas s of nonpara metric methods where the nu m ber of mix tur e comp onents are determined in a data-driven manner; the num- ber of co mp onents is p otentially infinite (see e.g. M¨ uller and Q uin tana, 2 004; Ras- m ussen, 2000). An infinite mixture is obtained by letting R → ∞ in the finite mixture model of Equation 3 .1 and repla cing the Dirichlet dis tribution prior o f the mixing propor tions π by a Dirichlet pro cess . The for mal probability distri- bution of the Dirichlet pro ces s can b e intuitiv ely derived with the so-ca lle d stick- br e aking pr esentation . Consider a unit length stick and a s tick-breaking pro cess , where the break po int β is sto chastically deter mined, following the b eta distribu- tion β ∼ B eta (1 , α ), where α tunes the exp ected breaking p oint. The pro cess can b e viewed a s consec utiv ely break ing off p or tions of a unit length s tic k to obtain an infinite seq ue nce of stick lengths π 1 = β 1 ; π i = β i Q i − 1 l =1 (1 − β l ), w ith P ∞ i =1 π i = 1 (Ishw aran and Ja mes, 2001). This defines the probability distribution Stic k( α ) ov er p otent ial partitionings of the unit s tick. A tr uncated stick-breaking representation co nsiders only the first T elements. Settin g the prior π ∼ Stick( α ), defined b y the stic k-breaking r epresentation in Equatio n 3.1 assigns a prior on the nu m ber of mixture comp onents and their mixing prop ortions that a re ultimately learned fro m the observed data. The prior helps to find a co mpromise b etw een increasing mo del complexity and likelihoo d of the obser v ations. T raditio nal appro a ches used to determine the num ber mixture comp onents are based on ob jective functions that p enaliz e increa s ing model complexity , for in- stance in cer tain v ariants of the K -means or in sp ectral clustering (see e.g. Hastie 3 If G i s a distri bution drawn f rom a D i richlet pro cess with the probabilit y measure P ov er the sample space, G ∼ DP( P ), then eac h finite partition { A k } k of the sample space i s di stributed as ( G ( A 1 ) , ... , G ( A k )) ∼ D ir ( P ( A 1 ) , ... , P ( A k )). 22 3.2. PROBABILISTIC MOD ELING P A RADIGM et al., 200 9). O ther mo del s election criteria include cro ss-v a lidation and com- parison of the mo dels in terms of their likeliho o d o r v arious informa tion-theoretic criteria that s eek a compromise b etw een mo del co mplex ity a nd fit (se e e.g. Gelman et al., 2 003). How ev er, the sample size ma y be insufficient for such a ppr oaches, and the mo dels may lack a rigor ous framework to acco un t for uncertainties in the o bserv a tions and mo del par ameters. Mo deling uncertaint y in the parameters while lear ning the mo del str ucture can lead to more robust inference in nonpara- metric probabilistic mo dels but als o adds inheren t c o mputational c o mplexity in the learning pro cess. 3.2.3 Ba y esian analysis The term ’Bay esian’ refers to interpretation of mo del para meters as v ariables. The uncertaint y over the par a meter v alues, arising from limited empirical evidence, is describ ed in terms o f pr o bability distributions. This is in co n trast to the tr aditional view where pa rameters have fixed v a lues with no distribution and the uncertaint y is ignore d. The Bayesian approach leads to a lear ning ta sk where the ob jective is to es timate the p osterior distribution p ( θ | X ) of the mo del parameter s θ , given the observ ations X . The p osterior is given by the Bayes’ rule (Ba y es, 176 3 ): p ( θ | X ) = p ( X | θ ) p ( θ ) p ( X ) . (3.3) The tw o key elements of the p osterior are the likeliho o d and the prior . The like- liho o d p ( X | θ ) describ es the proba bilit y of the observ ations, given the para meter v alues θ . The parameter s can a lso characterize a lternative mo del structures. The prior p ( θ ) enco des prior b eliefs ab out the mo del and r ewards solutions that match with the prior assumptions or yield simpler mo dels. Such regula rizing prop erties can be particula rly useful when training data is s carce a nd there is consider a ble uncertaint y in the pa rameter estimates. With strong, infor mative prio rs, new o b- serv a tions hav e little effect o n the p osterior . In the limit of lar ge sample size the po sterior conv erges to the ordinary likelihoo d p ( X | θ ). The Bay esian inference pro- vides a robust framework for taking the uncerta in ties into a ccount when the data is scar ce, as it often is in pr actical mo deling tasks. Moreov er, the Bay es’ rule pr o - vides a forma l framework for s equential up date of b eliefs based on accumulating evidence. The prior predictive dens it y p ( X ) = R p ( X , θ ) d θ is a no r malizing con- stant, whic h is indep endent of the parameters θ and can o ften b e ig nored during mo del fitting. The inv olv ed distributions can have c omplex non-sta ndard forms and limited empirical data can only pr ovide p artial evidence reg arding the different asp ects of the data- generating pro cess. Often only a subset of the para meters a nd other v aria bles a nd their interdependencies can be directly o bserved. The Bay esian ap- proach provides a framework for mak ing inferences on the unobs erved quantities through hierarch ical mo dels, where the proba bility distribution of eac h v a riable is characterized b y higher- le vel pa rameters, so-called hyp erp ar ameters . A similar reasoning can b e used to mo del the uncertaint y in the hyperpa rameters, until the uncertainties b ecome modele d at an appropria te detail. Prior informa tion ca n help to compens ate the lack of data on c e rtain asp ects of a mo del, and explicit mo dels for the nois e can characterize uncertaint y in the empirical o bs erv ations . Distr ibu- tions can also share parameter s, which pr ovides a basis for po o ling e v idence from m ultiple sources, as for insta nce in Publication 4. In man y applications o nly a 23 CHAPTER 3. ST A TISTICAL LEAR NING AN D EXPLORA TOR Y DA T A ANA L Y SIS subset of the parameters in the mo del are o f interest a nd the mo deling pro cess can b e consider ably simplified by marginalizing over the less interesting n uisance v aria bles to obta in an exp ectation over their potential v alues. The Bay esian para digm provides a principled framework for modeling the un- certaint y at all levels o f sta tistical inference, including the parameters, the o bserved and latent v ar iables and the mo del s tructure; a ll information of the mo de l is in- corp ora ted in the pos terior distribution, which summarizes empirical evidence and prior knowledge, and provides a complete description of the ex pected outcomes of the data-ge ne r ating pr o cess. When the data do es not contain s ufficient informa- tion to decide b et w een the alternative mo del struc tur es a nd parameter v alues, the Bay esian framework provides to ols to take expecta tions over all p otential mo dels, weigh ted by their rela tiv e evidence. A central challenge in the Bayesian analysis is that the mo dels often include analytically int ractable p osterior distributions, and lear ning o f the mo dels c an b e computationally demanding. Widely-used appro aches fo r estimating p osterio r dis- tributions include Markov Chain Monte Carlo (MCMC) metho ds and v ar iational learning. Sto chastic MCMC methods pr ovide a widely-used family of algor ithms to estimate intractable distr ibutions b y drawing rando m samples from these distri- butions (see e.g . Gelma n et al., 200 3); a sufficiently larg e p o ol of random sa mples will conv er ge to the underlying distribution, a nd sa mple sta tis tics can then be used to characterize the distribution. Howev er, sampling-based metho ds a re computa- tionally in tensive and s low. In v ariational learning, considered in Section 3.3.1, the intractable distributions a re approximated b y more conv enient tractable dis - tributions, which yields faster lear ning pro ce dure, but p otentially less a ccurate results. While analysis of the f ull p osterio r distribution will provide a co mplete description o f the uncer tainties regarding the parameters, simplified summar y sta- tistics, such as the mean, v ariance and qua ntiles of the posterio r ca n pr ovide a sufficient characteriza tion of t he posterior in man y practical applications. T he y can be obtained for instance by summar izing the output of sampling-ba sed o r v ari- ational metho ds. Moreov er, when the uncertaint y in the res ults can b e ignor ed, po in t estimates can provide s imple, interpretable summa r ies that are often useful in further biomedical analysis, as for instance in Publication 2. Point estimates are single optimal v alues with no distribution. How ev er, p oint estimates a re not nec- essarily sufficient for instance in biomedical diagnostics a nd other prediction tasks, where different outcomes a re as s o ciated with different costs a nd it may b e crucial to asse s s the probabilities of the alter na tive outcomes. F or further discuss io n on learning the Bay esian mo dels, see Section 3.3.1. In this thesis the Bayesian approach provides a for mal framework to p erform robust inference based on incomplete functional g enomics data sets a nd to incor- po rate prio r information of the models in the analysis. The Bayesian para dig m can alternatively b e interpreted as a philosophical p osition, where pro babilit y is viewed as a sub jective concept (Cox, 1 946), or considered a dir e c t consequence of making rationa l decisions under uncer tain t y (Berna rdo and Smith, 200 0 ). F or further co ncepts in mo del selection, compariso n and av er aging in the Bay esian analysis, see Gelma n et al. (20 03). F or applications in computationa l biology , s ee Wilkinson (2007 ). 24 3.3. LEA RNING AND INFER ENCE 3.3 L earning and inference The final stage in pro babilistic mo deling is to lear n the optimal statistical presen- tation for the data, giv en the model family and the o b jectiv e function. This section highlights central challenges a nd metho dolo gical issues in s tatistical learning . 3.3.1 Mo del fit t ing L e arning in probabilistic models often f o cuses on o ptimizing the mo del parame- ters θ . In addition, p o sterior distr ibution o f the latent v aria bles, p ( z | x , θ ), can b e calculated. Estimating the latent v ar iable v alues is called statistica l infer enc e . In the Ba y esian analysis, the model parameters can also be treated as latent v ari- ables with a pr ior pro bability densit y , in which case the distinction b et ween mode l parameters a nd la ten t v a riables will disa ppea r. A compre he ns ive characterization of the v ariables and their uncertain t y w o uld be achiev ed by estimating the full po sterior dis tr ibution. How ever, this can b e computationally very demanding, in particular w hen the p oster ior is not ana lytically tracta ble. The p os ter ior is o ften approximated with sto chastic or a nalytical pro cedures , such as sto chastic MCMC sampling metho ds or v ar iational a pproximations, and appro priate summar y sta- tistics. In ma n y practical settings, it is sufficient to summar iz e the full p osterior distribution with a p oint estimate. Poin t estimates do not characterize the uncer- tainties in the a nalysis result, but ar e often more conv enien t to in terpret than full po sterior distributions. V ario us optimization alg orithms are av aila ble to learn statistical mo dels, given the learning pr o cedure. The po ten tial challenges in the optimization include c om- putational c omplexity and the pre s ence of lo c al optima on complex probability density top olog ies, as w ell as unidentifiability of the mo dels. Finding a global op- tim um of a co mplex mo del can b e computationally exhaus tiv e, and it can b eco me int ractable with increasing sample size. In unidentifiable mo dels, the data does not contain sufficient infor mation to cho ose b etw een alternative mo dels with equa l statistical evidence. Ultimately , the uncer ta in t y in inf erence arises from limited sample size and the lack of computational reso urces. In the remainder of this section, let us c o nsider more closely the par ticular learning pro ce dur es central to this thesis: p oint es timates and v ariationa l approx- imation, and the s tandard optimization algor ithms used to le a rn such representa- tions. P oin t estimates Assuming indep endent and identically distr ibuted observ ations, the likelih o o d o f the data, given mo del par ameters, is p ( X | θ ) = Q i p ( x i | θ ). This pr ovides a prob- abilistic measure of mo del fit and the ob jective function to maximize. Maximiza- tion of the likelihoo d p ( X | θ ) with resp ect to θ yields a m aximum likeliho o d (ML) estimate of the model pa r ameters, and specifies an o ptimal mo del that b est de- scrib es the da ta. This is a sta ndard p oint estimate used in probabilistic mo deling. Practica l implementations typically op erate o n lo g-likeliho o d , the lo garithm of the likelihoo d function. As a monotone function, this yields the sa me optima , but has additional desirable proper ties: it factorizes the product in to a sum and is less prone to n umerical ov erflows during optimization. 25 CHAPTER 3. ST A TISTICAL LEAR NING AN D EXPLORA TOR Y DA T A ANA L Y SIS The maximum a p osteriori (MAP) estima te additionally takes prior info r ma- tion of the model parameters in to account. While the ML estimate maximizes the likeliho o d p ( X | θ ) of the observ ations, the MAP estimate maximizes the p os- terior p ( θ | X ) ∼ p ( X | θ ) p ( θ ) o f the mo del parameter s. The ob jective function to maximize is the log-likeliho o d l og p ( θ | X ) ∼ l og p ( X | θ ) + l og p ( θ ) . (3.4) The prio r is explicit in MAP estimation and the model co ntains the ML esti- mate as a sp ecial ca se; assuming lar g e sample size, or non-informativ e, unif orm prior p ( θ ) ∼ 1 , the likelihoo d of the data p ( X | θ ) will dominate and the MAP esti- mation b eco mes equiv a le n t to optimizing p ( X | θ ), yielding the traditiona l ML es ti- mate. The ML and MAP estimates are asymptotically cons is ten t appr oximations of the p osterio r distribution, since the po s terior will conv erge a p oint distribution with a large sample s ize. The computation and interpretation of p oint estimates is straightforward co mpared to the use of po sterior distributions in the full B ay e s ian treatment. The differences b etw ee n ML a nd MAP estimates hig hlight the role of prior information in the mo deling when training data is limited. V ariational i nference In certain modeling tasks the uncer ta in t y in the model para meters needs to be taken into a ccount. Then point estimates are not sufficien t. The uncertain t y is characterized b y the p osterio r distribution p ( θ | X ). How ever, the po sterior distri- butions a re often intractable and need to b e estimated by approximative metho ds. V ariational appr oximations provide a fast and pr incipled optimizatio n scheme (see e.g. Bis hop, 20 06) that yie lds only approximative solutions, but can a c c elerate po sterior inference b y or ders o f magnitude compared to sto chastic, sampling-based MCMC metho ds that can in principle provide exact so lutions, assuming that in- finite computationa l resourc e s are av aila ble. The p otential decrease in accuracy in v ar iational approximations is often acceptable, g iven the gains in efficiency . V aria tional approximation character iz es the uncertaint y in θ with a tra c table dis- tribution q ( θ ) that a pproximates the full, potentially intractable p osterior p ( θ | X ), V aria tional inference is fo r mu lated as an optimization p roblem where an in- tractable pos terior dis tribution p ( Z , θ | X ) is approximated by a more easily tract- able dis tribution q ( Z , θ ) by minimizing the KL– divergence betw een the t w o distri- butions. This is also shown to maximize a low er bo und of the marginal likeliho o d p ( X ), and subseq uen tly the likeliho o d of the data, yielding an approximation o f the ov erall mo del. The log- likeliho o d of the da ta can b e decomp osed into a s um of the low er bo und L ( q ) of th e observ ed data and the KL–divergence d K L ( q , p ) betw een the approximative and the exact p oster ior distr ibutions: l og p ( X ) = L ( q ) + d K L ( q , p ) , (3.5) where L ( q ) = R z q ( Z , θ ) l og p ( Z , θ , X ) q ( Z , θ ) ; d K L ( q , p ) = − R z q ( Z , θ ) l og p ( Z , θ | X ) q ( Z , θ ) . (3.6) The KL-divergence is non-neg ative, and eq ua ls to zero if and only if the a p- proximation and the e xact distribution a r e identical. Therefo r e L ( q ) giv es a 26 3.3. LEA RNING AND INFER ENCE low e r bound for the log -likelihoo d l og p ( X ) in Equation 3.5. Minimization of d K L with resp ect to q will provide an a nalytically tr actable approximation q ( Z , θ ) of p ( Z , θ | X ). Minimization of d K L will als o maximize the lower b ound L ( q ) since the log-likeliho o d log p ( X ) is indep endent o f q . The a ppr oximation t ypically assumes independent pa rameters and latent v ar iables, yielding a factorize d approximation q ( Z , θ ) = q z ( Z ) q θ ( θ ) based on tractable standard distributions. It is also po ssi- ble to factorize q z and q θ int o further comp onents. V ar iational appro ximations are used for efficien t lea rning of infinite multiv ariate Gaussian mixture mo dels in Publication 3. Exp ectation–Maximization (EM) The EM algorithm is a general pro cedur e for learning proba bilistic latent v a r iable mo dels (Dempster et al., 1 977), and a sp ecial case of v aria tional inference. The algorithm provides an efficient algorithm for finding p oint estimates for mo del parameters in laten t v ar iable models. T he ob jective of the EM algorithm is to maximize the marginal likelihoo d p ( X | θ ) = Z z p ( X , Z | θ ) d Z (3.7) of the obs erv ations X with re s pec t to the mo del pa rameters θ . Marginalizatio n ov er the probability densit y of the latent v aria bles provides an inference pro cedure that is robust to uncertaint y in the latent v a riable v alues. The algo r ithm itera tes betw een es tima ting the po sterior of the latent v ariables , and optimizing the mo del parameters (see e.g. Bishop, 2006). Given initial v a lues θ 0 of the mo del pa ram- eters, the exp e ctation step ev aluates the p osterior densit y of the latent v ar iables, p ( z | x , θ t ), keeping θ t fixed. If the p osterio r is not a na lytically trac ta ble, v aria- tional approximation q ( z ) can b e used to obtain a low er b ound for the likelihoo d in E q uation 3.7. The maximization step optimizes the mo del par ameters θ with resp ect to the following ob jective function: Q ( θ , θ t ) = Z z p ( Z | X , θ t ) log p ( X , Z | θ ) d Z . (3.8) This is the e x pecta tion of the c omplete-data lo g-likelih o o d l og p ( X , Z | θ ) over the latent v ar ia ble density p ( Z | X , θ t ), obtained from the previous exp ectation step. The new parameter estimate is then θ t +1 = ar g max θ Q ( θ , θ t ) . The expe ctation and ma ximization steps determine an iterative lear ning pro- cedure wher e the latent v ar iable density and mo del pa rameters ar e iteratively upda ted until co n vergence. The maximization step will also increa s e the tar get likelihoo d of Equa tion 3.7, but p otentially with a remark a bly smaller co mputa- tional cos t (Dempster et al., 1977 ). In contrast to the mar ginal likelihoo d in Equation 3.7, the complete-data lik eliho o d in Equation 3 .8 is logarithmized be - fore int egration in the maximization step. When the joint distribution p ( x , z | θ ) belo ngs to the exp onential family , the logarithm will ca ncel the exp o nent ial in algebraic manipulations. This can considerably simplify the max imiza tion step. When the likelihoo ds in the optimizatio n are of suitable for m, the iteration steps can b e so lved analytically , which can co nsiderably reduce r equired e v aluations o f 27 CHAPTER 3. ST A TISTICAL LEAR NING AN D EXPLORA TOR Y DA T A ANA L Y SIS the ob jectiv e function. Conv ergence is guara n teed, if the optimization can increase the likelihoo d a t each itera tio n. Howev er, the iden tification of a global optimum is not guaranteed in the EM algor ithm. Incorp ora ting prior informa tion of the parameter v alues through Bayesian pri- ors can b e used to avoid overfitting a nd fo cus the mo deling o n pa rticular features in the data, as in the reg ularized dep endency modeling framework o f Publica tion 4, where the EM algorithm is used to learn Gaussian latent v a r iable mo dels. Standard opti mization metho ds Optimization metho ds provide sta ndard to ols to implement selected lear ning pro- cedures. Optimization a lgorithms ar e used to identif y pa rameter v a lues that min- imize or maximize the ob jectiv e function, either globally , or in lo cal surroundings of the optimized v alue . Selec tio n of optimization metho d dep ends on smo oth- ness and contin uit y prop erties of the ob jective function, requir ed accur acy , and av a ilable res o urces. Gr adient-b ase d app r o ache s o ptimize the ob jective function by as s uming smo oth, contin uo us top olog y over the pr obability density where setting the der iv atives to zero will yield loca l optima. If a closed form solution is not a v ailable, it is of- ten possible to estimate gr adient dir e ctions in a given p oint. Optimization can then pro ceed by up dating the par ameters tow a rds the desir ed dir ection along the gradient, gradually impr oving the ob jective function v alue in subsequent gra dien t ascent steps. So-ca lled quasi-Newton metho ds use function v alues and gra dien ts to c haracterize the optimized manifold, and to optimize the parameters a lo ng the approximated g radients. An appropriate step length is ide ntified automatically based on the curv ature of the ob jection function sur fa ce. The Broyden-Fletcher- Goldfarb-Shanno (BF GS) (Br oyden, 197 0 ; Fletcher, 19 70; Goldfa r b, 1 970; Shanno, 1970) method is a q uasi-Newton approach used fo r standard optimization tasks in this thesis. 3.3.2 Generalizabilit y and o v erlearning Probabilis tic mo dels a re formulated in terms of pro babilit y dis tributions over the sample space and par ameter v alues. This forms the bas is for generaliza tion t o new, unobserved even ts. A g eneralizable mo del can des crib e ess en tial character- istics of the underlying pro ce s s that generated the observ atio ns ; a g e neralizable mo del is also able to characterize future observ a tions. Overle arning , or overfitting refers to mo dels that describ e the training da ta w ell, but do not gene r alize to new observ ations. Suc h mo dels describ e no t only the genera l pro cess es underlying the observ ations, but also noise in the particular observ ations. Avoiding ov erfitting is a central asp ect in mo deling. Overlearning is par ticularly likely when training data is scar ce. While ov erfitting could in principle be avoided by collec ting more data, this is often not feasible since the cos t of data collection can b e prohibitively large. Generalizability can b e measured by inv e stigating how accur a tely the mo del describ es new observ atio ns. A standar d approa ch is to split the da ta in to a tr aining set , us e d to learn the mo del, and a test set , used to mea sure mo del p erformance on unseen obser v ations that were not used for tra ining. In cr oss-valid ation the test is repeated with sev eral different le a rning and test sets to asses s the v ar iabilit y in the testing pro cedur e. Cross-v alidation is used for instance in Publicatio n 5 of 28 3.3. LEA RNING AND INFER ENCE this thesis. Bo otstr ap analysis (see, for instance, Efron and Tibshira ni, 1994) is another widely used approach to measur e mo del per formance. The obs e rved data is viewed as a finite re a lization of a n underlying pro bability density . New samples from the underlying density ar e obtained by r e-sampling the obser ved data p oints with replacement to sim ulate v ar iability in the o riginal data; obse r v ations from the more dense r e gions of the pro babilit y space b ecome re-s a mpled more often than rare even ts. E ach b o otstrap sa mple re semb les the probability density of the original data . Mo deling multiple data sets obtained with the bo o tstrap helps to estimate the sensitivity of the mo del to v ariations in the data. Bo otstrap is used to assess mo del per formance in Publication 6. 3.3.3 Regularization and mo del selection In general, increasing model complexity will yield more flex ible models , which ha v e higher descriptive p ow e r but a re, on the o ther hand, more likely to overfit. There- fore r elatively simple mode ls can often outp erform more complex mo dels in terms of genera lizability . A compro mise betw een simplicity and descriptive p ower ca n be obtained b y imp osing additio na l co nstraints or soft p enalties in the mo deling to prefer compact so lutions, but at the same time retain the descr iptiv e power of the original, flexible mo del family . This is called r e gularization . Regulariza tio n is par - ticularly imp ortant when the sample size is small, as demonstrated for instance in Publication 4, where explicit and theoretica lly principled regular ization is achiev ed by setting appropria te prior s on the mo del structur e and par ameter v alues. The priors will then affect the MAP estimate of the mo del parameters. One commonly used appr oach is to prefer sp arse solutions that allow o nly a s mall num b er o f the po ten tial para meters to b e employ ed a t the same time to mo del the da ta (see e.g. Archam beau and Bach, 200 8). A family o f probabilistic approaches to balance betw een mo del fit and mo del complexity is provided by info r mation-theoretic cr i- teria (see e .g . Gelman e t al., 2003). The Bayesian Information Criterion (BIC) is a widely used infor mation criterion that in tro duces a p enalty term on the num b er of mo del parameters to prefer s impler mo dels. The log-likeliho o d L of the data, given the mo del, is balanced by a measur e of mo del complex it y , q l og ( N ), in the final ob jective function − 2 L + q l og ( N ). Here q denotes the num b er o f mode l pa- rameters a nd N is the constant sample s ize of the investigated data set. The BIC has b een criticized since it do es not addre ss changes in prior dis tributions, and its der iv ation is based on asy mptotic considera tio ns that hold o nly approximately with finite sample siz e (see e.g. Bishop, 200 6). On the other hand, BIC pr ovides a principled regular ization pro cedure that is eas y to implement. In this thesis, the BIC has b een used to regular ize the algor ithms in Publication 3. 3.3.4 V alidation After learning a pr obabilistic mo del, it is necess ary to confirm the qua lit y of the mo del and verify p otential findings in further, independent exp eriments. V alida- tion r efers to a versatile s e t of approa ches used to inv estigate mo del p erforma nce, as well as in mo del cr iticis m, compa rison and selection. In ternal a nd externa l approaches provide t w o co mplementary c a tegories for mo del v a lidation. Int er- nal validation refers to pro cedur es to as sess mo del p erformanc e based on training data alone. F o r insta nc e , it is p ossible to estimate the sensitivity of the mo del to initialization, parameteriza tion, and v ar iations in the data , or con v ergence of 29 CHAPTER 3. ST A TISTICAL LEAR NING AN D EXPLORA TOR Y DA T A ANA L Y SIS the lear ning pro cess. Int ernal ana lysis can help to estima te the weaknesses a nd generaliza bilit y of the mo del, and to co mpare a lternative mo dels. B o otstrap a nd cross- v alidation ar e widely use d appr oaches for internal v alidation and the analysis of mo del p erformance (see e.g . Bishop, 2006). B o otstrap can provide informa tio n ab out the sensitivity o f the results to sampling e ffects in the data. Cross-v alidation provides informa tion a bo ut the mo del gener alization p erformance and ro bustness by comparing predictions of the mo del to real outcomes . External validation ap- proaches inv estigate mo del predictions and fit o n new, independent data s ets and exp eriments. Explora tory analy sis of high- thr oughput data s e ts o ften includes massive m ultiple testing, a nd provides potentially thousands of a utomatically gen- erated hypotheses. Only a small set of the initial findings can be in v estigated more closely b y human interven tion and costly lab or atory exp eriments. This highlights the need to prioritize the results and assess the uncertaint y in the mo dels. 30 Chapter 4 Reducing uncertai n t y in high-throughput microarra y studies As far as the laws of mathematics r efer to r e ality, they ar e not c ertain, as far as they ar e c ertain, they do n ot r efer to r e ality. A. Einstein (1956) Gene express ion microa r rays are currently the most widely used tech nology for genome-wide transc riptional profiling, and they co nstitute the main source of data in this thesis. An ov erview o f microa rray technology is provided in Section 2.3 .1. Microar r ay measurements a r e asso c ia ted with hig h levels of noise fro m technical and bio logical sour ces. Appro priate pre pr o cessing techniques can help to reduce noise a nd obtain reliable mea s urements, which is the cr ucial starting p oint for any data ana ly sis task. This c hapter presents the first main c ont ribution of the thesis, prepro ces sing techniques that utilize side infor mation in geno mic se quence databases and micr oarray data co lle c tions in o rder to improve the accur acy o f high- throughput gene expres sion data. The chapter is o rganized a s follows: Section 4 .1 gives a n ov erview of the v arious sources of nois e in high-throughput microa rray studies. Se c tion 4.2 introduce s a s trategy for noise reduction based on s ide infor- mation in external genomic sequence databas es. Section 4.3 extends this mo del b y describing a model- based approach that additionally combines statistical evidence across m ultiple micr oarray exp eriments in order to provide quantitativ e informa- tion of pro b e p erformance and utilizes this informa tion to improv e the r eliability of high-throug hput observ ations. The results ar e summar ized in Se c tion 4 .4. 4.1 S ou r c es of u n certain t y Measurement data obtained with novel high-throughput techn ologies comes with high levels of uncontrolled bio logical and technical v ariatio n. This is often called noise as it o bscures the meas ur ement s, and adds p otential bias and v ariance on the s ignal of interest. Biolog ical noise is asso ciated with natural biologica l v ar ia - tion b etw een cell p opulations, cellular pro cess e s and individuals. Single-nucleotide 31 CHAPTER 4. REDUCING UNCER T AINTY IN HIGH-THROUGHPUT MICRO A RRA Y S TUDIES po lymorphisms, alterna tiv e splicing a nd no n-sp ecific h ybridization add biological v aria tion in the data (Dai et al., 2 005; Zhang et al., 20 0 5). More technical sources of noise in the measurement pro cess include RNA extraction and amplification, exp eriment-specific v ariation, as well as platform- and la bo ratory - sp e cific effects (Choi et al., 2003 ; MAQC Consor tium, 20 06; T u e t a l., 2002 ). A significa nt source of nois e on gene expres s ion arrays comes from individ- ual prob es that are designed to measure the activit y of a giv en transcript in a biological sample. Figure ?? A shows prob e-level observ ations of differential g ene expression for a collection of prob es designed to target the same mRNA transcript. One of the prob es is highly contaminated and likely to a dd unrela ted v ar iation to the analysis . A num ber of factors affect prob e p erformance . F or instance, it has bee n re po r ted in P ublica tion 1 and elsewher e (Hwang et al., 2004 ; Mecham et al., 2004b) that a large po rtion of microa rray prob es may target unintended mRNA sequences. Mo reov er, althoug h the pr ob e s hav e been designed to uniquely hy- bridize with their intended mRNA target, r emark able cr oss-hybridization with the prob es b y single-nucleotide p olymorphisms (Dai et al., 2005 ; Sliwersk a et al., 2007) and o ther mRNAs with closely simila r sequences (Zhang et al., 2 005) hav e b een rep orted; high-affinity prob es with high GC-conten t may have hig her likelihoo d of cross- hybridization with nonsp ecific targets (Mei et al., 2003). Alternative splic- ing (MAQC Consor tium, 2006 ) and mRNA deg radation (Auer et al., 200 3) may cause differences betw een prob es targeting different p os itions o f the gene sequence. Such effects will contribute to pr ob e-level contamination in a prob e- and condition- sp ecific manner. How e ver, sour ces of pr ob e - level no ise are still p o or ly under sto o d (Irizarry et al., 2005; Li et al., 2005 ) despite their imp o rtance fo r express ion anal- ysis and prob e design. High levels of nois e set sp ecific challenges for analysis . Better understanding of the technical asp e cts o f the meas ur ement pro ce s s will lead to improv ed analytica l pro cedures and ultimately to more accura te biolog ical r esults (Reimers, 2010 ). Publication 2 provides computationa l to ols to in vestigate prob e p erfor mance and the relative contributions o f the v ario us so urces of pr ob e-level contamination on short oligonucleotide arr ays. 4.2 Pr epr o cessing microarra y data with side in- formation Pr epr o c essing of the r aw data obtained from the or iginal measurements ca n help to reduce noise a nd improv e comparability b etw een micr o array exp eriments. Pre- pro cessing can b e defined in ter ms o f statistical transformatio ns on the r aw data, and this is a central part o f data a nalysis in hig h-throughput studies. This sec- tion outlines the s ta ndard prepro cessing steps for shor t olig onucleotide arrays, the main sour c e of tr a nscriptional pro filing data in this thesis. How ev er, the genera l concepts also apply to other microarr ay platfor ms (Reimers, 20 10). Standard prepro cessing steps A num b er of prepro ces sing techniques for shor t oligonucleotide ar rays hav e b een int ro duced (Irizarry et al., 2006 ; Reimers, 2010). The standar d prepro ces sing steps in microarr ay analysis include qua lit y cont rol, background correctio n, normaliza- tion and summarization. 32 4.2. PREPROCESSING MICRO ARRA Y D A T A WITH SID E I NFORMA TION 0 50 100 150 −4 −2 0 2 Observations Signal log−ratio 00 00 00 00 00 00 00 00 00 00 00 00 11 11 11 11 11 11 11 11 11 11 11 11 000000 000000 000000 111111 111111 111111 ALL133 GEA133 ALL95 GEA95 133A 95A/Av2 Relative increase (%) 0 10 20 30 Mistargeted 5’/3’ position GC−content SNP 00000000000000000000000000 00000000000000000000000000 00000000000000000000000000 00000000000000000000000000 00000000000000000000000000 11111111111111111111111111 11111111111111111111111111 11111111111111111111111111 11111111111111111111111111 11111111111111111111111111 00 00 00 00 00 00 00 00 00 00 00 00 00 00 00 00 00 00 11 11 11 11 11 11 11 11 11 11 11 11 11 11 11 11 11 11 Relative increase (%) A B Observations Signal log−ratio ALL133 GEA133 GEA95 133A 95A/Av2 ALL95 Figure 4.1: A Example of a prob e set that cont ains a probe with high conta mination levels (dashed line) detected by the probabili stic RP A mo del. The prob e-lev el observ ations of differen- tial gene expressi on for the di fferen t pr obes tha t measure the same target transcript are indicated b y gra y l ines. The blac k li ne shows the estimated signal of the target transcript across a n um ber of conditions. B Increase in the av erage v ariance of the pr obes asso ciated with the inv estigated noise sources: mistargeted prob es having errors in the genomic al i gnmen t, m ost 5’/3’ prob es of eac h prob e set, GC-rich, and SNP-asso ciated pr obes. The v ar iances were estimated b y RP A and describ e the noise level of the prob es. The results are shown for the individual ALL and GEA data sets, and for their com bined results on b oth platforms (133A and 95A/Av2). c IEEE. Reprint ed with permi ssion from Publ i cation 2. Micr o arr ay quality c ontr ol is used to iden tify ar rays with remark able ex per i- men tal defects, and to remove them fro m subsequent analysis. The t ypical tests consider RNA degra dation lev els and a num b er of other summary statistics to guarantee that the array da ta is of r easonable qua lity . The arr ays that pa ss the microarr ay qua lit y control a re pr epro cessed further. Each ar ray t ypically has spa- tial biases tha t v ary smo othly a cross the arr ay , ar ising from technical facto rs in the exp eriment. Backgr ound c orr e ction is used to de tec t and r emov e s uch spatial effects from the arr ay data , and to provide a uniform ba ckground signal, enha nc- ing the comparability o f the prob e-level observ ations betw een differen t parts of the array . Moreov er, background co rrection can estimate the gener al noise level on the array; this helps to detect pro be s whose s ignal differs sig nificantly from the background noise. Robust m ulti-array av eraging (RMA) is one of the most widely used approaches for prepro ces s ing short oligo nucleotide arr ay data (Iriz a rry et al., 2003a ). The ba ckground corre c tion in RMA is based on a global mo del for prob e int ensities. The obser ved in tensity , Y , is mo deled as a sum of an exp onential signal comp onent, S a nd Gaussia n noise B . Background cor r ected data is then obtained as the expe c ta tion E B ( S | Y ). While background correction makes the observ ations co mparable within array , normalization is used to improv e the com- parability between arrays. Quantile normalizatio n is a widely used metho d that forces all a rrays to follow the same empirica l intensit y distribution (see e.g. Bo l- stad et a l., 2 003). Qua ntile no r malization mak es the measurements acros s different arrays comparable, assuming that the overall dis tribution of mRNA concentration is approximately the same in all cell p opula tions. This has proven to b e a feasible assumption in tra nscriptional profiling studies. As alwa ys, there are exceptions . F or instance, human brain tissues hav e systematic differences in g e ne expressio n compared to other or gans. O n short oligonucleotide ar r ays, a num ber of pro bes target the same transcript. In the final summarization step , the individual pro be - 33 CHAPTER 4. REDUCING UNCER T AINTY IN HIGH-THROUGHPUT MICRO A RRA Y S TUDIES level obser v ations of ea ch target transc ript are summarized into a single s ummary estimate of transcript activity . Standard algorithmic implementations ar e av aila ble for each prepro ces s ing step. Prob e-level prepro cessing metho ds Differences in pr ob e characteristics ca use systematic difference s in pr ob e p erfor- mance. The use of several pro bes fo r each targe t lea ds to more ro bust es timates on transcript activity but it is clear that probe quality ma y significantly affect the results of a microarr ay study (Iriz arry et al., 2003b). Widely used prepro- cessing a lgorithms utilize pro be- sp e cific par ameters to mo del pr ob e-sp ecific effects in the pr ob e summariza tion step. Some of the first a nd most well-known prob e- level pr epro cessing algorithms include dChip/MBEI (Li and W ong, 2 001), RMA (Irizarry et al., 20 03a), and gMOS (Milo et al., 2 003). T aking prob e-level effects int o account can cons ide r ably improv e the quality of a microa rray study (Reimers, 2010). Publications 1 and 2 incorp orate side infor mation of the prob es to prepro- cessing, and introduce improv ed prob e-level a nalysis metho ds for differential gene expression studies. In o rder to in tro duce pro be - level prepro cessing metho ds in more detail, let us consider the pr ob e summarization step of the RMA a lgorithm (Iriza r ry et a l., 2003a ). RMA has a Gaussian mo del for prob e effects with prob e-sp ecific m ean parameters and a sha red v ariance para meter for the prob es. The mea n parameters characterize prob e-sp ecific binding affinities that cause systema tic differ e nces in the s ignal levels captured by each prob e. Estimating the pr ob e-sp ecific effects helps to remove this effect in the final pr o be s et-level summary of the prob e-level observ ations. T o briefly outline the algo rithm, let us consider a collection o f pro bes (a pr ob eset ) that mea sure the e x pression level o f the s a me targe t transcript g in condition i . The pr ob e-level observ ations are mo deled as a sum of the true, underlying ex pression signa l g i , which is common to all prob es, prob e-sp ecific binding affinity µ j , and Gaussia n noise ǫ . A prob e-level observ ation for prob e j in condition i is then mo deled in RMA as s ij = g i + µ j + ǫ. (4.1) Measurements from multiple conditions are needed to estimate the prob e- sp ecific effects µ j . RMA and other mo dels that measure a bsolute gene expr ession hav e an imp orta n t drawback: the prob e affinity effects { µ j } ar e uniden tifiable. In order to o btain an ident ifiable mo del, the RMA algo rithm includes an a dditional constraint that the pro be affinity effects are zero on av erage: Σ j µ j = 0. This yields a well-defined alg orithm that has been shown to pro duce a ccurate measurements of gene expr ession in practica l s ettings. F urther ex tensions of the RMA algorithm include g c RMA, which has a more deta iled chemical mo del for the prob e effects (W u and Irizar ry, 2004), refRMA (Katz et a l., 200 6 ), whic h utilizes pr ob e-sp ecific effects derived from background da ta co llections, and fRMA (McCall et al., 2010), which also mo dels batch-specific effects in microarr ay studies. The estimatio n of uniden tifiable prob e affinities is a main challenge for most pr ob e-level prepro cess- ing models . RMA and other prob e-level models for sho rt oligonucleotide arrays hav e b een designed to estimate absolute e x pression lev els o f the g e nes. How ever, gene expres - sion studies are often ultimately targeted at inv estigating differ ential expr ession 34 4.2. PREPROCESSING MICRO ARRA Y D A T A WITH SID E I NFORMA TION levels , that is, differences in gene expres sion b etw een exp erimental conditions . Measurements o f differential expres s ion is obtained for instance by co mparing the expression lev els, obtained through the RMA algorithm o r other metho ds, b etw een different co nditions. Ho w ever, the summariza tion of the pr ob e -level v a lues is then per formed prior to the actual comparison. Due to the unidentifiabilit y of the prob e affinity par ameters in the RMA and o ther prob e-level models , this is p otentially sub o ptimal. Publication 1 demonstrates that reversing t he order, i.e., calculat- ing differential gene express ion already at the pr ob e level b efore prob eset-level summarization, le a ds to impro v ed estimates of differential gene expressio n. The explanation is that the pro cedure c ircumv ents the need to estimate the uniden tifi- able prob e affinity par ameters. This is formally describ ed in P ublication 2, w hich provides a probabilistic extension of the Prob e-level Expressio n Change Averag- ing (PECA) pro cedure of Publica tion 1. In PECA, a s tandard weigh ted average statistics summarizes the pr ob e level obs erv ations of differential gene expression. PECA do es not mo del prob e-sp ecific effects, but it is shown to outp erform widely used prob e-le vel prepr o cessing metho ds, such as the RMA, in estimating differen- tial ex pression. Publicatio n 2, consider ed in more detail in Section 4.3, provides an extended probabilistic framework that also mo dels prob e-sp ecific effects. Utilizing s ide inform ation in transcriptome databases Prob e-level pr epro cessing mo dels and microa rray a nalysis can b e further impr ov ed by utilizing external informa tio n o f the probes (Eisens tein, 2006; Hwang et al., 2004; Ka tz et a l., 200 6). Althoug h any given microarr ay is designed on most up- to-date sequence information av ailable, r apidly evolving genomic sequence data can reveal inaccuracies in prob e a nnotations when the bo dy of knowledge grows. In recent studies, including Publication 1, a re ma rk able n um ber o f prob es on v ar i- ous olig onucleotide arrays have b een detected not to uniquely matc h their int ended target (Hwang et al., 2004 ; Mecham et al., 200 4 a). A r emark a ble p ortion of pro bes on several po pular microa r ray platforms in human and mous e did not match with their intended mRN A targ e t, or were found to target unin tended mRNA tran- scripts in the Ent rez Nucleotide (Wheeler et al., 2 005) sequence databa s e in Publi- cation 1 (T able ?? ). The observ ations are in genera l concordant with other studies, although the exa ct figures v ar y a c cording to the utilized database and co mpari- son deta ils (Gautier e t a l., 2004; Mec ham et al., 2004 b). In this thesis, strategies are developed to improv e microar ray analy s is with background infor ma tion from genomic sequence databa ses, and with mo del-based analysis of micro array collec- tions. Prob e verification is increasing ly used in standar d pr epro cessing, a nd to con- firm the results of a microa rray study . Matching the pr ob e sequences of a given array to up dated ge no mic s e q uence da tabases and constructing an alter native in- terpretation of the array data based o n the most up-to-date genomic a nnotations has been shown to increase the accuracy and cross- platform consistency of mi- croar ray ana lyses in Publication 1 and elsewhere (Dai e t al., 20 05; Ga utier et al., 2004). Publication 1 combines prob e verification with a nov el prob e-level pr epro cess- ing metho d, PECA, to s ug gest a novel framework for compar ing and combining results across different microar ray platforms . While huge rep os itories of micro array data are av aila ble, the data for any particula r exper iment al condition is typically scarce, and coming fro m a n umber of different micro array platforms. Therefore 35 CHAPTER 4. REDUCING UNCER T AINTY IN HIGH-THROUGHPUT MICRO A RRA Y S TUDIES reliable approaches for in tegrating microarray data are v aluable. Integration of results ac r oss platforms has proven problema tic due to v arious s o urces of tec hnical v aria tion b etw een a rray technologies. Matchin g of prob e sequences b etw een mi- croar ray pla tforms has b een shown to incr ease the consistency of micr o array mea- surements (Hwang e t al., 20 04; Mecham et al., 20 04b). How ever, prob e matching betw een a rray platforms guarantees only technical compa rability (Irizar ry et a l., 2005). Prob e verification ag ainst exter nal sequence databases is needed to c o n- firm that the prob es a r e also bio logically accura te. This can also improv e the comparability across array pla tforms, as confirmed by the v alidation studies in Publication 1 (Figure ?? A). The PECA method of Publication 1 utilizes genomic sequence databases to reduce prob e-level noise by removing erro neous pro bes ba sed on up dated g enomic knowledge. The strategy r elies on exter nal infor mation in the databases and can therefore only remov e known sources of prob e-level contamination. Publication 2 int ro duces a probabilistic framework to mea sure prob e reliability directly based on microarr ay da ta co llections. The a na lysis can reveal b oth well-c haracterized and unknown sources of prob e-level con tamination, and leads to improv ed estimates of gene ex pression. This mo del, coined Robust Pro ba bilistic Averaging (RP A), also provides a theoretically justified framework for incorp ora ting prior knowledge of the prob es int o the analysis. Array type Num ber of pr ob es V erified prob es (%) HG-U133 P lus2.0 604,25 8 58.2 HG-U133A 247,96 5 82.5 HG-U95Av2 199,08 4 82.6 MOE430 2.0 496,46 8 68.2 MG-U74Av2 197,99 3 73.1 T able 4.1: The pr oportion of sequence-ve rified prob es on three popular human microarray plat- forms and t w o mouse platforms, as observ ed in Publi cation 1. Prob es that matc hed to mRNA sequences corresponding to unique genes (defined by a Gene ID iden tifier) in the En trez database are considered verified. A remark able p ortion of the prob es on the inv estigated arrays did not matc h the En trez transcript sequences, or had amb iguous targets. 4.3 M o del-based noise reduction Standard a pproaches for in v estigating prob e perfor ma nce t ypically r ely on external information, such a s g enomic sequence data (s e e Mec ham et al. 20 04b; Zhang et a l. 2005 a nd Publication 1) or physical mo dels (Naef a nd Magnas co, 2003 ; W u et al., 2005). Ho w ev er, suc h mo dels cannot reveal probes with uncharacteriz ed so urces of contamination, suc h as cross- h ybridization with alternatively spliced transcripts or closely related mRNA sequences. V ast collections o f micr oarray data are av a ila ble in public rep osito ries. These large- scale data sets con tain v aluable information of b oth biologic al a nd technical asp ects of ge ne expression s tudies. Publication 2 int ro duces a da ta-driven s tr ategy to extra ct and utilize prob e-level infor mation in microarr ay data collec tio ns. 36 4.3. MOD EL-BASED NOISE REDUCTION A B Correlation MAS RMA FARMS PECA RPA 0.2 0.3 0.4 0.5 0.6 0.7 0.8 Correlation Figure 4.2: A Effect of sequence verification on comparabili t y betw een microarr a y platforms. Correlations betw een RMA-prepro cessed tec hnical replicates on tw o arr a y platforms where the same s amples hav e been h ybri dized on the tw o array types. The P earson correlations were calculated f or each pair of arrays measuring the same biological sample. The gray l ines show correlations obtained with the differen t probe matching cr iteria. In the hESC array comparison, the b est matc h prob e sets con tained exactly the same probes on b oth array generations, whic h resulted in very high correlations. The adv an tag es of prob e verification and alternativ e mappings we re lar gest when arrays with differen t probe collections were compared in the mCPI, ALL and IM arra y comparisons. B Reproducibility of signal estimates in real data sets bet w een t he tec hnical replicates, i.e., the ’b est matc h’ prob e sets b et w een the HG-U95Av2 and HG-U133A platforms. The consistency was measured by the Pe arson correlation bet we en the pairs of arr a ys, to which the same sample was h ybridized. c Published by Oxford Unive rsity Pr ess. Reprinted with p ermissi on from Publication 1. The model, R obust Pr ob abilistic Aver aging (RP A) , is a probabilistic prepro- cessing pr o cedure that is based on explicit mo deling assumptions to a nalyze pro be reliability and q uant ify the uncertaint y in measurement data ba sed on gene ex- pression data co llections, indepe ndently of exter nal information of the probes . The mo del can b e viewed as a pro babilistic extension o f the prob e-level prepro cessing approach for differential gene expr ession studies presented in Publica tion 1. The explicit Bay esian formulation quantifies the uncer taint y in the mo del parameter s, and allows the incorp ora tio n of prio r infor mation concerning pr ob e reliability into the a nalysis. RP A provides estimates o f prob e reliability , and a pro bes et-level estimate o f differential gene express ion directly fr om expr ession da ta and indep en- dent ly of the nois e sour ce. The RP A mo del is independent of ph ysical models or external and consta ntly upda ted information such as genomic sequence data, but provides a fra mework for incorpor ating suc h prior infor mation of the probes in gene expression analysis. Other probabilistic methods for micr oarray pr e pr o cessing include BGX (Hein et al., 2005), g MO S (Milo et al., 200 3 ) and its extensions (Liu et al., 20 05). The key difference to the RP A pr o cedure of Publica tion 2 is that these metho ds are designed to provide pro bes et-level summaries o f a bs olute gene expression levels, and suffer from the same uniden tifia bility problem of prob e affinit y parameters as the RMA algo rithm (Iriz a rry et al., 20 03a). In contrast, RP A mo dels pro be- level es tima tes of differential gene expressio n. This remov es the unidentifiabilit y 37 CHAPTER 4. REDUCING UNCER T AINTY IN HIGH-THROUGHPUT MICRO A RRA Y S TUDIES issue, which is adv antageous when the ob jective is to compare gene ex pression levels be tw een exp erimental conditions . Another imp ortant difference is that the other prepro cessing metho ds do not provide explicit estimates of pro be- s pec ific parameters , or to ols to inv estigate pro be performance. Publica tion 2 assigns an explicit pr obabilistic measure of reliability to e a ch pro be. This g ives to ols to analyze prob e p erformance and to guide prob e design. Robust Probabil i stic Av eraging Let us now consider in more deta il the proba bilistic prepro ces sing fra mew ork, RP A, in tro duce d in Publication 2 . Prob e p erformance is ultimately determined by its ability to accurately meas ure the expressio n level o f the ta r get trans cript, which is unknown in practical situations. Although the per fo rmance of individua l prob es v aries, the collection of prob es designed to measur e th e same t ranscript will provide gr ound truth for asses sing pr o be p erfo r mance (Figure ?? A). RP A captures the shar ed signal o f the prob es within a pro bes et, and ass umes tha t the s hared signa l characterizes the expressio n of the co mmon targ et transcript of the probes. The reliabilit y of individual prob es is estimated with respect to the str ongest shared signal of the pro bes . RP A assumes no rmally distributed prob e effects, and qua n tifies pr ob e reliability based o n prob e v aria nce a round the prob eset-level sig nal across a large num ber o f arrays. This extends the formulation of the RMA model in Equation 4.1 by intro ducing an additional probe- sp e cific Gaussian noise comp onent: s ij = g i + µ j + ε ij . (4.2) In contrast to RMA, the v a riance is prob e-sp ecific in this mo del, a nd distributed as ε ij ∼ N (0 , τ 2 j ). The v ariance parameters { τ 2 j } are o f in terest in pro be reli- ability analysis; they r eflect the noise level of the prob e, in contrast to prob e- level prepr o cessing methods tha t fo cus on es tima ting the uniden tifiable mean pa- rameter of the Gaus sian noise mo del, corres po nding to prob e affinity (see e.g. Irizarr y et al., 20 03a; Li and W ong , 2001 ). In P ublication 2, prob e-level cal- culation o f differential ex pr ession av oids the need to mo del unidentifiable pr ob e affinities, the key pr ob e-sp ecific par ameter in other prob e-level prepro c essing meth- o ds. More fo rmally , the unidentifiable pr ob e affinity para meters µ . cancel out in RP A when the sig nal log- ratio b etw een a user-s pecified ’reference’ ar ray and the remaining arrays is computed for ea ch probe: the differential expre s sion signal betw een ar rays t = { 1 , . . . , T } and the reference array c f or pro be j is obtained by m tj = s tj − s cj = g t − g c + ε tj − ε cj = d t + ε tj − ε cj . In vector notation, the differential expres sion pro file of pr ob e j across the T arrays is then written a s m j = d + ε j , i.e., a noisy observ ation of the true underlying differe ntial ex pr ession signal d and prob e-sp ecific noise ε j . The unidentifiable pro be affinity parameters ca ncel o ut in the RP A mo de l of Publication 2. This can partly ex plain the previous empirical obs e rv ations that calculating differ en tial express ion already at prob e-level improves the analysis o f differential gene ex pr ession (Zhang et al., 2002 ; Elo et al., 20 05). How ever, t he previous mo de ls ar e non-pro babilistic prepro cess ing metho ds that do not a im at quantifying the uncertaint y in the probes. Use of a single parameter fo r probe effects in RP A also g ives mor e stra ightforw ard interpretations of pro be reliability . Posterior estimates of the mo del para meters are derived to es timate pr o be reliability a nd differential gene expression. The differential e x pression vector d = 38 4.3. MOD EL-BASED NOISE REDUCTION { d t } and the prob e-sp ecific v ariances τ 2 = { τ 2 j } are estimated simult aneously . The po sterior density of the mo del para meters is obtained from the likeliho o d o f the data and the prior according to Bay es’ rule (Equation 3.3) as p ( d , τ 2 | m ) ∼ p ( m | d , τ 2 ) p ( d , τ 2 ) . (4.3) T o obtain this po sterior, let us consider the lik eliho o d p ( m | d , τ 2 ) of the data and the prior p ( d , τ 2 ) of the mo del parameters. The no ise on the selected con trol array ε cj is a la ten t v aria ble, and mar g inalized out in the mo del to obtain the lik eliho o d: p ( m | d , τ 2 ) = Y tj Z N ( m tj | d t − ε cj , τ 2 j ) N ( ε cj | 0 , τ 2 j ) dε cj ∼ Y j (2 π τ 2 j ) − T 2 exp ( − P t ( m tj − d t ) 2 − [ P t ( m tj − d t )] 2 T +1 2 τ 2 j ) . (4.4) Let us a s sume indep endent pr iors, p ( d , τ 2 ) = p ( d ) p ( τ 2 ), flat non-informative prior p ( d ) ∼ 1 and c o njugate prior s for the v aria nce pa rameters in τ 2 (in verse Gamma function, see Gelman et al. 200 3). With these standard assumptions, the prior takes the form p ( d , τ 2 ) ∼ Y j I G ( τ 2 j ; α j , β j ) , (4.5) where α j and β j are the s hape and s cale par a meters of the inverse Gamma dis - tribution. Prio r information of the prob es ca n be incorp or ated in the ana lysis through these par a meters. Prob e-level differen tial expr ession is then de s crib ed by tw o s ets of parameter s; the differential gene express ion vector d = [ d 1 . . . d T ], and the prob e-sp ecific v aria nces τ 2 = [ τ 2 1 . . . τ 2 J ]. High v ariance τ 2 j indicates that the prob e-level observ a tion m j is strongly deviated from the e s timated true sig nal d . Denoting ˆ α j = α j + T 2 and ˆ β j = β j + 1 2 P t ( m tj − d t ) 2 − 1 2 ( P t ( m tj − d t )) 2 T +1 , the po sterior of the mo del parameters in Equation 4.3 takes the form p ( d , τ 2 | m ) ∼ Y j ( τ 2 j ) − ( ˆ α j +1) exp ( − ˆ β j τ 2 j ) . (4.6) The for m ulation allows estimating the uncertaint y in the expr ession estimates and prob e-level parameters. In prac tice, a MAP p o int estimate of the par ameters, obtained by maximizing the p os terior, is o ften sufficient . In the limit of a large sample size ( T → ∞ ), the mo del will conv erge to estimating ordinar y mean and v aria nce parameters . With limited sa mple sizes that are typical in micro a rray studies the prior parameters pr ovide reg ularization tha t makes the probabilistic formulation mo re robust to ov e rfitting and lo cal o ptima, compar ed to direct esti- mation of the mean and v ar iance pa rameters. Moreov er, the probabilis tic ana ly sis takes the uncertaint y in the data and mo del parameter s in to account in a n explicit manner. The mo del also provides a principled framework for incorp or ating prior know- ledge prob e reliability in microar ray prepro cess ing thr ough the pr ob e-sp ecific hy- per parameters α, β . Estimation and use of pro be- spe c ific effects from exter nal microarr ay data collections has b een previously suggested in the con text o f the 39 CHAPTER 4. REDUCING UNCER T AINTY IN HIGH-THROUGHPUT MICRO A RRA Y S TUDIES refRMA metho d by K a tz et al. (2006), w he r e such side information was shown to improv e gene ex pression estima tes . The RP A metho d o f Publication 2 provides an alternative proba bilistic treatment. Mo del v ali dation The proba bilistic RP A mo del introduced in Publicatio n 2 w as v alidated b y compar- ing the prepro ces sing p erfor mance to other prepr o cessing metho ds, a nd addition- ally by compar ing the estimates o f prob e-level noise to known sources of prob e-level contamination. The compa rison metho ds include the F ARMS (Ho chreiter, 2006), MAS5 (Hubb ell et al., 2002), PE CA (Publication 1 ), and RMA (Iriza rry et al., 2003a ) prepro cessing algorithms. F ARMS has a more detailed mo del for probe effects than the other metho ds, and it contains implicitly a similar pr ob e-sp ecific v aria nce par ameter than o ur RP A mo del. F ARMS is based o n a fa c tor analysis mo del, and is defined as s ij = z i λ j + µ j + ε ij , wher e z i captures the underlying gene expr e ssion. In co n trast to RMA and RP A that hav e a single pr ob e-sp ecific parameter, F ARMS has three pro b e-s pecific parameter s { λ j , µ j , ε ij } . MAS5 is a standard prepro ces sing algorithm provided b y the array manuf acturer. The algo- rithm p erfo rms lo cal ba c kground cor rection, utilizes so-ca lle d mismatc h prob es to control for non-sp ecific hybridization, and scales the data from each array to the same average intensit y level to improv e comparability across ar rays. MAS5 sum- marizes prob e-level obser v ations of absolute gene expression lev els using robust summary sta tistics, T ukey biweigh t estimate, but unlik e F ARMS, RMA and RP A, MAS5 do es not mo del pr ob e-sp ecific effects. The prepro ce s sing pe rformance of these metho ds was inv es tig ated in spike-in exp eriments where cer tain targ et trans c r ipts measured by the ar ray ha ve been spiked in a t known concentrations, as well as o n rea l data sets. The results from the spike-in expe riments w ere compared in terms of r eceiver o pe r ating characteris- tics (ROC). The s tandard RMA, P ECA (Publica tio n 1) a nd RP A (Publication 2) had comparable p erformance in spike-in data, and they outp erfor med the MAS5 (Hubbell et al., 20 0 2) a nd F ARMS (Ho chreiter, 2 006) prepr o cessing a lgorithms in estimating differential gene expres sion. On rea l data sets, PECA a nd RP A out- per formed the other metho ds, providing higher repro ducibility b etw een technical replicates measured on different microa rray pla tforms (Figure ?? B). In contrast to s ta ndard prepro ces sing a lgorithms, RP A provides explicit qua n- titative estimates of prob e p e rformance. The mo del has bee n v alidated on widely used human whole- genome a rrays by co mparing the estimates of pr ob e r eliability with k nown prob e-level erro r sources: error s in pro be - genome alignment, int erro- gation p osition of a prob e on the ta rget s equence, GC- conten t, and the presence of SNPs in the prob e targ e t sequences; a go o d mo del for assess ing pr ob e relia bil- it y should detect prob es con taminated by the kno wn error so urces. The r esults from our analy s is ca n b e used to characterize the relativ e contribution of differ- ent sources of prob e-level noise (Figure ?? B). In g eneral, the prob es with known sources of contamination were more noisy than the other prob es, with 7-39% in- crease in the av erage v ariance, a s detected b y RP A. An y single source of error seems to e xplain only a fraction o f the most highly c o nt aminated prob es. A large po rtion (35- 60%) of the detec ted least r eliable pro bes were not a s so ciated with the inv es tig ated known noise so urces. This suggests that previous methods that r e- mov e prob e-level noise based on external information, such as g enomic a lignments will fail to detect a sig nificant p ortion of p o orly p erforming pro bes . The RP A 40 4.4. CON CLUSION mo del o f Publica tion 2 provides rigo rous alg o rithmic to ols to investigate the v ari- ous prob e-level erro r sour ces. Better under standing of the factors affecting prob e per formance can adv anc e pro be design and contribute to re ducing pr ob e-related noise in future genera tio ns of gene expressio n ar rays. 4.4 Conclus ion The contributions presented in this Chapter provide improv ed prepro cessing strate- gies for differ en tial g ene expression studies. The introduced techniques utilize prob e-level analysis, as well as side information in sequenc e and microa rray data- bases. Pro be- level s tudies hav e led to the establishment o f prob e verification and alternative microarray interpretations as a standard step in microarr ay prepro- cessing and a nalysis. The alterna tiv e interpretations for microar ray data based on upda ted geno mic sequence data (Gautier et al., 2 004; Dai et a l., 2005 ) are now im- plement ed a s routine to ols in p opular pr epro cessing algor ithms such as the RMA, or the RP A metho d of Publication 2. The pro be- le vel analy s is strategy has b een recently extended to exo n a rray con text, where express ion levels of alternative splice v ariants of the sa me ge nes are compare d under pa rticular exp er imen tal co n- ditions. The prob e-level a pproach has shown sup erior pr epro cessing per fo rmance also with ex on ar rays (Laa ja la et al., 20 09). A conv enien t access to the algo- rithmic to ols developed in P ublications 1 and 2 for micro array prepro cess ing a nd prob e-level analysis is pr ovided b y the accompanied op en sour c e implementation in BioConductor . 1 1 h ttp://www.bioconductor.org/pac k ages/release/bioc/html/RP A.html 41 Chapter 5 Global anal ysis of the h uman transcriptome When we tr y to pick out anything by itself, we find that it is b oun d fast by a thousand i nvisible c or ds that c annot b e br oken, to every thing in t he universe. J. Muir (1869) Measurements of tra nscriptional activity provide only a par tial vie w to physio- logical pro cesses, but their wide av aila bilit y provides a unique reso urce for inv esti- gating g ene activity at a genome- a nd organism- w ide sca le. V ersatile and ca refully controlled gene expr ession atlases hav e beco me a v ailable for no rmal human tissues, cancer as well as for other diseases (see , for instance, Kilpinen et al., 200 8 ; Lukk et al., 2010 ; Roth et al., 200 6; Su et a l., 2004). These da ta sources con tain v aluable information ab out share d and unique mechanisms b et w een dispa rate conditions, which is not av aila ble in smalle r and mo re sp ecific exp eriments (Lage et al., 20 08; Scherf et al., 20 00). While standard meth o ds for gene expressio n analysis have fo cused on comparisons b etw een particular conditions, versatile transcr iptome at- lases allow for g lo bal organism-w ide characteriza tio n of tr anscriptional activ ation patterns (Levine et al., 2 006). Nov el methodologic a l approaches are needed in order to realize the full p otential o f these information sources, as ma n y tradi- tional metho ds for expres sion analys is ar e not applicable to versatile lar ge-scale collections. This chapter provides an ov er view to current appr oaches fo r global transcriptome analysis in Section 5.1 and introduces the second main contribution of the thesis, a novel explo ratory approach that ca n be us ed to inv es tigate con text- sp ecific res po nses in genome-sca le interaction net w orks a c r oss orga nism-wide col- lections of measur ement data in Section 5.2. The conclusions ar e summariz ed in Section 5.3. 5.1 S tandard approac hes Global observ ations of tra nscriptional a c tivit y reflect known and previously un- characterized cell- biological pr o cesses. Exploratory analysis of the tra ns criptome can provide resear ch h ypotheses and materia l for more deta iled inv estigations. 42 5.1. S T AN DARD APPRO A CHES Widely-used standard approaches for globa l transcr iptome analys is include v ar ious clustering, dimensiona lity r e ductio n and visualization techniques (see e.g. Hutten- how e r and Hofmann, 2010 ; Polanski a nd Kimmel, 2007; Quack en bush, 2001). The large data collec tio ns op en up new p oss ibilities to inv estigate functional related- ness betw een physiologica l conditions, disease s tates, as w ell a s ce llula r pro ce s ses, and to dis cov er pre v iously unc haracterized co nnec tions a nd functional mechanisms (Bergmann et al., 2004 ; Kilpinen et al., 20 08; Lukk et al., 20 1 0). Gene expression studies hav e traditionally fo cused on the analy sis o f relatively small and tar g eted data sets, such as pa rticular diseases or cell types . A typical ob- jective is to detect genes, or gene g roups, that are different ially expresse d b etw een particular conditions, for insta nce to predict disea s e outcomes, or to identify po ten- tially unknown disea se subtypes. The incr easing av aila bilit y of larg e and versatile transcriptome collections that may cover thousands of exp e rimental conditions al- lows g lobal, data-dr iven analysis, and the formulation o f nov el resear ch questions where the tr aditional analysis metho ds a re often insufficient (Huttenho w er and Hofmann, 2010 ). A v ariety of a pproaches hav e been prop osed and inv estigated in the recent years in the glo bal tra nscriptome ana lysis co n text. An actively studied mo deling problem in transcriptome ana lysis is the disc overy of tr anscriptio nal mo du les , i.e., ident ification of coherent g ene groups tha t show c o o rdinated transcriptional re- sp onses under particular conditions (Segal et al., 20 03a, 200 4; Stuart et al., 20 0 3). Mo dels have also b een pr op o sed to predict gene r e g ulators (Segal et a l., 20 03b), and to infer cellular pro cesses and net works based on transcriptional activ ation patterns (F riedman, 2 004; Seg al et al., 2 003c). An increasing n um ber of mo dels are b eing developed to integrate tra nscriptome measur ement s to other sources o f genomic information, such as r egulation and interactions be tw een the genes to detect and characterize cellular pro cess es and disease mechanisms (Baras h a nd F riedman, 2002; Chari et a l., 2010 ; V a ske et al., 2010). Findings from tra nscrip- tome analysis ha v e p otential biomedical implica tions, as in Lam b e t al. (2006 ), where chemically per tur bed canc e r cell lines were screened to enhance the detec- tion of drug targ e ts based o n share d functional mech anisms b etw ee n dispa rate conditions, or in Sørlie et al. (200 1), whe r e cluster ana lysis o f cancer patients based on genome-wide tra nscriptional pr ofiling exp eriments led to the discovery of a novel br east cancer subt ype . In the remainder o f this s e c tion, the mo deling approaches that are particularly clo s ely r elated to the contributions o f this thesis are considered in more detail. In v esti g ating known pro cesses A p opular strategy for geno me-wide g ene expr ession analysis is to consider known biological pro ces ses and their activ ation patterns across diverse colle c tio ns mea- surement data fro m v a rious ex per imen tal conditions. Bio medical databases con- tain a v a riety of informa tion concerning genes and their int eractions. F or in- stance, the Gene Ontology da ta base (Ashburner et al., 200 0) provides functional and mo lecular classifications for the g enes in human and a num b er of other org a n- isms. O ther categ ories are based on micr o-RNA reg ulation, chromosomal lo cations, chemical p erturbatio ns and other featur es (Subramania n et al., 2 005). Join t anal- ysis of functionally rela ted genes c a n increase the statistical p ow er of the a nalysis. So-called gene set - b ase d appr o aches are typically designe d to test differ e n tial ex- pression betw een tw o particular conditions (Go e man a nd Buhlmann, 2 007; Nam 43 CHAPTER 5. GLOBAL ANA L Y SIS OF THE HUMAN TRANSCRIPTOME and Kim, 2008), but they can als o be used to build g lo bal maps of tra nscriptional activity o f the known process es (Levine et al., 2006). How ev er, gene set-based approaches typically igno re more detailed informatio n of the interactions b etw ee n individual genes. Path w a y and interaction data bases contain more detailed infor - mation concerning molecula r interactions and ce ll- biological pro c esses (Ka nehisa et al., 2 0 08; V as trik et al., 20 07). Network-b ase d metho ds utilize r elational infor- mation of the genes to guide e x pression analysis. F o r instance, Draghici et al. (2007) demonstra ted that taking into account asp ects of pathw ay top o logy , such as g e ne and interaction types, can improve the e s timation of pathw a y a ctivity b e- t ween tw o predefined conditions. Another recent appro a ch which utilizes pathw a y top ology in inferring path wa y activity is P ARADIGM (V aske et al., 201 0 ), which also integrates other sources o f genomic infor mation in path w a y a nalysis. Ho w- ever, these metho ds hav e been designed for the analysis of particular exp er imen tal conditions, r ather than comprehensive ex pression atlas es. MA TISSE (Ulitsky and Shamir, 2007 ) is a netw ork-bas ed approach that se a rches fo r functionally rela ted genes that a re connected in the netw o r k, a nd have corr elated express ion profiles across many conditions. The p otential shortcoming of this approa ch is that it a s - sumes global co rrelation across all conditions b etw een the interacting ge ne s , while many genes ca n hav e multiple, context-sensitiv e functional roles. Different condi- tions induce different resp onses in the same genes, a nd the definition of ’g ene set’ is v ague (Montaner et al., 20 09; Nacu et al., 200 7). Therefore metho ds have been suggested to identify ’key condition-r esp onsive genes’ of pre defined ge ne sets (Lee et al., 2008), or to decomp ose predefined pathw a ys into smaller and mo r e sp ecific functional mo dules (Chang et al., 2009 ). These appr o aches rely on predefined functional cla ssifications for the g enes. The data-driven analysis in Publication 3 provides a complementary approach wher e the gene sets are lear ned directly from the data, guided by prior knowledge of genetic in teractions. This avoids the need to r efine sub optimal anno tations, and enables the discov ery of ne w pro cess es. The findings demons trate that simply mea suring whether a gene set, or a netw ork, is differentially ex pressed betw een particular conditions is often no t s ufficien t for measuring the activity of cell-biolo gical pro cesses . Since gene function and inter- actions ar e r egulated in a co nt ext-sp ecific manner, it is impor tant to additiona lly characterize how, and in which conditio ns the expre s sion changes. Global analy s is of transc riptional activ ation patterns interaction netw o rks, in tro duced in Publica- tion 3, can address such questions. Biclustering and subs p ace clustering Approaches that a re based on previously c ha r acterized genes and proc e sses are biased tow ards well-ch aracter iz e d phenomena. This limits their v alue in de nov o discov ery of functional patterns. Unsup ervised metho ds provide too ls for such analysis, but o ften with an increased computationa l co st and a higher pro po rtion of false p o sitive findings. Cluster analysis is widely used fo r unsup ervised analysis of g ene expressio n data, providing tools for class discov ery , gene function prediction and for visualiza- tion purp oses. Examples of widely used clustering approaches include hierar chical clustering a nd K-means (see e.g. Polanski and Kimmel, 2 0 07). Clustering of pa- tien t samples with similar expression profiles has led to the discov ery of novel can- cer subtypes with biomedica l implications (Sørlie et al., 20 01); cluster ing of genes with c o o rdinated activ ation patterns can b e used, for instance, to pr edict no vel 44 5.1. S T AN DARD APPRO A CHES functional asso ciations for p o orly characterized g enes (Allo cco et al., 2004). The self-orga nizing map (Kohonen, 1982, 2001) is a r elated approach that provides e ffi- cient to ols to visualize high-dimensional data on low er-dimensional displays, with particular applications in transcr iptional profiling studies (T amay o et al., 1999; T¨ or¨ onen et al., 1 9 99). The standard clustering metho ds ar e ba sed on compariso n of glo bal expressio n patterns, and therefor e are r elatively co arse to ols for a nalyz- ing larg e transcriptome collections. Different genes resp ond in different w ays, a s well a s in different co nditio ns. Therefore it is problematic to find clus ters in high- dimensional data space s , such as in who le-genome expressio n pr ofiling studies ; different gene gro ups can reveal different relationships b etw een the s amples. De- tection of sma lle r, coher en t subspaces with a par ticular str ucture can b e useful in biomedical applications , where the ob jectiv e is to identif y sets of interesting g enes for further analysis . Both genes a nd the a sso ciated conditions may b e unknown, and the learning ta s k is to detect them from the data. This can help, for instance, in identif ying res p ons es to drug treatments in particular genes (Ihmels et a l., 2002; T anay et al., 2002 ), or in identifying functionally coherent tra nscriptional mo dules in gene expressio n databases (Segal et al., 200 4; T anay et al., 2005). Subsp ac e clustering metho ds (Parsons et al., 2004) provide a family of a lgo- rithms that can b e used to identify subse ts of dependent features revealing coher- ent clustering for the samples; this defines a subspa ce in the original feature space. Subspace cluster ing mode ls ar e a sp ecial case of a more ge neral family of biclus- tering alg orithms (Madeira and O liveira, 2004). Closely related mo dels are also called co-cluster ing (Cho et al., 2004 ), two-w a y clustering Gad et al. (2000 ), a nd plaid mo dels (La zzeroni and O wen, 2002). Biclustering metho ds provide genera l to ols to detect co-reg ulated gene gr o ups and asso cia ted co nditio ns fro m the da ta, to provide compact summaries and to aid in terpretation of tra nscriptome data collections. Bicluster ing mo dels enable the discov ery of gene expr ession signa- tur es (Hu et al., 2006) that hav e emerged as a cent ral concept in global expressio n analysis context. A signa tur e describ es a co-e xpression state of the g enes, a sso- ciated with pa r ticular co nditions. Established signatures hav e b een fo und to be reliable indicators of the ph ysio logical state of a cell, and commer c ial signatur es hav e become av ailable for ro utine clinical practice (Nuyten a nd v an de Vijver, 2008). How ever, the establis hed signatures are t ypically desig ned to provide op- timal classification p erformance b et w een t w o par ticular conditions. The problem with the c la ssification-ba s ed signatures is that their asso ciations to the underly ing ph ysiologic a l pro ces ses ar e no t well understo o d (Lucas et al., 20 09). In Publica- tion 3 the under standing is enhanced b y der iving trans c riptional signature s tha t are explicitly connected to well-c haracterized pro ces ses thro ug h the netw ork. Role of side information Standard cluster ing mo dels ig nore prior infor mation of the data, which c ould b e used to super vise the analysis, to connect the findings to known pro cesses, as w ell as to improve sca lability . F or insta nce, standar d model- based feature s election, or subspace cluster ing techniques would consider all po ten tial connec tions b etw een the genes or features (Law et a l., 200 4; Roth and La nge, 2 004). Without addi- tional constraints o n the solution space they can t y pically handle at most tens or h undreds of features, which is often insufficient in high-throughput g enomics applications. Use of side information in cluster ing can help to guide unsupe rvised analysis, for instance based on k nown or p otential interactions b etw e en the genes. 45 CHAPTER 5. GLOBAL ANA L Y SIS OF THE HUMAN TRANSCRIPTOME This has bee n shown to improv e the detec tio n of functionally coherent gene gr oups (Hanisch et al., 2002; Shiga et al., 2007; Ulitsky and Shamir, 2007; Zh u et al., 2005). How ev er, while these metho ds provide to ols to cluster the genes, they do not mo del diff erences betw een co nditio ns. Extensions of biclustering models that can utilize relational information of the genes include cMonkey (Reiss et al., 2006) and a mo dified version of SAMBA biclustering (T anay et al., 200 4). How- ever, cMo nkey and SAMBA are application-or iented too ls that rely on additional, organis m-sp e cific informa tion, and their implemen tation is currently not av aila ble for most or ganisms, including that of the human. F urther application-o riented mo dels for utilizing side information in the disco v ery of transcriptional mo dules hav e r ecently b een prop osed fo r instance b y Sav age et al. (2010) and Suthram et al. (2010). Publication 3 in tro duces a c o mplemen tary metho d where the exhaustively large search space is limited with side information concer ning known rela tions b e- t ween the genes, derived from genomic interaction databases . This is a general algorithmic approach whose applicability is no t limited to pa rticular organisms. Other approac hes Prior information on the cellular net works, regulatory mechanisms, and ge ne func- tion is often av aila ble, and can help to co nstruct more detailed mo dels of gene function and netw ork analysis, as well as to summarize functional asp ects of ge- nomic data collections (Huttenhow er et a l., 200 9; Segal et a l., 2003 b; T roy ansk aya, 2005). V ersatile trans criptome colle ctions also enable network r e c onstruct ion , i.e., de novo discov ery (Lezon et a l., 200 6; Myers et a l., 2 005) and augmen tation (Nov ak and Jain, 2006) of genetic in teraction net works. Other metho dologica l appr oaches for global tra nscriptome analys is are provided by pr obabilistic laten t v ariable mo d- els (Rog ers et al., 200 5; Segal et al., 2 0 03a), hierarchical Dirichlet pro cess algo- rithms (Gerb er et al., 2 007), as well as matrix and tensor computations (Alter and Golub, 2005 ). These metho ds pr ovide further mo del-based to ols to identify and characterize tr anscriptional programs b y decomp os ing gene express io n data sets int o smaller, functionally cohere n t comp onents. 5.2 G lobal mo deling of transcriptional activit y in in teraction net w orks Molecular interaction net w orks cover tho usands of ge nes, proteins a nd small mo- lecules. C o o rdinated reg ulation of gene function through molecula r in teractions determines cell function, and is reflected in tr a nscriptional activity of the genes. Since individual proce s ses and their transcriptional resp onses are in general un- known (Lee et al., 2008; Mon taner et a l., 2 0 09), data-driven detection of condition- sp ecific resp onses can provide an efficient proxy for identifying distinct transcr ipt- ional states of the netw o rk with p otentially distinct functional r oles. While a nu m ber o f metho ds hav e b een pro po sed to co mpa re netw ork activ ation patterns betw een pa rticular conditions (Draghici et al., 20 0 7; Ideker et a l., 2 002; Cabusora et a l., 200 5 ; Noirel et al., 2008), or to use netw ork information to detect function- ally r e lated g ene gro ups (Segal et al., 2003 d; Shiga et al., 2 007; Ulitsky and Shamir, 2007), genera l-purp ose alg orithms for a global analysis of context-specific netw ork activ ation patterns in a genome- and or ganism-wide s cale hav e b een missing. 46 5.2. GLOBA L MODELING OF TRANSCRIPTIONA L ACTIVITY IN INTERACTION N ETW ORKS Figure 5.1: Organism-wi de analysis of transcriptional responses i n a human path w a y inte raction net w ork revea ls ph ysiologically coheren t activ ation patterns a nd condition-specific regulation. One of the subnet w orks and its condition-specific resp onses, a s detected by the NetResp onse algorithm i s sho wn in the Fi gur e. The expressi on of eac h gene is vis ualized with resp ect to its mean level of expression across all s ampl es. c The Author 2010. Published b y Oxford Universit y Press. Reprinted with p ermis s ion from Publication 3. Publication 3 int ro duces and v alida tes t w o ge ne r al-purp ose a lgorithms that provide to ols for global mo deling o f transcriptiona l resp onses in interaction net- works. The mo tiv ation is similar to biclustering approa c hes that detect function- ally coherent ge ne g roups that show coordina ted resp ons e in a subset of condi- tions (Madeira and Oliveira, 2004 ). T he net w ork ties the findings more tightly to cell-biologic a l pro cesses, fo cusing the ana lysis and improving interpretability . In contrast to previous net work-based biclustering models for glo ba l transcripto me analysis, such as cMonkey (Reis s et al., 20 06) o r SAMBA (T anay et al., 2004), the a lgorithms intro duced in Publication 3 ar e gener al-purp ose to ols, and do not depe nd on orga nis m-sp ecific annotations. A tw o-step approac h The fir st a pproach in Publicatio n 3 is a straig h tforward extension of net w ork-based gene clustering metho ds. In this tw o-step approach, the functionally coherent sub- net works, and their condition-sp ecific resp onses a re detected in separate steps. In the first step, a netw ork-based clustering metho d is used to detect functiona lly co - herent subnetw o rks. In Publication 3, MA TISSE, a state- of-the-art algorithm de- scrib ed in Ulitsky and Shamir (2007), is used to detect the subnetw orks. MA TISSE finds connected subgraphs in the netw ork tha t hav e high internal corr elations b e- t ween the genes. In the second step, condition-sp ecific r e spo nses of each iden tified subnetw or k ar e sea rched for by a nonpa rametric Gaus sian mixture mo del, which allows a data-driven detectio n of the resp onse s . Howev er, the t w o-step approach, coined MA TISSE+, can b e sub optimal for detecting subnet works with particular condition-sp ecific resp onses. The main contribution of Publicatio n 3 is to intro- duce a second g eneral-purp os e alg o rithm, coined NetResponse, where the detection of condition-sp ecific r esp o nses is used as the explicit key criterion for subnetw ork search. 47 CHAPTER 5. GLOBAL ANA L Y SIS OF THE HUMAN TRANSCRIPTOME The NetR esp onse algorithm The netw ork-based search pro c e dur e in tro duced in Publica tion 3 sear ch es for lo- cal subnetworks , i.e., functionally coherent net w ork modules where the interacting genes show co or dinated res po nses in a subset of conditions (Figur e 5.1 ). Side in- formation of the gene interactions is us ed to guide mo deling, but the algorithm is indep endent o f predefined class ific a tions for genes o r mea surement co nditions. T rans criptional res po ns es of the netw ork ar e describ ed in terms of subnetw o rk activ ation. Reg ulation of the s ubnet w ork g e nes can inv olv e s im ultaneous activ a- tion and repressio n of the genes: sufficie nt amounts of mRNA for key proteins has to be av ailable while interfering genes ma y need to b e silenced. The model assumes that a giv en subnetw ork n can have multiple transc riptional states, as- so ciated with different physiological co n texts. A transcr iptional s tate is r eflected in a unique expr ession signatur e s ( n ) , a vector that descr ibe s the expr ession levels of the subnetw o rk g e nes, a sso ciated with the pa rticular transcr iptional state. Ex- pression of so me genes is regula ted at pr ecise lev els, whereas other genes fluctuate more freely . Given the sta te, e xpression o f the subnetw ork genes is mo deled as a noisy obser v ation of the transcr iptional state. With a Gaussian noise mo del with cov a riance Σ ( n ) , the obser v ation is describ ed b y x ( n ) ∼ N ( s ( n ) , Σ ( n ) ). A given sub- net work can hav e R ( n ) latent transcriptiona l states indexed by r . In practice, the states, including their n um ber R ( n ) , are unknown, a nd they hav e to b e estimated from the data. In a sp ecific measurement co ndition, the subnetw ork n can be in any one of the laten t ph ysiologica l states indexed by r . Asso ciations b etw een the observ ations and the underlying transcr iptional states a re unknown and they are treated as la ten t v a riables. Gene expres sion in subnet work n is then mo deled with a Gaussian mixture mo del: x ( n ) ∼ R ( n ) X r =1 w ( n ) r p ( x ( n ) | θ r ) , (5.1) where each co mponent distribution p is a ssumed to be Gaussian with para meters θ r = { s ( n ) r , Σ ( n ) r } . In prac tice , we assume a diag onal cov aria nc e matr ix Σ ( n ) r , leav- ing the dep e ndenc ie s b etw e en the g enes unmo deled within each transcriptional state. Use o f diagonal cov a riances is justified by consider able ga ins in computa- tional efficiency when the detection of distinct resp onses is of pr imary interest. It is p ossible , how ev er, that such simplified mo del will fail to detect certain subnet- works wher e the transcr iptio nal le vels of the genes hav e s trong linear dep endencies within the individual transcr iptio nal states; signaling cascades could b e e xpec ted to manifest such activ ation patterns, for insta nc e . More detailed mo de ls o f tr an- scriptional a ctivit y could help to disting uish the individual states in particular when the tra nscriptional states a re partially overlapping, but with increa s ed com- putational cos t. A particular transcriptiona l resp onse is then characterized with the triple { s ( n ) r , Σ ( n ) r , w ( n ) r } . This defines the shap e, fluctuatio ns a nd frequency of the associa ted t ranscriptiona l sta te of subnetw ork n . A p oster ior proba bilit y of each latent state can b e calculated for each mea surement sample from the Bayes’ rule (Equa tion 3.3). The p os ter ior proba bilities can be in terpreted a s soft co mpo- nent mem ber ships for the sa mples. A hard, deterministic assignment is obtained by selecting for ea ch sample the comp onent with the highest pos terior pr obability . The r emaining task is to identify the subnet works having such distinct tran- scriptional states. Det ection of the distinct states is now use d as a sea rch criterion 48 5.2. GLOBA L MODELING OF TRANSCRIPTIONA L ACTIVITY IN INTERACTION N ETW ORKS for the subnetw orks. In order to achiev e fast computation, an agg lomerative pr o ce- dure is used wher e in teracting genes are gradua lly mer ged int o larger subnetw orks. Initially , each gene is assigned in its o wn singleto n s ubnet w ork. Agglomer ation pro ceeds b y a t ea ch step merging t he t w o neig h bo ring subnet works where joint mo deling of the genes lea ds to the highest impr ov e ment in the ob jective function v alue. Joint modeling of dependent genes reveals co o rdinated res p ons es and im- prov es the likeliho o d o f the data in compariso n with indep endent mo dels, giving the first criterion for merging the subnetw orks. How ev er, increasing subnetw or k size tends to increase model complexity and the p oss ibilit y of ov erfitting, since the nu m ber of samples remains c o nstant while t he dim ensionality (subnetw ork size) increases. T o comp ensate for this effect, the Bayesian information criterio n (see Gelman et al., 2003) is us ed to p enalize increasing mo del complexity and to de- termine optimal subnetw ork size. T he final cost function for a subnet work G is C ( G ) = − 2 L + q l og ( N ), where L is the (marginal) log - likelihoo d of the data, given the mixture mo del in Eq ua tion 5.1, q is the n um ber of pa r ameters and N denotes sample size. The algorithm then compa res indep endent a nd joint mo dels for each subnetw or k pair that has a direct link in the netw ork, and merg es at each step the subnetw or k pair G i , G j that minimizes the cost ∆ C = − 2( L i,j − ( L i + L j )) + ( q i,j − ( q i + q j )) log ( N ) . (5.2) The iter ation contin ues unt il no improv emen t is o btained by mer ging the sub- net works. The combination o f mo deling techniques yields a sca lable algor ithm for genome- and org anism-wide in v estigations: First, the a nalysis fo cuses on those parts o f the data that a re supp orted by known interactions, which increase s mod- eling p ow er and co ns iderably limits the search s pace. Second, the agglomer ative scheme finds a fast a ppr oximativ e solution where at each step the subnetw ork pair that leads to the highest improv emen t in cost function is merg e d. Third, an ef- ficient v ariational appr oximation is used to le arn the mixture mo dels (Kurihara et al., 2 007b). Note that the algo rithm does not necessarily identify a glo bally optimal so lutio n. Ho w ev er, detection of physiologically coherent and r e pro ducible resp onses is often sufficient for practical applicatio ns. Global view on net w ork activ ation patterns The NetResp onse algor ithm in troduced in P ublication 3 was a pplied to inv es tig ate transcriptiona l activ atio n patterns o f a pathw ay interaction netw ork of 18 00 genes based on the KEGG databas e of metab olic pathw ays (Kanehisa et a l., 2008 ) pro- vided by the SP IA pack age (T a rca et a l., 2009 ) across 353 g ene express ion samples from 65 tissues. The tw o algo rithms pro po sed in Publica tio n 3 , MA TISSE+ and NetRespo nse w ere shown to outp erform a n unsup ervised biclustering approach in terms of repro ducibility of the finding. The in tro duced NetRepo nse algor ithm, where the de tec tio n of tra nscriptional resp onse pa tterns is used as a sea rch crite- rion for subnetw ork identification, w as the b est-p erfo rming metho d. The algorithm ident ified 10 6 subnetw orks with 3- 20 genes, with distinct transc riptional resp onses across the conditions . O ne of the subnetw orks is illustrated in Figur e 5.1; the other findings ar e provided in the supplement ary materia l o f Publica tion 3. The detected trans criptional r esp onses were physiologically co her ent, sugge sting a p o- ten tial functional role. The r epro ducibility o f the re s po nses was confir med in a n independent v a lidation data set, where 8 0% of the pr edicted res po nses were de- tected ( p < 0 . 05 ). The findings highlight context-specific re g ulation of the g enes. 49 CHAPTER 5. GLOBAL ANA L Y SIS OF THE HUMAN TRANSCRIPTOME Some resp onses are shared by many conditions , while others are more s pecific to particular contexts such as the imm une system, muscles, or the brain; rela ted ph ysiologic a l co nditions often exhibit similar netw ork activ a tion patterns . Tiss ue relatedness ca n b e measured in terms of sha red transcr iptional resp onse s of the subnetw or ks, giving an a lternative formulation o f the tissue connectome map sug- gested b y Grec o et al. (2008 ) in or der to highlig ht functional connec tivit y b etw een tissues based on the num ber of shared differentially expr essed genes. In P ublica- tion 3, shared netw ork resp onses are used instead of shared gene count. The use of co-reg ula ted gene g roups is exp ected to b e mor e robust to noise than the us e of individual g enes. The analysis provides a globa l view on netw o rk activ ation acr oss the nor mal h uman b o dy , and ca n b e used to formulate novel hypotheses of gene function in previously unexplored contexts. 5.3 Conclus ion Gene function and interactions a re often sub ject to condition- s pec ific regulation (Liang et al., 2006; Ra ch lin et al., 2006), but these hav e been typically studied only in pa rticular exp erimental conditions. Org anism-wide analysis can p oten- tially reveal new functional co nnections and help to formulate novel hypotheses of gene function in previously unexplo red co n texts, and to detect highly sp ecialized functions that a r e sp ecific to few co nditions. Changes in c ell-biologica l condi- tions induce changes in the expres sion lev els of co -regulated genes, in order to pro duce specific physiological responses, typically affecting o nly a small part of the net work. Since individual pro ce sses and their transcriptiona l r esp onses a re in general unknown (Lee et al., 20 08; Montaner et al., 20 09), data-driven detectio n of condition-sp ecific resp onses can provide an efficien t pr oxy for identifying distinct transcriptiona l states of the netw ork, with p otentially distinct functional roles. Publication 3 provides efficient mo del- based to ols for global, or ganism-wide dis- cov ery and character iz a tion o f context-sp ecific trans c riptional activity in genome- scale in teraction netw orks, indep endently o f predefined classifica tions for genes and c o nditions. The netw ork is used to bring in prior information of gene func- tion, which would b e missing in unsup ervised mo dels, and allows da ta -driven de- tection of co o rdinately regulated gene sets a nd their context-specific r esp onses. The algorithm is readily applicable in any or g anism wher e gene expre ssion a nd pairwise interaction data, including pa th w ays, pro tein in teractions and regulatory net works, are av ailable. It has therefore a considerably larg er scop e than pr evious net work-based models fo r glo ba l trans criptome analys is, which r e ly on or ganism- sp ecific annotations, but lack implemen tations for most organisms (Reiss et al., 2006; T anay et al., 200 4). While biomedical implications of the findings req uir e further investigation, the results highlight shar ed a nd repro ducible resp onses be t w een physiological condi- tions, and provide a global view of transcriptiona l activ a tion patterns across the normal human b o dy . Other po ten tial a pplica tions for the metho d include larg e- scale screening of dr ug r esp onses and disease subtype discovery . Implementation of the algor ithm is freely av ailable thr ough BioConductor. 1 1 h ttp://bioconductor.org/pac k ages/dev el/bioc/html/netresponse.html 50 Chapter 6 Human transcriptome and other la y ers of genomic information The way to de al with the pr oblem of big data is to b e at it sen s eless with other big data. J. Quack en bush (2006) This chapter pre s ent s the third main contribution of the thesis, co mputational strategies to in teg rate mea surements of h uman tr anscriptome to o ther la y ers of genomic informa tion. Geno mic, tra nscriptomic, pro teomic, epigeno mic and o ther sources of measurement data characterize differ en t asp ects of genome o rganiza - tion (Hawkins et al., 2010; Montaner and D opazo, 2 0 10; Sara e t al., 201 0 ); an y single source provides only a limited view to the cellular system. Understanding functional orga nization of the genome and ultimately the cell function requires int egration of data from the v ar ious levels of genome org anization and mo del- ing of their dynamical interplay . Such an holistic approa ch, which is also called systems biolo gy , is a key to understanding living o rganisms, which are “rich in emergent prop erties b ecause fo rever new groups of pro p er ties e merge a t every level of int egration” (Ma yr, 2004). Combining evidence acro ss multiple sources can help to discov er functiona l mechanisms and interactions, which a re not seen in the individual data sets, and to increase statistical p ow er in nois y a nd inc o m- plete high- throughput exp eriments (Huttenho wer and Hofmann, 201 0; Reed et a l., 2006). Int egration o f heterog eneous genomic data comes with a v ariety o f technical and metho dolo gical challenges (Hwang et al., 2005; T roy ansk ay a, 20 05), and the particular mo deling a pproaches v ary acco rding to the analysis ta sk and particular prop erties of the inv estigated measurement s ources. In tegrative studies have b een limited by p o or av a ilability of co-o ccur r ing g e no mic obser v ations, but suitable data s e ts a re now b ecoming increasing ly av ailable in b oth in-house and public biomedical data r epo sitories (The Cancer Ge no me Atlas Research Netw o rk, 2008 ). New observ atio ns hig hlight the need for nov el, in tegrative appro aches in functional genomics (Co e et al., 200 8). Recent studies hav e prop osed for instance metho ds to int egrate epigenetic mo difications (Sadiko vic et al., 2 008), micro-RNA (Qin, 200 8), 51 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION transcription factor binding (Sav age et al., 20 10), a s w ell as protein expressio n (Johnson et al., 20 08). Given the complex sto chastic nature of biological sys tems, computational efficiency , r obustness against uncer taint y and int erpretability of the results are k ey issues. Prior infor ma tion o f biolo gical s ystems is o ften incomplete, and sub ject to high levels o f uncontrolled v ariation and complex interdependencies betw een differe nt parts of the cellular system (T royansk ay a , 20 0 5). These issues emphasize the need for principled appro a ches requiring minimal prior k nowledge ab out the data , as w ell as minimal mo del fitting pr o cedures. Sec tio n 6.1 gives an ov erview of the standar d mo dels for high-throug hput da ta integration metho ds, which hav e close connections to the mo deling appr oaches develop ed in this work. 6.1 S tandard appr oac hes for genomic data in te- gration The in tegrative appro aches can be ro ug hly classified in thre e categ ories: meth- o ds that (i) combine s tatistical ev idenc e a cross related studies in order to obtain more accura te infer ences of ta r get v aria bles, (ii) utilize side informa tion in or der to g uide the analysis o f a single, primar y data sourc e , and (iii) detect and char- acterize depe ndencie s betw een the measurement sourc es in order to discover new functional connections be tw een the differ e nt lay ers o f geno mic infor mation. The contributions in Chapters 4 and 5 are asso ciated with the first t w o categor ie s; the contributions presented in this chapter, the regula r ized dep e nde ncy detection fra- mework of Publication 4, and asso ciative clustering of Publications 5 a nd 6 , b elong to the third category . 6.1.1 Com binin g statistical evidence The fir st gener a l category o f metho ds for geno mic data in tegration c onsists o f ap- proaches where evidence acro ss similar studies is combined to increa se statistica l power, for instance by comparing and in tegrating data from indep endent micr oar- ray exp eriments targeted at studying the same disease. In Publications 2 a nd 3, joint ana lysis of a larg e num ber of commensurable micr oarray exp eriments, w her e the obser ved data is directly comparable b etw e en the arrays, helps to increase statistical p ow er a nd to reveal weak, shared signa ls in the d ata that can not b e detected in more restricted exp erimental setups and sma ller datas ets. How ever, the related obser v ations a re o ften not dire c tly compar a ble, and fur- ther metho dological to ols ar e needed for integration. M eta-analysis provides to ols for s uc h a nalysis (Ramas amy et a l., 2 008). Meta-analy sis forms part of the microa r - ray a nalysis pro ce dur e intro duce d in Publica tion 1, where methods to integrate related microar ray measurements acr oss different ar ray platfor ms ar e develop ed. Meta-analys is emphasizes shared effects b etw een the studies ov er sta tistical sig - nificance in individua l e x per imen ts. In its standard form, meta-ana lysis a ssumes that eac h individual study mea sures the same t arget v ariable with v arying lev- els of noise. The analysis starts fro m identifying a measure of effe ct size based on differences, means , or other summar y statistics o f the observ ations such as the Hedges’ g , used in P ublication 1. W eighted av eraging of the effect sizes pr o- vides the final, co m bined re sult. W eig ht ing a ccounts for differences in reliability of the individual studies, for instance b y emphasizing studies with la r ge sample size, or low measur ement v ar iance. Averaging is ex pected to yield mo re a ccurate 52 6.1. S T AN DARD APPRO A CHES FOR GENOMIC DA T A INTEGRA TION estimates o f the tar get v a riable than individual studies. This ca n b e particular ly useful when se veral studies with small sample sizes are av ailable for instance from different lab or atories, which is a co mmon setting in microarr ay ana lysis context, where the data sets pr o duce d by individual la bo ratories ar e routinely dep osited to s hared communit y databa ses. Ultimately , the quality of meta- a nalysis results rests on the quality of the individua l studies. Modeling choices, such as the choice of the effect size measure and included studies will affect the analys is outcome. Kernel metho ds (see e.g . Sch¨ olk opf and Smola, 2 002) provide ano ther widely used approach for integrating statistical evidence a cross multiple, potentially het- erogeneo us measurement sourc es. Kernel metho ds op erate on similarity matrices, and provide a natural fr amework fo r combining statistical evidence to detect sim- ilarity and patterns that a re suppo rted by multiple obs erv ations . The modeling framework also allows for efficient mo deling o f nonlinea r feature s paces. Multi-task le arning refers to a class of appro a ches wher e m ultiple, related mo d- eling tasks a re solved sim ultaneously by com bining statistical p ow er across the related tasks. A typical task is to improve the accur acy of individual classifiers by taking adv an tage o f the p otential dep endencies betw een them (se e e.g. Ca ruana, 1997). 6.1.2 Role of side information The s econd categ ory of a pproaches for genomic data integration consists of meth- o ds that are asymmetric b y nature; int egration is used to suppor t the analy s is of one, primar y data so urce. Side information can b e used, for ins ta nce, to limit the sea rch space and to fo cus the ana lysis to av oid overfitting, s peed up co mpu- tation, as well as to obtain po ten tially mo re sensitive a nd accur ate findings (see e.g. Eise ns tein, 2006 ). One strategy is to imp ose ha rd constra in ts on the mo del, or mo del family , ba sed on s ide information to targe t sp ecific resea rch questions. In gene expr e ssion context, functional class ifications or known interactions b etw een the genes ca n b e used to co nstrain the analysis (Go eman and Buhlmann, 20 07; Ulitsky a nd Shamir, 2009). In factor analysis and mixed effect mo dels, clinical an- notations of the samples help to fo cus the mo deling on par ticular conditions (s e e e.g. Carv a lho et al., 2008). Ha rd co ns traints rely heavily o n the a ccuracy o f side information. Soft, or probabilis tic approa ches can take the uncerta int y in side in- formation into ac c o unt , but they are computatio nally more demanding. Examples of such methods in the co ntext of transcriptome analy sis include for instance the sup e rvised biclustering models, suc h as cMonkey and modified SAMBA, as w e ll as other metho ds that g uide the ana lysis with a dditional infor mation of genes a nd regulator y mechanisms, such as transcription factor bin ding (Reiss et al., 200 6; Sav a g e et al., 201 0 ; T anay et a l., 2004 ). Publicatio n 3 uses gene in teraction net- work as a hard constr a int for modeling transcriptiona l co-r egulation of the genes , but the condition-sp ecific r esp onses o f the detected gene g roups a re identifi ed in an unsuper vised manner. A c o mplemen tary appr o ach for utilizing side information of the ex per imen ts is provided by m u lti-way le arning . A clas s ical example is the a na lysis of v aria nce (ANO V A), w her e a single data set is modeled by decompos ing it into a set of basic, underlying effects, which c haracteriz e the data optimally . The effects are asso ci- ated with multiple, p otentially o v erlapping attributes of the measurement s a mples, such as disease state, g ender a nd age, whic h are known prior to the analy sis. T ak- ing such prior knowledge of sys tema tic v aria tion betw een the s a mples into account 53 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION helps to increase mo deling p ow er and can reveal the attr ibute-spe cific effects. An int eresting subta s k is to mo del the interactions b etw ee n the a ttr ibutes, so-called inter action effe cts . These are manifested only with par ticular combinations of attributes, and indicate dep endency b etw een the attributes. F or instance, simul- taneous cigarette smo king and asb estos exp osure will c o nsiderably increase the risk of lung cancer, compared to any of the tw o risk factors alone (see e.g. Nymar k et al., 200 7). F actor analysis is a closely related appro a ch where the attributes, also called factors , ar e not given but instead estimated fro m the data. Mixe d effe ct mo dels combine the sup ervised and unsup ervised approaches by incorp orat- ing bo th fixe d and r andom effe cts in the mo del, co rresp onding to the known and latent attributes, resp ectively (see e.g . Ca rv alho et al., 2008). The standard factor- ization a pproaches for individual data s ets ar e related to the dependency -seeking approaches in Publications 4-6 , wher e co-o cc ur ring data sources are decomp os e d in an unsup ervised manner into comp o nent s that are maximally infor mative of the comp onents in the other data set. 6.1.3 Mo deling of m utual dependency Symmetric models for dep endency detection form the third ma in ca teg ory of meth- o ds for g enomic da ta integration, as well as the ma in topic o f this chapter. De- pendenc y mo deling is used to distinguish the shar e d signal from dataset-sp e cific v aria tion. The shar e d effects are informativ e o f the commonalities and interactions betw een the obser v ations, and are often the main fo cus of interest in integrativ e analysis. This mo tiv ates the developmen t of metho ds that can allo cate co mputa- tional resource s efficiently to mo deling of the shar ed features and interactions. Multi-view le arning is a genera l ca tegory of appro aches for symmetric dep en- dency mo deling tasks. In m ulti-view lear ning, m ultiple measur emen t sour ces are av a ilable, and each source is considered as a different view o n the s a me ob jects. The task is to enhance mo deling p erformance by combining the complementary views. A class ical e x ample of such a model is canonical correla tion analys is (Hotelling , 1936). Related approaches that have recently b een a pplied in functional g enomics include for instance probabilistic v ariants of meta-ana lysis (Choi et al., 2 0 07; Con- lon et al., 2007), generalized singula r v alue decomp osition (see e.g. Alter e t a l., 2003; Ber ger et al., 200 6 ) and simult aneous non-nega tiv e ma tr ix facto rization (Badea, 2008 ). The dep endency mo deling a pproaches in this thesis make a n explicit distinc- tion b etw ee n statistical repr esentation of da ta and the mo deling task. Let us denote the r e presentations of tw o co -o ccurring multiv ariate observ a tions, x and y , with f x ( x ) and f y ( y ), resp ectively . The selected repre sent ations dep end o n the application task. The repres en tation ca n b e for instance used to p erform feature selection as in c anonic al c orr elation analysis (CCA) Hotelling (1936), capture non- linear features in the da ta as in kernelized versions of CCA (see e.g. Y amanishi et al., 2003), or partition the data a s in informatio n b ottleneck (F riedman et al., 2001) and asso cia tive clustering (Publica tions 5-6 ). Statistic al indep endenc e of the representations implies that their joint probability density c a n b e de c ompo sed as p ( f x ( x ) , f y ( y )) = p ( f x ( x )) p ( f y ( y )). Deviations from this assumption indicate sta- tistical dependency . The representations can ha ve a flexible par ametric fo r m which can be optimized by the dep endency modeling algorithms to identify dep endency structure in the data. Recent examples of s uch dep endency-maximizing methods include probabilistic 54 6.1. S T AN DARD APPRO A CHES FOR GENOMIC DA T A INTEGRA TION canonical correla tion analy sis (Bach and Jordan, 2 005), which has close theoretical connection to the regularized mo dels in troduced in Publication 4 , and the ass o- ciative cluster ing principle introduced in Publications 5-6. Canonica l co rrelations and contingency table analys is fo r m the metho do logical background for the co n- tributions in Publications 4-6. In the remainder o f this sectio n these t w o sta ndard approaches for dep e ndenc y detection are consider ed more closely . Classical and probabilis tic canonical correlation analysis Canonical co rrelation analys is (CCA) is a clas sical method for detecting linea r depe ndencie s betw een t w o multiv ariate random v ariables (Hotelling, 19 36). While ordinary correla tion characterizes the a sso ciation stre ngth b etw ee n tw o vectors with paired scalar observ a tions, CCA assumes pair e d vectorial v alues , and gener - alizes co rrelation to mult idimensional sources by sear c hing for maximally cor relat- ing lo w-dimensional repre s en tation of the tw o sources, defined by linear pro jections Xv x , Yv y . Mult iple pro jection comp onents can be obtained iter atively , by finding the most co rrelating pro jection first, and then consecutiv ely the next o nes after removing the dependencie s explained b y the prev ious CCA co mpone nts; the low e r - dimensional r epresentations ar e defined by pr o jections to linear h yper planes. The mo del can be for m ulated as a generalized eigenv alue problem that ha s an analytical solution with t w o useful prop erties : the result is in v ariant to line a r tr ansforma- tions of the data, and the solution for any fixed n um ber o f compo nent s maximizes m utual inf ormation betw e e n the pro jections for Gaussia n data (Kullbac k, 19 59; Bach a nd Jorda n, 2002). Extens ions of the cla s sical CCA inc lude g e neralizations to multiple data sour ces (Kettenring, 1971 ; B ach and Jor dan, 200 2), regula rized solutions with non-negative and sparse pro jections (Sigg et al., 2007 ; Archam b ea u and Bach, 2 008; Witten et al., 20 09), and non-linear extensions, fo r instance with kernel methods (Bach and Jorda n, 2002; Y ama nishi et al., 2003 ). Direct o pti- mization of corre la tions in the cla s sical CCA provides an efficient way to de tect depe ndencie s betw een data sources, but it la cks an e xplicit mo del to deal with the uncertaint y in the data and mo del parameter s. Recently , the cla s sical CCA was shown to corr esp o nd to the ML solution of a particular genera tive mo del where the tw o data sets are assumed to stem fr om a shared Gaussian la tent v ariable z and normally distributed data-set-sp ecific noise (Bach and Jo r dan, 2 005). Using linear assumptions, the mo del is formally defined as x ∼ W x z + ε x y ∼ W y z + ε y . (6.1) The manifestation of the shar ed signa l in each data set can be different. This is pa- rameterized by W x and W y . Assuming a standar d Gaussian model for the share d latent v ar iable, z ∼ N ( 0 , I ) and data set-sp ecific e ffects where ε x ∼ N ( 0 , Ψ x ) (and r esp ectively for y ), the cor relation-max imizing pro jections of the traditional CCA introduced in Section 6.1 can b e r etrieved from the ML solution of the mo del (Archam b eau et al., 2006; Bach and Jorda n, 20 05). The mo del decomposes the observed co -o ccurring data sets into shar e d and data set-s p e cific comp onents based on explicit mo deling ass umptions (Figure 6.1). The dataset-sp ecific e ffects can a lso be describ ed in ter ms of latent v ariables as ε x = B x z x and ε y = B y z y , a llowing the co nstruction of more detailed mo dels for the dataset-s pecific effects (Kla mi and 55 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION Kaski, 2008). The shared signal z is treated a s a latent v a r iable and marginalize d out in the model, providing the marginal likelihoo d for the obser v ations: p ( X , Y | W , Ψ) = Z p ( X , Y | Z , W , Ψ) p ( Z ) d Z , (6.2) where Ψ deno tes the block-diago na l matrix o f Ψ x , Ψ y , and W = [ W x ; W y ]. The probabilistic form ulation of CCA has opened up a w a y to new pr obabilistic ex- tensions that can treat the mo deling ass umptions and uncertainties in the data in a more explicit and robust manner (Archam beau et al., 2006 ; Klami a nd Kaski, 2008; Klami et al., 2010). The general formulation provides a flexible mo deling framework, wher e differ- ent mo deling as sumptions ca n b e used to a dapt the mo dels in different applications. The connection to classic a l CCA assumes full cov aria nces for the datase t- spec ific effects. Simpler mo dels for the da taset-sp ecific effects will not distinguish b etw een the shared and margina l effects as effectively , but they have few er mo del param- eters that can p otentially reduce overlearning and speed up computation. It is also p ossible to tune the dimensionality of the shar ed latent sig na l. Lear ning o f low e r-dimensional mo dels ca n b e faster and p otentially les s pr one to o v erfitting. Int erpretation of simpler mo dels is also more s traightforw ard in many a pplications. The proba bilistic formulation allows rigor ous treatment of uncerta in ties in the data and mo del par ameters also with sma ll sample sizes that ar e common in biomedica l studies, a nd allows the incorp or ation of prior information through Bay esian prio rs, as in the reg ularized dep endency detection framework in troduced in P ublication 4. y x x y z z z Figure 6.1: A gr aphical representat ion of the generative shared laten t v ariable mo del in Equa- tion (6.1). The latent source z is shared b y observ ations x and y . The other effects that are specific to each observ ation are c haracte rized by z x and z y , respectively . Gray shading indicates observ ed v ariables. Con tingency table analysis Contingency table analys is is a class ical approach used to study asso ciations b e- t ween co-o ccur ring ca tegorical observ ations. The co-o ccur rences a r e represe n ted by cro ss-tabulating them o n a c ont ingency table , the rows and columns of which corres p ond to the fir st and second s et o f features, r esp ectively . V arious tests are av a ilable for measuring dependency betw een t he ro ws and columns of the table Y ates (1 934); Agresti (1992), including the classical Fisher test (Fisher , 1934 ), a standard to ol for measuring sta tistical enrichmen t of functional ca tegories in gene cluster a nalysis (Hosack et al., 2003 ). While the classic al contingency table ana l- ysis is used t o meas ure dependency betw een co-o ccurring v ar iables, mo re recent approaches use contingency tables to de r ive ob jective functions for dep endency ex- ploration tasks. The as s o ciative clustering principle introduced in Publica tions 5 -6 is an example of such approa ch. 56 6.2. R EGULARIZED DEPENDENCY DETECTION Other appr oaches that use contingency table dependencie s as ob jectiv e func- tions include the i nformation b ott lene ck (IB) principle (Tishb y et a l., 1999 ) and discriminative clustering (DC) (Sinkkonen et al., 200 2; Kaski et al., 2005). These are a symmetric, de p endency- seeking appr oaches that can be used to discov er clus- ter str ucture in a primary data such that it is maximally info r mative of ano ther, discrete auxilia ry v aria ble . The dep endency is r epresented on a contingency ta- ble, and ma x imization o f contingency table dependencies provides the ob jectiv e function for clustering . While the standar d IB operates on discrete data, DC is used to discover cluster structure in contin uous-v alued data. The tw o approaches also employ different ob jective functions. In classica l IB, a disc rete v aria ble X is clustered in such a wa y that the c lus ter assignments beco me ma ximally infor ma- tive of another discrete v aria ble Y . The complexity of the cluster assignments is controlled b y minimizing the mutual information b etw een the cluster indices and the original v ariables. The task is t o find a partitioning ˜ X that minimizes the cost L ( p ( ˜ X | X )) = I ( ˜ X ; X ) − β I ( ˜ X ; Y ) , where β controls clustering res olution. In DC, mut ual informatio n is replaced by a Bay es factor b etw een the tw o hypothe- ses of dep endent a nd indep endent margins. T he Bay es factor is asymptotically consistent with mutual information, but pr ovides an unbiased estimate for limited sample size (see e.g . Sinkkonen et al., 200 5). The standa rd infor mation b ottleneck and discriminative clustering ar e asy mmetric metho ds that trea t o ne of the data sources as the primary target of analys is . In cont rast, the dependency maximization approaches consider ed in this thes is , the associa tive clus tering (AC) and regulariz ed versions of canonical co rrelation analysis a re s ymmetric and they operate exclusiv ely on contin uo us-v alued da ta. CCA is no t based on contingency table analy sis, but it has close connectio ns to the Gaussian IB (Chechik et al., 200 5) that seeks maxima l dep endency b etw een t wo s ets of no rmally distributed v ar iables. The Gaussian IB retrieves the same subspace as CCA for one of the data sets. How ev er, in co n trast to the s ymmetric CCA mo del, Gaussian IB is a directed metho d that finds dep endency- maximizing pro jections for only one of the tw o da ta sets . The second dep endency detection approach considered in this thesis, the asso ciative clustering, is pa rticularly related to the symmetric IB that finds t wo se ts of clusters, one for ea ch v a riable, which are optimally co mpressed presentations o f the origina l data, and at the sa me time maximally informative of each other (F riedma n et al., 2 001). While the ob jective function in IB is derived from m utual information, AC uses the Bay es factor as an ob jective function in a similar manner as it is used in the asymmetr ic discrimina tiv e clustering. Another key difference is tha t while the symmetr ic IB op erates on discrete data, A C e mploys c ont ingency table analy sis in or der to discov er cluster structure in contin uous-v alued da ta s pa ces. 6.2 Regularized dep end enc y detection Standard unsup ervis ed methods for dep endency detection, such a s the cano nical correla tion analysis or the symmetric information bottleneck, seek maximal dep en- dency b etw ee n tw o data sets with minimal assumptions ab out the dep endencies. The unconstr ained mo dels inv olve high degr ees of freedo m when a pplied to hig h- dimensional genomic o bserv atio ns. Such flexibilit y ca n eas ily lea d to ov erfitting, which is even worse for more flexible nonparametr ic or nonlinear, kernel-based de- pendenc y discov e r y metho ds. Several w ays to r egularize the solution hav e b een 57 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION suggested to ov ercome asso ciated pr oblems, fo r instance b y imp osing sparsity con- straints on the solution space (Bie and Mo or, 200 3; Vino d, 19 76). In many applica tions prior informa tion of the dependenc ie s is av aila ble , or particular t ypes of dep endency are relev ant for the a nalysis tas k. Suc h prior infor- mation can b e used to reduce the degrees o f freedom in the mo del, and to regula rize depe ndency detectio n. In the ca ncer g ene discovery application o f P ublication 4 , DNA m utations are systematica lly cor related with tra nscriptional activity of the genes within the affected regio n, and identification o f such r egions is a biomedi- cally relev a nt re s earch task. Pr ior knowledge of c hromosoma l distances b etw een the observ ations can improve the detection of the relev a nt s patial dependencie s . How ever, pr incipled approaches to inco rp orate such prior information in depen- dency mo deling ha v e b een missing. Publication 4 intro duces regular ized mo dels for depe ndency detection based on classical canonical corre lation ana lysis (Hotelling, 1936) and its pr obabilistic formulation (Ba ch and Jorda n, 2 0 05). The mo dels are extended by inco rp orating appropr iate prior ter ms, which ar e then used to reduce the degrees of freedom based on prior biologic a l knowledge. Correlation-based v arian t In order to in tro duce the r egularized dep endency detection framework of Publica- tion 4, let us star t by considering r e gularization of the c lassical co rrelation-ba sed CCA. This searches for arbitrary linear pro jection vectors v x , v y that maximize the correlatio n b etw een the pro jections of the data sets X , Y . Multiple pr o jectio n comp onents can be obtained iterativ ely , b y finding the most correlating pro jec- tion fir st, and then consec utiv ely the next ones a fter re moving the dependencies explained by the previous CCA comp onents. The pro c e dure will iden tify maxi- mally dep endent linear subspaces o f the inv es tig ated data sets. T o regularize the solution, Publication 4 couples the pro jections thro ugh a trans fo rmation matrix T in such a wa y that v y = Tv x . With a co mpletely unconstraine d T the mo del reduces to the classical unconstrained CCA; suitable constra in ts on can b e used to regularize dep e ndenc y detection. T o enfor c e regulariza tion o ne could for instance prefer solutio ns for T that are close to a given tr ansformation matr ix, T ∼ M , or imp ose more gener a l constraints on the structure of the tra nsformation ma trix that would prefer particular rota- tional or other linear relationships. Suitable constra in ts dep end on the particular applications; the solutions can be made to prefer par ticular t ypes of depe ndency in a soft manner by appro priate p enalty ter ms . In Publication 4 the completely unconstrained CC A mo del has bee n compar ed with a fully regula rized mo del with T = I ; this enco des the biological assumption that prob es with small chromosomal distances tend to capture more similar signal b et w een gene expressio n and copy nu m ber measurements than prob es w ith a lar ger chromosomal distance; the pro- jection vectors c haracterize this r elationship, and are therefore exp ected to hav e similar for m, v x ∼ v y . Utilization o f other, more general cons traints in related data in tegration tasks provides a pro mis ing topic for future studies. The cor r elation-base d treatment provides an int uitive and easily implement able formulation for regular ized dep endency detection. How e ver, it lacks an explicit mo del for the s hared and data - sp ecific eff ects, and it is likely that s ome of the dataset-sp ecific effects ar e ca ptured by the cor relation-max imizing pro jections. This is sub optimal for characterizing the shared effects, and motiv ates the proba- bilistic treatment. 58 6.2. R EGULARIZED DEPENDENCY DETECTION Probabilistic de p endency dete ction with simi larit y constraints The probabilistic a pproach for reg ularized dep endency detection in Publication 4 is bas ed on a n explicit mo del of the data - generating pro ces s formulated in Equa - tion (6 .1 ). In this mo del, the transforma tion matrices W x , W y sp ecify how the shared latent v ariable Z is manifested in each data set X , Y , r e spe c tively . In the standa rd mo del, the relations hip b etw een the tra nsformation matrices is not constrained, and the algo r ithm sear ches for ar bitrary linear transformatio ns that maximize the likelihoo d of the observ ations in Equa tion (6.2). The proba bilistic formulation op ens up p ossibilities to guide dep endency search thro ugh Bay esian priors. In Publicatio n 4, the standard pro babilistic CCA mo del is extended by inco rp o- rating additional pr io r terms that r egularize the rela tionship by repa rameterizing the tra nsformation matrices as W y = TW x , a nd setting a prior o n T . The trea t- men t is ana logous to the correlation-based v ariant, but now the transformation matrices oper ate o n the laten t components, rather t han the observ ations. This allows to distinguis h the s hared and dataset-s p ecific effects more explicitly in the mo del. The task is then to lear n the optimal para meter matrix W = [ W x ; W y ], given the constra in t W y = TW x . The Bay es’ rule gives the mo del likelihoo d p ( X , Y , W , Ψ ) ∼ p ( X , Y | W , Ψ ) p ( W , Ψ ) . (6.3) The likeliho o d term p ( X , Y | W , Ψ ) can b e ca lculated based on the mo del in E qua- tion (6.1 ). This defines the ob jective function for standar d probabilistic CCA, which implicitly a s sumes a flat prior p ( W , Ψ ) ∼ 1 for the model par a meters. T he formulation in Equation (6.3 ) makes the choice of the pr ior ex plicit, allowing mo di- fications o n the prior term. T o o btain a tractable prior , le t us as sume that the prior factorizes as p ( W , Ψ ) = p ( W ) p ( Ψ ). The first term ca n b e further decomp osed as p ( W ) ∼ p ( W x ) p ( T ), a ssuming indep e ndent prio rs for W x and T . A conv enien t and tractable prior for T is provided by the matrix nor mal dis tr ibution: 1 p ( T ) = N m ( T | M , U , V ) . (6.4) F or computational s implicit y , let us assume indep endent rows and columns with U = V = σ T I . The mean matrix M c a n b e used to emphasize certain t ypes of depe ndency betw een W x and W y . Assuming uninformative, flat priors p ( W x ) ∼ 1 and p ( Ψ ) ∼ 1, as in the standar d probabilistic CCA model, and denoting Σ = WW T + Ψ , the ne g ative log-likeliho o d of the mo del is − l og p ( X , Y , W , Ψ ) ∼ l og | Σ | + T r Σ − 1 ˜ Σ + k T − M k 2 F 2 σ 2 T . (6.5) This is the ob jective function to minimize. Note that this has the same form as the ob jective function of the standard probabilis tic CCA, except the additional p enalty term k T − M k 2 F 2 σ 2 T arising from the pr io r p ( T ). This yields the cos t function employ ed in Publication 4. In our cancer gene disco very applica tio n the choice M = I is used to enco de the biological pr ior constrain T ≈ I , which s tates that the obs e r v ations with a sma ll chromosomal distance should on av erage show similar res po ns es in the in tegrated data se ts, i.e., W x ≈ W y . The reg ularization stre ngth can b e tuned 1 N m ( T | M , U , V ) ∼ ex p − 1 2 T r { U − 1 ( T − M ) V − 1 ( T − M ) T } where M is the m ean matrix, and U and V denote r ow and column co v ariances, resp ectiv ely . 59 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION with σ 2 T . A fully reg ularized mo del is obtained with σ 2 T → 0. When σ 2 T → ∞ , W x and W y bec ome indep endent a pri ori , yielding the ordinary pr obabilistic CCA. The σ 2 T can b e used to reg ularize the so lution b etw een these tw o extre mes . Note that it is pos sible to incorp ora te als o other types of prior informa tio n concer ning the depe ndencies into the mo del through p ( T ). The mo del parameter s W , Ψ a re estimated with the EM alg orithm. The regular iz ed version is not analytically tractable with resp ect to W in the general case, but can b e optimized with standard gradient-based optimization techniques. Spec ia l cases of the mo de l have ana ly tical solutions, which can sp eed up the mo del fitting pro cedure. In particular , the fully regula rized and unconstrained mo dels, obtained with σ 2 T = 0 and σ 2 T = ∞ resp ectively , hav e clo sed-form solutions for W . Note that the current formulation assumes that the regular ization parameters M , σ 2 T are defined prior to the a na lysis. Alterna tively , these parameter s could b e optimized based on externa l cr iteria, such as ca ncer gene detection p erforma nce in our application, o r lea rned from the d ata in a fully Bayesian treatment these parameters would b e tre a ted as latent v ar ia bles. Incorpor ation of additional prior information of the data set-sp ecific effects thro ugh priors on W x and Ψ provides promising lines for further work. 6.2.1 Cancer gene discov ery wit h dep endency detection The regula rized mo dels pr ovide a principled framework for studying asso ciations betw een transcriptio nal activit y and o ther regulator y la y ers of the genome. In Publication 4, the mo dels are used to inv es tig ate cancer mechanisms. DNA copy nu m ber changes ar e a key mechanism for cancer, and integration of copy n um ber information with mRNA expr ession mea surements can r eveal functional e ffects of the m utations. While causation may b e difficult to grasp, study of the dependen- cies can help to identify functionally active m utations, a nd to provide candidate biomarkers with p otential diagnostic, prognostic and clinical impact in cancer studies. The mo deling task in the c a ncer gene discov ery application of Publication 4 is to identify chromosomal regions that show exceptionally high le vels of dependency betw een g ene copy num ber and transcriptiona l levels. The mo del is used to detect depe ndency within lo cal chromosoma l regions that ar e then compared in order to identify the exc eptional reg ions. The dep endency is qua n tified within a given region by comparing the strength of shared and da ta s et-sp ecific sig na l. High scores indicate r egions where the sha r ed s ignal is par ticularly high relative to the data-set-sp ecific effects. A sliding - window appro ach is used to screen the genome for dependencie s . The regions are defined by the d closes t prob es around each gene. Then the dimensionality of the mo dels s tays constant, which allows direct compariso n of the dependency measures be tw een the re gions without additiona l adjustment terms that w o uld be otherwise needed to comp ensa te for diff erences in mo del complexity . Prior informa tion of the depe ndencies is us e d to regularize cance r gene detec- tion. Chromoso mal g ains and losses ar e likely to b e p o sitively correlated with the expression levels of the affected g enes within the s ame chromosomal region or its close proximit y; copy num b er gain is likely to increas e the ex pression of the ass o- ciated genes wher eas deletion will block ge ne expression. The prior infor mation is enco ded in the mo del by s e tting M = I in the pr ior term p ( T ). T his a ccounts for the exp ected p ositive co rrelations b etw een gene expres sion and c o py n um ber 60 6.2. R EGULARIZED DEPENDENCY DETECTION within the inv estigated chromosomal re g ion. Reg ularization based on such prior information is shown to improv e cancer ge ne detection p erfo r mance in Publica- tion 4, where the regular ized v a r iants outper formed the unconstrained mo dels. A genome-wide screen of 51 g a stric cancer patients (Myllyk angas et al., 20 0 8) reveals clear asso ciatio ns betw een DNA copy num b er changes and transcr iptional activity . The Figur e 6 .2 illustrates dep endency detection o n chromosome arm 17q, where the regular ized mo del reveals hig h dep endency b etw e e n the t w o data sources in a known cancer-a s so ciated r egion. The reg ularized and unconstra ine d mo dels w ere c o mpared in ter ms o f r eceiver-op e rator c haracteris tics calculated by comparing the o rdered gene list fro m the dep endency screen to an exp ert-cur a ted list o f known genes a sso ciated with gastr ic cancer (Myllyk anga s et al., 20 0 8). A large prop ortion of the most significa nt findings in the whole- genome analysis were known cancer genes; the r e maining findings with no known asso ciatio ns to gas tric cancer are promising candidates for further study . Biomedical in terpretation of the mo del pa rameters is also stra ightf orward. A ML es timate o f the latent v ariable v alues Z c haracterizes the strength o f the shar e d signal b etw e e n DNA mut ations and transcr iptional activity for each patient. This allows ro bust identification of small, p otentially unknown patient subgroups with shared amplificatio n effects. These would remain p otentially undetected when comparing patient g roups defined bas ed o n existing clinical a nno tations. The pa- rameters in W can down weigh signal from p o orly p e rforming pr ob es in e a ch data set, or prob es that measure genes whose transcriptio na l levels a re no t functionally affected by the copy num b er change. This provides to ols to distinguish b etw een so-called driver mutations having functional effects from less active p assenger mu- tations, which is an impor ta n t task in cancer studies. On the other hand, the mo del can combine statistical p ow er acro ss the adjace nt measurement prob es, and it captures the stronges t shar ed signal in the t w o sets of observ ations. This is useful since g ene expressio n a nd copy num ber da ta are typically c haracterize d by high levels of biologic a l and measurement v ariation and s ma ll sa mple siz e. Related approac hes Int egration of chromoso ma l ab err ations a nd tra nscriptional activity is an actively studied data in tegration task in functional ge no mics. The firs t s tudies with stan- dard statistical tests were carried out by Hyman et al. (20 0 2) and Phillips et al. (2001) when simultaneous genome-wide obs e rv ations of the tw o data so urces had bec ome av aila ble. The mo deling approaches utilized in this context ca n b e r oughly classified in reg ression- ba sed, c o rrelation- based a nd latent v aria ble appro aches. The reg r ession-bas ed models (Adler et al., 2006; Bicciato et al., 2 009; v an Wierin- gen and v an de Wiel, 2009) characterize alterations in gene expre s sion levels based on copy num b er o bserv atio ns with multiv ar iate regress ion or clos e ly r elated mod- els. The co rrelation- based approa ch es (Gonz´ ale z et al., 2009; Sc h¨ afer et al., 200 9; Soneson et al., 2010) provide symmetric mo dels for dep endency detectio n, based on correlatio n and related statistical mo dels. Many o f these methods als o reg- ularize the solutions, t y pic a lly based on sparsity co nstraints and non-negativity of the pro jections (Lˆ e Cao et al., 2009; W aaijenborg et al., 2008; Witten et al., 2009; Parkhomenko et al., 200 9 ). The correla tion-based approach in P ublica tion 4 int ro duces a complement ary approach for reg ularization that constrains the re- lationship b etw een subspaces where the co rrelations are estima ted. The la ten t v aria ble mo dels by Berg er et a l. (200 6); Shen et al. (20 09); V as ke et al. (2010 ), 61 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION 30 40 50 60 70 80 −2 −1 0 1 2 3 4 30 40 50 60 70 80 −2 0 2 4 30 40 50 60 70 80 0.05 0.10 0.15 Nucleotide position (Mbp) Dependency Signal Signal Gene copy number (17q) Gene expression (17q) Dependency score Figure 6.2: Gene expression, copy num ber si gnal, and the dep endency score along the chromo- some arm 17q obtained wi th the regularized laten t v ariable framework in Equation 6. 5. Kno wn cancer-associated genes fr om an exp ert-curated l ist are mar k ed with bl ac k dots. and Publication 4 are ba sed on explicit mo de ling assumptions concerning the data- generating pro cesses. The iCluster alg orithm (Shen et al., 2009 ) is closely related to the latent v ariable mo del co nsidered in Publica tion 4. While our mo del detects contin uo us dep endencies, iCluster uses a discrete latent v a riable to partition the samples into distinct subgroups. T he iCluster mo del is regularize d by sparsit y constraints on W , while we tune the relationship b etw een W x and W y . Mor e- ov er, the mo del in P ublication 4 utilizes full cov aria nce matrices to mo del for the dataset-sp ecific effects, whereas iCluster us es diagona l cov aria nces. The mo re de- tailed model for dataset-s pecific effects in our mo del sho uld he lp to distinguis h the shared signal mo re accurately . Other latent v ar iable a pproaches include the iterative method bas ed on genera lized singular-v alue decomp osition (Berger et al., 2006), and the pr obabilistic factor graph mo del P ARADIGM (V aske et al., 2010 ), which additionally utilizes pathw a y top ology infor ma tion in the mo deling. Exp erimental compariso n b e t w een t he related integrative approaches can be problematic s ince they ta r get rela ted, but different resea rch questions where the biological ground truth is often unknown. F or insta nce, some metho ds utilize pa- tien t c la ss information in order to detect cla ss-sp ecific alterations (Sch¨ afer et al., 2009), other metho ds p erform de novo class discov ery (Shen et al., 2009 ), provide to ols for g e ne prior itization (Sa la ri et al., 2010), or guide t he analysis with ad- ditional functional informa tio n o f the genes (V aske et al., 2010 ). The algo rithms int ro duced in Publication 4 are particularly useful for gene prior itization and cla ss discov ery purpo ses, where the target is to identify the mos t pro mis ing ca ncer gene candidates for further v alidation, or to detect p otent ially nov el cancer subtypes. 62 6.3. A SSOCIA TIVE CLUSTERING How ever, while an incr easing n um ber of metho ds are released as c o nv eniently a c- cessible algo r ithmic to ols (Salari et al., 2010; Shen et al., 2009 ; Sc h¨ afer e t al., 2009; Witten et al., 2 009), implementations of most mo dels are not av ailable for compariso n purp oses. O p en sour ce implementations of the dep endency detection algorithms dev eloped in this thesis have bee n released to enhance transparency and repro ducibility of the c omputational exp eriments and t o encoura ge further use of these mo dels (Huo vilainen and Lahti, 2010 ). 6.3 A sso ciativ e clustering F unctions of human genes are often studied indirectly , by studying mo del org a n- isms suc h as the mo use (D avis, 2004; Jo yce and Palsson, 2006). Orthologs are genes in different species that originate from a single gene in the last common ancestor of thes e spe cies. Suc h genes have o ften retained iden tical bio logical roles in the present-da y organisms, and are likely to shar e the function (Fitch, 197 0). Mutations in the genomic DNA sequence a r e a key mechanism in ev olution. Con- sequently , DNA s equence similarity can pro vide h ypotheses of g ene function in po o rly annotated sp ecies. An exce ptio nal level o f conserv a tion may highlight c rit- ical physiological similar ities betw een sp ecies, whereas divergence can indicate sig- nificant evolutionary changes (Jorda n et al., 20 05). Inv estigating evolutionary co n- serv a tion and div ergence will po ten tially lead to a deep er understanding o f what makes each sp ecies unique. Evolutionary changes prima rily target the struc tur e and sequence of geno mic DNA. How ev er, not a ll changes will lea d to phenotypic differences. On the other hand, sequence similarity is not a guarantee of func- tional similarity b ecause s mall changes in DNA can p otent ially hav e r emark a ble functional implications. Therefore, in a ddition to inv estigating str u ctur al c onservation of the ge nes at the seque nce level, a nother level of in vestigation is needed to study functional c on- servation of the ge ne s and their regula tion, which is reflected at the transcriptome (Jim´ enez et a l., 2002; Jor da n et al., 200 5). T ranscriptiona l re gulation of the gene s is a key r e gulatory mechanism that can hav e remar k able phenotypic co nsequences in highly mo dular cell- biological systems (Hartw ell et al., 1999 ) even when the original function of the regula ted genes would remain intact. Systematic compar ison of tra nscriptional activity b etw een different sp ecies would provide a straig htforward str ategy for inv estigating conserv ation of gene regulation (Be r gmann et al., 2004; Enar d et al., 20 02; Zhou a nd Gibson, 2004). How ever, direct comparison of individual genes b etw een s pecie s ma y not b e o p- timal for discovering subtle and complex dep endency structures. The a sso ciative clustering principle ( A C), in troduced in Publications 5-6, pro vides a fra mework for detecting g roups o f ortho logous genes with exceptional levels of c onserv a tion and div ergence in transcr iptional ac tivit y bet ween tw o sp ecies. While standard depe ndency detection metho ds for contin uo us data, suc h as the generalized sin- gular v a lue decomp osition (see e.g . Alter et al., 20 0 3) or canonical cor relation analysis (Hotelling, 19 3 6) detect g lobal linear depe ndencie s b etw een observ ations, A C sea rches for dep endent, lo cal gro upings to reveal gene groups with exc e ptio nal levels of cons erv ation a nd divergence in transc riptional activity . The model is free of par ticular distributional assumptions ab out the data, whic h helps to allo c ate mo deling resour ces to detecting dep endent subgr oups when v aria tio n within each group is less r elev ant for the analysis . The rema inder of this section provides 63 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 0000 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 1111 000000000000 000000000000 000000000000 000000000000 000000000000 111111111111 111111111111 111111111111 111111111111 111111111111 CONTINGENCY TABLE gene 1 gene 3 PAIRED DATA Y gene 2 TREE CO−OCCURRENCE 1 Repeated AC (by bootstrap) CO−OCCURRENCE INTERESTING .034 1.5 0.05 ... X gene 5 gene 3 gene 1 gene 7 gene 3 gene 1 gene 7 gene 2 gene 1 gene 3 gene 7 gene 3 gene 1 gene 7 gene 5 ... N 2 ... gene 9 gene 9 gene 3 gene 1 gene 9 gene 8 gene 2 gene 4 EXPRESSION SPACE HUMAN EXPRESSION SPACE MOUSE 0000 0000 0000 0000 0000 1111 1111 1111 1111 1111 + + + + + + + CLUSTERS gene 7 gene 3 gene 1 MARGIN 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 + + + + + RELIABLE CO−OCCURRENCES EXPRESSION MOUSE GENE HUMAN GENE CLUSTERS OVERLAPPING 2.4 1.6 5.2 ... EXPRESSION + + + + Figure 6.3: Principle of asso ciativ e clustering (A C). AC p erforms simultaneous clustering of t w o data sets, consisting of pair ed observ ations, and seeks to maximi ze the dep endency b etw een the t w o sets of clusters. The clusters ar e defined by cluster cent roids in eac h data space. The cluster- ing results are represent ed on a cont ingency table, where cl usters of the t w o data sets corresp ond with the rows and columns of the con tingenc y table, respectively . These are called the mar- gin clusters of the contingenc y table. The table cells are called cross clusters and they con tain orthologous genes from the t wo data sets. The cluster cen troids are optimized to produce a con- tingency table with maximal dependency b et w een the mar gin cluster coun ts. Cross clusters that sho w significan t deviation from the null hypothesis of independent margins indicate dependency . In order to enhance the r eliability of the results, the clustering is repeated with sli ghtly differing bo otstrap samples. Then reliable co-o ccurrences are identified f rom a co-o ccurrence tree with a specified threshold. F requent ly co-occurring orthologues are selected for further analyzes. an ov erview of the ass o ciative clustering principle and its a pplication to studying evolutionary divergence b etw een sp ecies. The asso ciativ e clustering principle The pr inciple of asso c ia tive clustering (AC) is illustrated in Fig ure ?? . AC p er- forms simultaneous cluster ing of tw o data sets to reveal ma ximally dep endent cluster structure betw een t wo sets of observ a tio ns. The clusters are defined in each data s pa ce by V or onoi p ar ameterizatio n , where the clusters ar e defined b y cluster centroids to pro duce connected, internally homog eneous cluster s. Let us denote the t wo sets of clus ters by { V ( x ) i } i , { V ( y ) j } j . A given da ta p oint x is then assigned to the cluster corresp onding to the nea rest cent roid m i in the feature space, with re s pec t to a giv en dis ta nce measure 2 d . This divides the space int o non-ov erlapping V or onoi r e gions . The reg ions define a clustering for all p oints of the da ta spa ce. The a sso ciation b etw een the clusters of the tw o data sets c a n b e 2 x ∈ V ( x ) i if d ( x , m i ) ≤ d ( x , m k ) for all k . 64 6.3. A SSOCIA TIVE CLUSTERING represented on a c o nt ingency table, where the rows a nd c o lumns corresp ond to clusters in the fir st and second da ta set, resp ectively . T he clusters in each data set are called mar gin clust ers . Each pair of co-o ccurring observ ations ( x i , y i ) maps to one marg in cluster in ea c h data set, a nd each contingency table cell co rresp onds to a pair of margin clusters. These are called cr oss clusters . A C searches f or a maximally dep endent cluster structure by optimizing the V oro noi centroids in the tw o da ta spaces in such a wa y that the dep endency b e- t ween the con tingency table marg ins is maximized. Let us denote the num b er of samples in cross cluster i, j by n ij . The co rresp onding marg in cluster coun ts are n i · = P j n ij and n · j = P i n ij . The observed sample frequencies ov er the contingency table ma rgins and cross-clus ters are assumed to follo w multinomial distribution with latent parameters θ i , θ j and θ ij , resp ectively . Assuming the mo del M I of indep endent mar gin clusters , the ex pected sample frequency in ea ch cross cluster is given by the outer pro duct of mar gin cluster fre q uencies. The mo del M d of dep endent mar gin clusters devia tes from this a s sumption. The Bayes factor (BF) is used to compare the tw o h ypo theses of dep enden t a nd indep endent mar - gins. This is a rigoro usly justified appro ach for mo de l compariso n, which indicates whether the obser v ations provide sup erior evidence for either mo del. Evidence is calculated over a ll p otential v alues of the mo del pa rameters, ma rginalized ov er the latent frequencies . In a sta ndard setting, the Bayes factor would be us e d to com- pare evidence b etw een the dep endent and indep endent marg in cluster mo dels for a given clustering solution. AC uses the B ayes fa ctor in a no n-standard manner; as an ob jective function to maximize by o ptimizing the c luster centroids in each data space; the centroids define the margin clusters and consequently the marg in cluster depe ndenc ie s. The centroids are optimized with a conjugate-g radient a lgorithm after smo o th- ing the cluster b orders with contin uous pa rameterizatio n. The hyperpa rameters n ( d ) , n ( x ) , a nd n ( y ) arise from Dir ich let pr iors of the tw o mult inomial mo dels M I , M D of independent and dependent margins, resp ectively . Setting the hyperpa - rameters to unity yields the classica l hypergeo metric measure o f contingency table depe ndency (Fisher , 1934 ; Y a tes, 1934 ). Wit h lar ge sample size, the lo garithmic Bay es factor appro aches m utual informatio n (Sinkkonen et a l., 20 0 5). The Bayes factor is a desira ble choice esp ecially with a limited sample size since a marginaliza- tion over the la ten t v aria bles makes it ro bust ag ainst uncertaint y in the par ameter v alues, and b ecaus e finite contingency table co un ts would give a biased e stimate of mutual informatio n. The num b er of c lusters in each da ta s pace is sp ecified in adv ance, typically based on the desired level of r esolution. Nonpara metric exten- sions, where the num ber of marg in clusters would b e infer r ed automatically from the data form o ne po tent ial topic for fur ther studies; a closely rela ted appr o ach was recently prop osed in Roger s et al. (201 0). Publication 6 introduces an additional, bo o tstrap-based pro cedure to a ssess the re lia bilit y of the findings (Figur e ?? ). The analy sis is rep eated w ith similar , but not identical tra ining da ta s e ts obta ined by sampling the origina l data with replacement. The most frequently detected dependencies a re then investigated more clos e ly . The a nalysis will emphasize finding s that are not sensitive to small v aria tions in the obse r ved da ta. 65 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION Comparison m etho ds Asso ciative cluster ing was compared with tw o alternative methods: standard K- means o n each of the t w o data sets, a nd a combination of K -means and informa- tion b ottleneck (K- IB). K-mea ns (see e.g. Bishop, 2 006) is a c la ssical cluster ing algorithm that provides homo geneous, connected clusters based on V orono i pa- rameteriza tio n. Homogeneit y is desirable for interpretation, since the data p oints within a giv en cluster ca n then b e conv e nien tly summarized by the c lus ter cen- troid. On the other hand, K-means co ns iders each data se t indep endent ly , which is sub optimal for the dependency mo deling ta s k. The t w o sets o f clusters o btained by K-means, o ne for each data space, can then b e prese n ted on a contingency table a s in asso ciative clustering. The second comparison metho d is K-IB intro- duced in Publica tion 5. K-IB uses K-means to partition the tw o co-o ccurr ing, contin uo us-v alued data sets into discrete atomic regio ns wher e each da ta p oint is assigned in its own singleto n cluster. This gives t w o sets of ato mic clusters that are mapp ed on a larg e contingency ta ble, fille d with frequencies of co-o ccurring data pa irs ( x k , y k ). The table is then compressed to the desir ed size b y aggre- gating the marg in clusters with the symmetric IB algo rithm in or der to maximize the dep endency betw een the contingency table margins (F riedman et al., 2001 ). Aggrega ting the atomic c lusters pr ovides a flexible clustering appr oach, but the resulting clus ters are no t necess arily homog e neous and they are therefore difficult to in terpret. A C compared fav orably to the other metho ds. While AC outp erfor med t he standard K-means in dependency mo deling, the cluster homog eneit y was not sig- nificantly reduced in A C. The cross cluster s from K-IB (Sinkk o nen et al., 2003) were more dep enden t than in A C. On the other hand, A C pro duced more easily in- terpretable lo calized clusters, a s mea sured by the sum of intra-cluster v ariances in Publication 6. Homogeneity makes it po ssible to summar ize clusters conv enient ly , for instance b y using the mea n expression p rofiles of the cluster samples, as in Figure 6.4B. While K-means sear ch es for maximally homo geneous cluster s a nd K- IB sear ch es for maxima lly dependent clusters , AC finds a successful co mpromise betw een the goals of dependency and homogeneity . 6.3.1 Exploratory analysis of transcriptional dive rgence be- t w een species Asso ciative clustering is used in Publications 5 and 6 to in vestigate conser v ation and divergence o f transc r iptional activity of 2 818 ortholo gous human-mouse gene pairs acro s s a n orga nis m-wide collectio n of transcriptiona l pr o filing data cov ering 46 and 45 tissue types in h uman and mouse , resp ectively (Su et al., 20 02). A C takes as input tw o gene expres sion matrice s with orthologo us genes, o ne for each species, and r e turns a dependency-maximizing c lus tering for the orthologo us g ene pairs. Int erpretation o f the results focus es on unexp ectedly la rge or small cr oss clusters revealed by the contingency table ana lysis o f a sso ciative clustering. Compared to plain correlatio n- based comparis ons b et w een the g ene expres sion pro files, A C can reveal additional cluster str ucture, where g enes with similar expr ession profiles are clustered toge ther, and a s so ciations betw een the tw o s pecies are inv estigated at the level of such detected g ene groups. The dep endency b etw een each pair of margin clusters ca n b e c haracteriz ed by comparing the resp ective ma r gin cluster centroids that provide a compac t summary of the samples within e a ch cluster . 66 6.4. CON CLUSION A B Mouse clusters Human clusters 5 10 15 20 0 500 1000 1500 Mouse Man Testis Figure 6. 4: A The con tingency table of asso ciative clustering highligh ts orthologous gene groups in human (rows) and mouse (columns) with exceptional l ev els of conserv ation (y ello w) or div er- gence (blue) in transcriptional activit y betw een the tw o species. B Average expression profiles of a highly conserved group of testis-sp ecific genes across 21 tissues in man and mouse. c IEEE. Reprint ed with permi ssion from Publ i cation 6. Biologica l interpretation of the findings, based on enr ichmen t of Gene Ontology (GO) categories (Ash burner et al., 20 00), revealed g enes with strong ly conserved and p otentially diverged trans criptional activity . The mo st highly e nriched cat- egories were asso ciated with r ibo somal functions, the high conserv ation o f which has also b e en s uggested in earlier studies (Jim´ enez et al., 20 02); rib osomal g enes often require co ordinated effort of a large group of genes, and they function in cell maintenance tasks that ar e cr itica l for sp ecies surviv al. An exceptional lev e l o f conserv ation was a ls o observed in a group of testis-sp ecific g e nes, yielding novel functional hypotheses for ce rtain po o r ly a nnotated genes within the same cro s s- cluster (Fig ure 6.4). T r anscriptional divergence, on the other hand, w as detected for instance in genes related to embry onic developmen t. While gener al-purp ose dep endency exploration to ols may not be optimal for studying the sp ecific issue of transcriptiona l conser v ation, such to ols can reveal de- pendenc y with minimal prior knowledge ab out the data. This is useful in functiona l genomics exp eriments where little prior knowledge is av aila ble. In P ublications 5 and 6, asso c ia tive clustering has b een additionally applied in inv estigating dep en- dencies betw een tr a nscriptional activity a nd tra nscription fac tor binding, a nother key regulato ry mechanism of the genes . 6.4 Conclus ion The mo dels introduced in P ublications 4-6 provide g eneral explorato ry to ols for the discovery and analysis of sta tistical dep endencies b etw een co-occur ring data sources and to ols to g uide mo deling through Bayesian priors. In particula r, the mo dels consider linear dep endencies (P ublication 4) and cluster-ba sed dep endency structures (Publications 5-6) betw een the data sources. The models are r eadily applicable to data integration tasks in functional genomics. In particular, the mo d- els hav e been a pplied to inv estigate dep e ndenc ie s b e t w een chromosomal m utations and transc r iptional activity in cancer, and evolutionary divergence o f transcript- 67 CHAPTER 6. HUMAN TRAN SCRIPTOME AND OTHER LA YERS OF GENOMIC INFORMA TION ional activity b etw een human and mouse. Biomedica l studies pr ovide a num ber of other p otential applica tions for such g e neral-purp ose metho ds. An increasing nu m ber of co-o ccur ring observ ations acr oss the v ario us re g ulatory lay er s of the genome are a v ailable concerning epigenetic mec hanisms, micro-RNAs, p olymor- phisms a nd other genomic feature s (The Cancer Geno me Atlas Research Net w ork, 2008). Sim ultaneous obser v ations provide a v alua ble reso urce for in vestigating the functional prop erties that emerg e from the interactions betw een the different lay- ers of genomic infor ma tion. An op en sour c e im plemen tation in Bio Conductor 3 provides a ccessible c o mputational to ols for related data integration tasks, helping to guara n tee t he utilit y of the developed mo dels for the co mputatio nal biology communit y . 3 h ttp://www.bioconductor.org/pac k ages/release/bioc/html/pin t.h tml 68 Chapter 7 Summary a nd conclusions Mathematics is biolo gy’s next micr osc op e, only b etter; biolo gy is math- ematics’ next physics, only b etter. J.E. Cohen (2004) F ollowing the initial sequencing of the h uman genome (International h uma n genome sequencing consortium, 20 01; V enter et a l., 2001), t he understa nding of structural a nd functional organizatio n o f genetic infor mation has extended ra pidly with the accumulation of r esearch data. This has op ened up new challenges and opp ortunities for making fundamental discoveries ab out liv ing org anisms and cre- ating a holistic picture ab out genome organizatio n. The increas ing need to or- ganize the lar ge volumes of g enomic data with minimal h uman interv en tion has made computatio n an incre a singly central element in mo dern scie ntific inquiry . It is a par adox of our time that the his to rical scale o f da ta in public and propr ietary rep ositories is only revealing how incomplete our knowledge of the eno rmous com- plexity of living sy s tems is. The pa rticular challenges in data- in tensive genomics are asso cia ted with the complex and p o or ly ch aracter iz ed natur e of living sys tems, as well as with limited av a ilability of observ a tions. It is p ossible to s olve so me of these challenges by c ombin ing statistical p ow er acro ss multiple exp eriments, a nd utilizing the wealth o f ba ckground information in public rep ositor ies. Ex plorator y data ana lysis can help to provide r e search hypothes e s and mater ial for more de- tailed in v estigations ba s ed on larg e-scale ge nomic observ ations when little prior knowledge is av ailable co ncerning the under lying phenomena; mo dels tha t a re ro- bust to uncertaint y and able to automatically adapt to the data, can fac ilita te the discov ery of nov el biolog ical hypo thes e s. Statistical lea rning and probabilistic mo dels provide a natural theoretical framework for such analysis. In t his thesis, genera l-purp ose exploratory data analysis metho ds ha v e b een developed for org anism-wide analysis of the human transcr iptome, a cen tral func- tional la yer of the genome. Integrating evidence across m ultiple so urces o f genomic information can help to reveal mechanisms that co uld not be inv estigated ba s ed on smaller and mo re tar geted experiments; this is a central asp ect in all contributions. In par ticula r, metho ds have b een developed (i) in order to impr ov e meas ur ement accuracy of high-throug hput obser v ations, (ii) in order to mo del transcriptional activ ation patterns and tissue r elatedness in geno me-wide interaction netw orks at an o rganism-wide sca le, and (iii) in order to integrate mea surements of the human transcriptome with other lay ers of genomic informa tion. These res ults contribute 69 CHAPTER 7. SUMMAR Y AND CONCLUSI ONS to some of the ’gra nd challenges’ in the g enomic era by developing strateg ies to understand cell-biolog ical systems, g enetic cont ributions to human health and e vo- lutionary v aria tion (Collins et al., 2003 ). The c o mputational exp eriments in this thesis ha v e b een carried out based on publicly a v ailable, anonymized data sets that follow commonly accepted ethical standa rds in biomedica l res e a rch. Op en access implemen tations o f the key alg o rithms hav e b een provided to g uarantee wide ac- cess to these to ols and to spark new research be yond the or iginal applications presented in this thesis. Metho dological extensions a nd application of the dev elop ed algo rithms to new data in tegration tasks in functional genomics and in other fields provide a promis - ing line for future studies. The m etho ds develop ed in this thesis are readily ap- plicable in g enome-wide scr eening studies in cancer and p otentially o ther dise ases. Increasing amounts of co-o ccurr ing data concerning v ario us asp ects of the genome hav e b ecome av ailable, including gene- and micr o-RNA expression, structural v a ri- ation in the DNA, epigenetic mo difica tions and gene re g ulatory net works. It is ex- pec ted that with small mo dificatio ns the in tro duced metho dolog y can be applied to study further asso cia tio ns b et w een these and other lay er s of g enome org aniza- tion, as well as t heir co n tributions to human health. The fundamental res e arch challenges in contemporar y genome biology provide a wide ar r ay of applications for statistical learning and explora tory analy sis, a nd a rich source of ideas for metho dological research. 70 71 Bibliograph y A. S. A d ler, M. Lin , H. Horlings, D. S. A. Nuy ten, M. J. v an de Vijver, and H. Y. Chang. Genetic regulators of large-scale transcriptional signatures in cancer. Natur e Genetics , 38:421– 430, 2006. A. Agresti. A survey of ex act inference for contingency tables. Statistic al Sc ienc e , 7: 131–153 , 1992. B. Alb erts, A . Johnson, J. Lewis, M. Raff, K. Rob erts, and P . W alter. Mole cular Biolo gy of the Cel l . Garland Science, New Y ork , fourth ed ition, 2002. D. B. A llison, X. Cui, G. P . Page, and M. Sabrip our. Microarra y data analysis: from disarra y to consolidation and consensus. Nat ur e Re views Genetics , 7:55–65, 2006. D. J. Allocco, I . S. Kohane, and A. J. Butte. Quantifying the relati onship b etw een co-expression, co-regulation and gene function. BMC Bioi nformatics , 5:18, 2004. O. Alter and G. H. Golub. Reconstructing t h e p athw ays of a cellular system from genome- scale signals by using matrix and tensor comput ations. Pr o c e e dings of the National A c ademy of Scienc es, USA , 102:17559–1 7564, 2005. O. Alter, P . O. Bro wn, an d D . Botstein. Generalized singular va lue d ecomposition for comparativ e analysis of genome-scale exp ression data sets of t w o differen t organisms. Pr o c e e dings of the National A c ademy of Scienc es, USA , 100:3351–3356 , 2003. C. Archam b eau and F. Bac h. Sparse p robabilistic pro jections. In D. Koller, D. Sch uur- mans, Y. Bengio, and L. Bottou, ed itors, A dvanc es in Neur al Information Pr o c essing Systems 21 , pages 73–80. MIT Press, Cam bridge, MA, 2008. C. Archam beau, N . Delannay , and M. V erleysen. Robust probabilistic p ro jections. In W. Cohen and A . Mo ore, editors, Pr o c e e dings of the 23r d I nternat ional c onfer enc e on machine le arning , volume 148, pages 33–40. ACM , Pittsburgh, P ennsylv ania, 2006. M. Ashburner, C. A. Ball, J. A. Blak e, D . Bots tein, H. Butler, J. M. Cherry , A. P . Davis, K. Dolinski, S. S. D wigh t, J. T. Eppig, M. A. Harris, D . P . Hill, L. Issel-T arver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwa ld, G. M. Rubin, and G. Sherlock. Gene ontology: tool for the unification of biology . Natur e Genetics , 25:25–2 9, 2000. H. A uer, S . Lyianarachc hi, D . Newsom, M. I. Kliso v ic, G. Marcucci, and K . Kornack er. Chipping a wa y at the chip b ias: R NA d egradation in microarra y analysis. Natur e Genetics , 35:292–293 , 2003. F. R. Bach and M. I. Jordan. Kernel indep endent comp onent analysis. Journal of Machine L e arning R ese ar ch , 3:1–4 8, 2002. 72 BIBLIOGRAPHY F. R. B ac h and M. I. Jordan. A probabilis tic in terpretation of canonica l correlation analysis. T ec hnical rep ort, Department of Statistics, Universit y of California, Berkeley , 2005. L. Badea. Extracting gene expression profiles common to colon and pancreatic adeno car- cinoma using sim ultaneous n onnegativ e matrix factorizatio n. In R. B. Altman, A. K. Dunker, L. Hunter, T. Murray , and T. E. Klein, editors, Pr o c e e dings of t he Pacific Symp osium on Bi o c omputing (PSB’08) , pages 267–278 . W orld Scientific, U SA, 2008. P . Baldi and S. Brun ak. Bioi nformatics: the machine le arning appr o ach . Bradford, London, third edition, 1999. T. Bammler et al. Standardizing global gene expression analysis b etw een lab oratories and across platforms. Natur e Metho ds , 2:351–3 56, 2005. N. Bannert and R . Kurth . Retro elements and the human genome: New p ersp ectives on an old relation. Pr o c e e dings of the National A c ademy of Scienc es , 101(S2):14572–145 79, 2004. A.-L. Barab´ asi and Z. N. Oltv ai. Net w ork biology: un derstanding the cell’s functional organization. Natur e R eviews , 5:101– 113, 2004. Y. Ba rash and N. F riedman. Con t ext-sp ecific baye sian clustering for gene expression data. Journal of Computational Bi olo gy , 9:169–191, 2002. V. Barb our, B. Cohen, and G. Y amey . Why bigger is n ot yet b etter: The problems with huge datasets. PL oS Me di cine , 2:e55, 2005. T. Barrett, D. B. T roup, S. E. Wilhite, P . Led oux, D. Rudnev , C. Ev angelista, I. F. Kim, A. S ob olev a, M. T omashevsky , K. A. Marshall, K. H. Phillippy , P . M. S h erman, R. N. Muertter, and R . Edgar. NCBI GEO: arc hiv e for high-throughp u t functional genomic data. Nuclei c A cids R ese ar ch , 37:D885–90, 2009. T. Ba yes. S tudies in the history of probability and statistics: IX. Thomas Bay es’ essa y To w ards solving a problem in the doct rine of chances. Biometrika , 45:296–315 , 1763. Prin ted in 1958. D. G. Beer, S. L. R. Kardia, C.-C. Huang, T. J. Giordano, A. M. Levin, D. E. Misek, L. Lin, G. Chen, T. G. Gharib, D. G. Thomas, M. L. Lizyness, R. Kuick, S. Hay asak a, J. M. G. T aylor, M. D . Iannettoni, M. B. Orringer, and S. H anash. Gene-exp ression profiles predict su rviv al of patients with lung adeno carcinoma. Natur e Me dicine , 8: 816–824 , 2002. S. Ben-David and M. Ack erman. Meas ures of clustering quality: A working set of ax- ioms for clustering. In D. Koller, D. Schuurmans, Y. Bengio, and L. Bottou, editors, A dvanc es in Neur al Information Pr o c essing Syst ems 21 , pages 121–128. M IT Press, Cam bridge, MA, 2008. D. A. Benson, I . Karsch-Mizrac h i, D. J. Lipman, J. Ostell, and E. W. Say ers. GenBank. Nucleic A ci ds Res e ar ch , 38:D46–51, 2010. J. A. Berger, S . Hautaniemi, S. K. Mitra, and J. A stola. Jointly analyzing gene expression and cop y num ber data in breast ca ncer using data reduction m o dels. IEEE/ACM T r ansactions on Computational Biol o gy and Bioinformatics , 3:2–16, 2006. S. Bergmann, J. Ihmels, and N. Bark ai. Similarities and differences in genome-wide expression data of six organisms. PL oS Bi olo gy , 2:85–93 , 2004. 73 BIBLIOGRAPHY J. M. Bernardo and A. F. M. Smith. Bayesian The ory . John Wiley & Sons Ltd, Chich- ester, England, 2000. R. Berouk h im, C. H. Mermel, D . Porter, G. W ei, S. Rayc haudhuri, J. Donov an, J. Bar- retina, J. S. Boehm, J. Dobson, M. Urashima, K. T. Mc Hen ry , R. M. Pinch b ac k, A . H. Ligon, Y.-J. Cho, L. Haery , H. Greulich, M. Reich, W. Winckler, M. S. La wrence, B. A. W eir, K . E. T anak a, D. Y. Chiang, A. J. Bass, A . Loo, C. H offman, J. Prensner, T. Liefeld, Q. Gao, D. Y ecies, S. Signoretti, E. Maher, F. J. Kay e, H. S asaki, J. E. T epp er, J. A. Fletcher, J. T ab ernero, J. Baselga, M.-S. Tsao, F. Demichelis, M. A. Rubin, P . A. Janne, M. J. Daly , C. Nu cera, R. L. Levine, B. L. Eb ert, S. Gabriel, A. K. Rustgi, C. R. Antonescu, M. Ladan yi, A. Letai, L. A. Garra w a y , M. Loda, D. G. Beer, L. D. T rue, A. Ok amoto, S . L. Pomero y , S. Singer, T. R. Golub, E. S. Lander, G. Getz, W. R. Sellers , and M. M eyers on. The landscap e of somatic copy-num ber alteration across human cancers. Natur e , 463:899–90 5, 2010. A. Bhattacharj ee, W. G. Richards, J. S taunton, et al. Classification of human lung carcinomas by mRNA exp ression profiling reveals distinct adeno carcinoma sub classes. Pr o c e e dings of the National A c ademy of Scienc es, USA , 98:13790–1379 5, 2001. S. Bicciato, R. Spinelli, M. Zampieri , E. Mangano, F. F errari, L. Beltrame, I . Cif ola, C. Pea no, A. S olari, and C. Battaglia. A computational pro cedure to identify significan t o verl ap of differentia lly expressed and genomic imbal anced regions in cancer datasets. Nucleic A ci ds Res e ar ch , 37:5057–507 0, 2009. T. D. Bie and B. D. Mo or. On the regularization of canonical correlation analysis. In S.-I. Amari, A. Cic hocki, S. Makino, and N. Murata, editors, Pr o c e e dings of the Inter- national Confer enc e on I ndep endent Comp onent Analysis and Blind Sour c e Sep ar ation (ICA2003) . Nara, Japan, April 1–4 2003. BioP AX workgroup. BioP AX - Biolo gi c al Pathways Exchange L anguage , 2005. Level 2, V ersion 1.0 Docu mentatio n. C. M. Bishop. Patt ern r e c o gni tion and machine le arning . Springer, Singap ore, 2006. J. Blak e. Bio-ontologi es – fast and furious. Natur e Biote chnolo gy , 22:773–774 , 2004. B. M. Bolstad, R. A. Irizarry , M. Astrand, and T. P . Sp eed. A comparison of normaliza- tion metho ds for high d ensit y oligon u cleotide arra y data based on va riance and bias. Bioinformatics , 19:185–193 , 2003. A.-L. Boulesteix. Over-optimism in bioinformatics researc h. Bi oinformatics , 26:437, 2010 . T. Bo veri. Zur F r age der Entstehung maligner T umor en . V erlag von Gustav Fisc her, Jena, 1914. J. R. Bradford, Y. Hey , T. Y ates, Y. Li, S. D. Pepper, and C. J. Miller. A comparison of massively parallel nucleotide sequen cing with oligon u cleotide microarra ys for global transcription profiling. BMC Genomics , 11:282, 2010. U. M. Braga-Neto and E. T. A. Marques. F rom functional genomics to fun ctional im- munomics: New chall enges, old p roblems, b ig rew ards. PL oS Computational B i olo gy , 2:e81, 2006. A. Brazma, P . H ingamp, J. Q u ac ken b ush, G. Sh erlock, P . S p ellman, C. Sto eck ert, J. Aach, W. A nsorge, C. A. Ball, H . C. Causton, T. Gaasterland, P . Glenisson, F. C. P . Holstege, I. F. Kim, V. Marko witz, J. C. Matese, H. Parkinson, A. R obinson, U. S ark ans, S. Sch u lze-Kremer, J. Stew art, R. T a ylor, J. Vilo, and M. V ingron. Min- im um information ab out a microarray exp erimen t (MIAME) – to w ard stand ards for microarra y d ata. Natur e Genetics , 29:365– 371, 2001. 74 BIBLIOGRAPHY A. Brazma, M. Kresty anino v a, and U . Sark ans. Standards for systems biology . Natur e R eviews Genetics , 7:593– 605, 2006. M. R. Brent. Steady progress and recent b reakthroughs in the accuracy of automated genome annotation. Nat ur e R eviews Genetics , 9:62–73, 2008. T. A. Brown. Genomes . G arland Science, UK, third edition, 2006. C. G. Broyden. The conv ergence of a class of double-rank m in imization algorithms, I I: The new algorithm. IMA Journal of Applie d Mathematics , 6:222–2 31, 1970. A. Butte. T he use and analysis of microarra y data. Natur e R eviews , 1:951–960, 2002. L. Cabusora, E. Su tton, A. F ulmer, and C. V. F orst. Differential netw ork expression during drug and stress resp onse. Bi oinformatics , 21:2898– 2905, 2005. G. A. Calin and C. M. Croce. MicroRNA signatures in human cancers. Natur e Re views Canc er , 6:857 –866, 2006. V. J. Carey and V. S tod d en. R eprod ucible Research Concepts and T o ols for Cancer Bioin- formatics. In M. F. O chs, J. T. Casagrande, and R . V . Da vuluri, editors, Biome dic al Informatics for Canc er R ese ar ch , pages 149–175. Springer US, Boston, MA, 2010. P . Carninci. Is sequencing enligh tenment e nding the dark age o f the transcriptome? Natur e Metho ds , 6:711–713, 2009. S. B. Carroll. Ge netics and t h e making of homo sapiens. Natur e , 422:849– 857, 2003. R. Caruana. Multita sk learning. Mach ine L e arning , 28:41–75, 1997. C. M. Carv alho, J. Chang, J. E. Lucas, J. R. Nevins, Q. W ang, and M. W est. High- dimensional sparse factor modeling: Applications in gene expression genomics. Journal of the Amer ic an Statistic al Asso ciation , 103:1438 –1456, 2008. J. T. Chang, C. Carv alho, S. Mori, A. H. Bi ld, M. L. Ga tza, Q. W ang, J. E. Lucas, A. P otti, P . G. F ebb o, M. W est, and J. R. Nevin s. A genomic strategy to elucidate mod u les of oncogenic pathw ay signaling netw orks. Mole cular Cel l , 34:104 –114, 2009. R. Chari, B. P . Co e, E. A. V ucic, W. W. Lo ckwood, and W. L. Lam. An integrativ e multi- dimensional genetic and epigenetic strategy to iden tify ab errant genes and pathw a ys in cancer. BMC Systems Biolo gy , 4:67, 2010. G. Chec hik, A . Glob erson, N. Tish b y , and Y. W eiss. Information Bottlenec k for Gaussian v ariables. Jour nal of Machine L e arning R ese ar ch , 6:165 –188, 2005. R. J. Cho, I. S. Dh illon, Y. Guan, and S . S ra. Minimum sum-squared residue co-clustering of gene ex pression d ata. In M. W. Berry , U . Day al, C. Kamath, and D. Skillicorn, editors, Pr o c e e dings of the 4th SIAM International Confer enc e on Data Mining , pages 114–125 . Florida, US A, 2004. H. Choi, R . Shen, A. M. Chinn aiy an, and D. Ghosh. A l atent var iable approach for meta-analysis of gene expression data from multiple microarra y experiments. BMC Bioinformatics , 8:364, 2007. J. K. Choi, U . Y u, S. Kim, and O. J. Y oo. Combining multiple microarray studies and mod eling interstudy va riation. Bioinf ormatics , 19:i84 –90, 2003. G. M. Churc h. The p ersonal genome p ro ject. Mole cular Systems Biolo gy , 1:30, 2005. 75 BIBLIOGRAPHY G. R . Cochrane and M. Y. Galperin. The 2010 Nucleic A cids Research Database I ssue and online Database Collection: a communit y of data resources. Nucleic A cids Res e ar ch , 38:D1–4, 2010. B. P . Coe, R. Chari, W. W. Lockwood, an d W. L. L am. Ev olving strategies for global gene expression analysis of cancer. Journal of Cel lular Physiolo gy , 217:590– 597, 2008. J. E. Cohen. Mathematics is biology’s next microscope, only b etter; biology is mathe- matics’ next physics, only b etter. PL oS Biolo gy , 2:e439, 2004. F. S. Collins, E. D. Green, A . E. Gut t mac her, and M. S. Guyer. A vision for the future of genomics researc h. Natur e , 422:835–84 7, 2003. E. Conlon, J. Song, and A. Liu. Bay esian meta-analysis mo dels for microarra y d ata: a comparativ e study . BMC Bioinf ormatics , 8:80, 2007. T. F. Consortium. The T ranscriptional Landscap e of the Mammalian Genome. Sc ienc e , 309:155 9–1563 , 2005. R. T . Co x. Probabilit y , frequency and rea sonable expectation. Americ an Journal of Physics , 17:1–13, 1946. F. Cric k. C entra l dogma of molecular biology . Natur e , 227:561–563, 1970. M. Dai, P . W ang, A. D . Boyd, G. Kosto v, B. Athey , E. G. Jones, W. E. Bunney , R. M. Myers , T. P . Speed , H. Akil, S. J. W atson, and F. Meng. Evolving gene/transcript d efi- nitions significantly alter the interpretation of GeneChip data. Nucleic A cids R ese ar ch , 33:e175 , 2005. C. Darwin. On the Origin of Sp e cies by Me ans of Natur al Sele ction . Murra y , Lon don, 1859. R. H. Davis. The age of mo del organisms. Natur e R eviews Genetics , 5:69– 76, 2004. A. P . Dempster, N. M. Laird, and D. B. R ubin. Maximum lik elihoo d from incomplete data via the EM alg orithm. Journal of the R oyal Statistic al So ciety, Series B , 39:1 –38, 1977. J. L. DeRisi, V. R . Iyer, and P . O. Brown. Exploring the metab olic and genetic control of gene expression on a genomic scale. Scienc e , 278:680–686 , 1997. J. D own w ard. Cancer biology: Signatures guide drug choice. Natur e , 439:274 –275, 2006. S. Draghici, P . Khatri, A. L. T arca, K . Amin, A. Done, C. V oic hita, C. Georgescu, and R. Romero. A sy stems biolog y approach for pathw a y level analysis. Genome R ese ar ch , 17:1537 –1545, 2007. J. T. Dudley , R. Tibshirani, T. Deshpande, and A. J. Butte. Disease signatures are robust across tissues and exp erimen ts. Mole cular Systems Biol o gy , 5:307, 2009. B. Efron and R. Tibshirani. A n Intr o duction to the Bo otstr ap . Chapman & Hall/CR C Monographs on S tatistics & App lied Probability , USA, 1994. M. B. Eisen, P . T. Sp ellman, P . O. Brow n, and D. Botstein. Cluster analysis and displa y of genome-wide expression patterns. Pr o c e e dings of the National A c ademy of Scienc es, USA , 95:14863– 14868, 1998. M. Eisenstein. Mo re than just ’doing the math’. Natur e Metho ds , 3:420–420, 2006. 76 BIBLIOGRAPHY L. L. Elo, L. Lah ti, H. Skottman, M. Ky l¨ aniemi, R . Lahesmaa, and T. Aittok allio. Inte- grating prob e-level expression changes across generations of Aff ymetrix arra ys. Nucleic A cids R ese ar ch , 33:e193, 2005. W. En ard, P . Kh aito vic h, J. Klose, S. Z¨ ollner, F. Heissig, K. Giav alisco, P . Nieselt-Struw e, E. Muchmore, A. V arki, R. Ra vid, G. M. Doxiadis, R. E. Bontrop, and S . P¨ a¨ abo. Intra- and inter-specific v ariati on of primate gene expression p atterns. Scienc e , 296:340–343 , 2002. C. Espinosa-Soto and A . W agner. Sp ecialization Can Drive the Evo lution of Mo dularity. PL oS Computational Biolo gy , 6:e1000719 , 2010. D. Ev ank o. H ac king the genome. Nat ur e M etho ds , 3:495– 495, 2006. D. Ev ank o. S upplement on v isualizing biological data. Nat ur e M etho ds , 7(S1), 2010. L. F euk , A. R . Carson, and S. W. Scherer. Structural v ariation in th e human genome. Natur e R eviews Genetics , 7:85–97, 2006. R. A. Fisher. Statistic al Metho ds for R ese ar ch Work ers . Oliver and Bo yd, Edinburgh, fifth edition, 1934. W. M. Fitch. D istinguishing homologous from analogous proteins. Systematic Zo olo gy , 19:99–1 13, 1970. R. Fletcher. A new approach to v ariable metric algorithms. The Computer Journal , 13: 317–322 , 1970. P . Flicek, B. L. A ken, B. Ballester, K. Beal, E. Bragin, S . Brent, Y. Chen , P . Clapham, G. Coates, S. F airley , S. Fitzgerald, J. F ernandez-Banet, L. Gordon, S. Gr¨ af, S. Haider, M. Hammond, K. How e, A. Jenkinson, N. Johnson, A. K¨ ah¨ ari, D. Keefe, S. Keenan, R. Kinsella , F. Kokocinski, G. Koscieln y , E. Kulesha, D. La wson, I. Longd en , T. Mas s- ingham, W. McLaren, K. Megy , B. Overduin, B. Pritchard, D. Rios, M. Ruffier, M. Sch uster, G. S later, D. Smedley , G. Spudich, Y. A. T ang, S. T rev anion, A. Vilella, J. V ogel, S. White, S. P . Wilder, A. Zadissa, E. Birney , F. Cunningham, I. Dunh am, R. Durbin, X. M. F ern´ andez-Su arez, J. H errero, T. J. P . Hubbard, A. P ark er, G. Proc- tor, J. Smith, and S. M. J. Searle. Ensembl’s 10th y ear. Nucleic A cids R ese ar ch , 38:D557–5 62, 2010. J. A. F oekens, Y. W ang, J. W. Martens, E. M. Berns, and J. G. Klijn. The use of ge- nomic to ols for th e molecular understanding of breast cancer and to guide p ersonalized medicine. Drug Di sc overy T o day , 13:481–48 7, 2008. N. F riedman and D. Koller. Being Bay esian ab out netw ork structu re: A Bay esian ap- proac h to structure disco very in Ba yes ian net w orks. Machine L e arning , 50:95–126, 2003. N. F riedman. Inferring cellular netw ork s using probabilistic graphical mod els. Scienc e , 303:799 –805, 2004. N. F riedman, O . Mosenzon, N. S lonim, and N. Tishb y . Multiv aria te information b ottle- neck. In J. S. Breese and D. Koller, editors, Pr o c e e dings of the 1 7th Confer enc e on Unc ertainty in Artificial I ntel ligenc e (UAI) , p ages 152–161. Morgan Kaufmann Pub- lishers, San F rancisco, CA, 2001. C. F urusaw a and K . K aneko . Zipf ’s law in gene ex pression. Physic al R eview L etters , 90:0881 02, 2003. 77 BIBLIOGRAPHY G10K COS consortium. Genome 10K: a p roposal to obtain whole-genome sequence for 10,000 vertebrate sp ecies. The Journal of Her e dity , 100:65 9–674, 2009. G. Gad, E. Lev in e, and E. Domany . Coupled t w o-w a y clustering analysis of gene microar- ra y data. Pr o c e e di ngs of the National A c ademy of Scienc es, USA , 9 7:12079 –12084 , 2000. J. Gagneur, H. S inha, F. Perocchi, R. Bourgon, W. Hub er, and L. M. Steinmetz. Genome- wide allele- and strand-sp ecific exp ression profiling. Mole cular Systems Biolo gy , 5:274, 2009. L. Gautier, M. Moller, L. F riis-Hansen, and S. Knudsen. Alternativ e mapping of p robes to genes for A ffymetrix c hips. BMC Bioinf ormatics , 5:111 , 2004. A. Gelman, J. B. Carlin, H. S. S tern, and D. B. R ubin. Bayesian Data A nalysis . Chapman & Hall/CR C, Bo ca R aton, FL, USA , second ed ition, 2003. G. K. Gerb er, R. D. Dow ell, T. S . Jaakkola, and D. K. Gifford. Au tomated discov ery of functional generalit y of human g ene expression programs. PL oS Computational Biolo gy , 3:e148, 2007. D. Gershon. DNA microarrays: More than gene ex p ression. Natur e , 437:1195–119 8, 2005. E. R. Gibney and C. M. Nolan. Epigenetics and gene expression. Her e dity , 105:4–13, 2010. J. J. Go eman and P . Buhlmann. Analyzing gene exp ression data in terms of gene sets: metho d ological issues. Bioinformatics , 23:980 –987, 2007. D. Goldfarb. A family of v ariable-metric meth ods derived by va riational means. M athe- matics of Computation , 24:23–26 , 1970. I. Gonz´ alez, S. D´ ejean, P . Martin, O. Gon¸ calv es, P . Besse, an d Baccini A . Highlighting relationships b etw een heterogeneous biological data th rough graphical displa ys based on regularized canonical correlation analysis. Journal of Biol o gic al Systems , 17:173– 199, 2009. D. Greco, P . Somervuo, A. D. Lieto, T. Raitila, L. Nitsch, E. Castr ´ en, and P . Auvin en. Physio logy , pathology and relatedness of human tissues from gene expression meta- analysis. PLo S One , 3:e1880, 2008. I. Gu yon and A. Elisseeff. An in trod u ction to v ariable and feature selection. Journal of Machine L e arning R ese ar ch , 3:1157 –1182, 2003. D. H anisc h, A. Zien, R. Zimmer, and T. Lengauer. Co-clustering of biological netw orks and gene expression data. Bioinformatics , 18:S145–154 , 2002. L. H. Hartw ell, J. J. Hopfield, S. Leibler, and A. W. Murray . F rom molecular to modu lar cell biology . Natur e , 402:C47 –52, 1999. T. Hastie, R. Tibshirani, and J. F riedman. The Elements of Statistic al L e arning . S pringer, New Y ork, second edition, 2009. R. D. Hawkins, G. C. Hon, and B. Ren. Next-generation genomics: an integrativ e approac h. Nat ur e R eviews Genetics , 11:476 –486, 2010. S. Heb er and B. Sick. Qu alit y assessmen t o f A ff ymetrix GeneChip data. OMICS: A Journal of Inte gr ative Biolo gy , 10:358–368, 2006. 78 BIBLIOGRAPHY A.-M. K. Hein, S. Richardson, H. C. Causton, G. K. Ambler, and P . J. Green. BGX: a fully Ba y esian integrated app roac h to the analysis of Affy metrix GeneChip data. Biostatistics , 6:349–373, 2005. N. L. Hjort, C. Holmes, P . M¨ uller, and S. G. W alker, editors. Bayesian nonp ar ametrics . Cam bridge Un iversit y Press, USA, 2010. S. Ho c hreiter, D.-A. Clevert, and K. Obermay er. A new summarization meth o d for affymetrix prob e level data. Bi oi nformatics , 22:943– 949, 2006. J. D. Hoheisel. Microarra y tec hnology: b eyond transcript profiling and genotyp e analysis. Natur e R eviews Genetics , 7:200–21 0, 2006. D. Hosac k, G. Dennis Jr., B. Sherman, H. Lane, and R. Lempicki. Identifying b iological themes within lists of genes with EASE. Ge nome Biolo gy , 4:R70, 2003. H. Hotelling. Relati ons b etw een tw o sets of va riates. Biometrika , 28:321–377, 1936. Z. Hu, C. F an, D. Oh, J. Marron, X. He, B. Qaqish, C. Liv asy , L. Carey , E. Reynolds, L. Dressler, A. Nob el, J. Park er, M. Ew end, L. S a wy er, J. W u , Y . Liu, R. Nanda, M. T retiako v a, A. Orrico, D. Dreher, J . Palazzo, L. Pe rreard, E. Nelson, M. Mone, H. H ansen, M. Mullins, J. Quack en bush, M. Ellis, O. Olopade, P . Bernard, and C. Perou. The molecular p ortraits of b reast tumors are conserved across microarra y platforms. BM C Genomics , 7:96, 2006. E. H ubb ell, W.-M. Liu, and R. Mei. Robust estimators for expression analysis. Bioin- formatics , 18:1585–159 2, 2002. O.-P . Hu ovilainen and L. Lahti. pint: P airwise integration of functional genomics data. Computer program. BioConductor, 2010. M. E. Hu rles, E. T. Dermitzakis, and C. Tyler-Smith. The functional impact of stru ctural v ariation in humans. T r ends in Genetics , 24:238 –245, 2008. C. H uttenhow er and O. Hofmann. A quick guide to large-scale genomic data mining. PL oS Computational Biolo gy , 6:e1000779 , 2010. C. Hu ttenhow er, E. M. Haley , M. A. Hib b s, V. Dumeaux, D. R. Barrett, H. A. Coller, and O . G. T roya nsk aya . Exploring the human genome with functional maps. Genome R ese ar ch , 19:1093–1106, 2009. D. Hw ang, A. G. Rust, S. Ramsey , J. J. Smith, D. M. Leslie, A. D. W eston, P . de A tauri, J. D. A itc hison, L. H oo d, A. F. Siegel, and H . Bolouri. A data in tegration metho dology for systems biology. Pr o c e e dings of the National A c ademy of Scienc es, USA , 102:17296– 17301, 2005. K.-B. Hw ang, S. W. Kong, S. A. Greenb erg, and P . J. Park. Combining gene expression data from different generations of oligon ucleotide arrays. BMC Bioinf ormatics , 5:159, 2004. E. Hyman, P . Kauraniemi, S. Hautaniemi, M. W olf, S . Mousses, E. Rozenblum, M. Ringner, G. Sauter, O. Monni, A. Elk ahloun , O.-P . Kallioniemi, and A. K allio- niemi. Impact of DNA Amplification on Gene Exp ression Pa tterns in Breast Cancer. Canc er R ese ar ch , 62:6240–624 5, 2002. T. Id eker, O. O zier, B. Schw ik o wski, and A. F. Siegel. Disco vering regulatory and sig- nalling circuits in molecular interactio n netw orks. Bioi nformatics , 18:S233–2 40, 2002. 79 BIBLIOGRAPHY J. Ihmels, G. F riedlander, S. Berg mann, O. Sarig, Y. Ziv , and N. Bark ai. Revealing mod u lar organization in the yeast transcriptional netw ork . Natur e Genetics , 31:370– 377, 2002. International Human Genome Sequencing Consortium. Finishing the euchromatic se- quence of the human genome. Nat ur e , 431:93 1–945, 2004. International human genome sequencing consortium. Initial sequencing and analysis of the human genome. Natur e , 409:860–921, 2001. J. P . A. I oannidis, D. B. A llison, C. A. Ball, I. Coulibaly , X. Cui, A. C. Culhan e, M. F alc hi, C. F urlanello, L. Game, G. Jurman, J. Mangion, T. Meh ta, M. Nitzb erg, G. P . Page, E. Petretto, and V. v an No ort. Rep eatabilit y of published microarra y gene exp ression analyses. Natur e Genetics , 41:149–155 , 2009. R. A . Irizarry , B. M. Bolstad, F. Collin, L. M. Cop e, B. H obbs, and T. P . Sp eed. Sum- maries of Affymetrix GeneChip p rob e level data. Nucleic A cids R ese ar ch , 31:e15, 2003a. R. A. Irizarry , B. H obbs, F. Collin, Y. D. Beazer -Barcla y , K . J. Antonellis , U. Scherf, and T. P . Sp eed. Exploration, normaliza tion, and summaries of high density oligon ucleotide arra y prob e level data. Biostatistics , 4:249–264, 2003b. R. A. Irizarry , D. W arren, F. Sp encer, I. F. Kim, S. Biswal, B. C. F rank, E. Gabrielson, J. G. N. Gar cia, J. Geoghegan, G. Germi no, C. Griffin, S. C. Hilme r, E. H offman, A. E. Jedlick a, E. Kaw asaki, F. Martinez-Murillo, L. Morsberger, H. Lee, D. Petersen, J. Quac ken b ush, A. Scott, M. Wilson, Y . Y ang, S. Q. Y e, and W. Y u . Multiple- laboratory comparison of microarray platforms. Nat ur e M etho ds , 2:345–350, 2005. R. A. I rizarry , Z. W u, and H. A . Jaffee. Comparison of A ffymetrix GeneChip ex pression measures. Bioi nformatics , 22:789– 794, 2006. H. Ishw aran and L. F. James. Gibbs sampling metho ds for stic k-breaking p riors. Journal of the Amer ic an Statistic al Asso ciation , 96:161–1 73, 2001. A. K. Jain an d R. C. D ub es. Algorithms for Clustering Data . Prentice Hall, Englew o o d Cliffs, New Jersey , 1988. J. L. Jim´ enez, M. P . Mitchell, and J. G. Sgo uros. Microarra y analysis of orthol ogous genes: conser v ation of t h e translational mac hinery across species at t he sequence and expression level. Genome Biolo gy , 4:R4, 2002. J. M. Johnson, S. Edw ards, D. Shoemaker, and E. E. Schadt. Dark matter in the genome: evidence of widespread transcription detected by microarra y tiling exp eriments. T r ends in Genetics , 21:93–102 , 2005. L. J. Johnson and P . J. T rick er. Epigenomic plasticity within p opulations: its evolutionary significance and p otential. Her e dity , 105:113–121, 2010. N. Johnson, V. Sp eirs, N. J. Curtin, and A. G. H all. A comparativ e stu dy of genome- wide SN P , CGH microarra y and p rotein exp ression analysis to exp lore genotypic and phenotypic mechanis ms of acquired anties trogen resistance in breast cancer. Br e ast Canc er R ese ar ch and T r e atment , 111:55 –63, 2008. I. K. Jordan, L. Mari˜ no-R amirez, and E. V. Ko onin. Ev olutionary significance of gene expression divergence Gene , 345:119–1 26, 2005. A. R. Joyce and B. O. Palss on. The mo del organism as a system: integrating ’omics’ data sets. Nat ur e R eviews Mole cular Cel l Biolo gy , 7:198–210, 2006. 80 BIBLIOGRAPHY M. Kanehisa, M. Araki, S. Goto, M. H attori, M. Hirak aw a, M. Itoh , T. Kataya ma, S. Kaw ashima, S. Okud a, T. T okimatsu, and Y . Y amanishi. KEGG for linking genomes to life and th e en vironment. Nuclei c A cids R ese ar ch , 36:D480–484 , 2008. M. Kanehisa, S. Goto, M. F u rumic hi, M. T anab e, and M. Hirak aw a. KEGG for represen- tation and analysis of molecular netw orks involving diseases and drugs. Nucleic A cids R ese ar ch , 38:D355–360, 2010. S. Kaski, J. Sinkkonen, and A . Klami. Discriminative clustering. Neur o c omputing , 69: 18–41, 2005. S. Katz, R. A. Irizarry , X . Lin, M. T ripput i, and M. W. Porter. A summ arization approach for Affy metrix GeneChip data using a reference training set from a large, b iologica lly diverse database. BMC Bioi nformatics , 7:464, 2006. J. Kettenring. Canonical analysis of several sets of v ariables. Biometrika , 58:433–451 , 1971. S. Kilpinen, R. Autio, K. Ojala, K. Iljin, E. Buc her, H. Sara, T. Pisto, M. Saarela, R. I . S kotheim, M. Bjorkman, J.-P . Mpindi, S. Haapa-Paananen, P . V ainio, H. Ed- gren, M. W olf, J. Astola , M. Nees, S. H autaniemi, and O. Kallioniemi. Systematic bioinformatic analysis of ex pression levels of 17,330 human genes across 9,783 samples from 175 types of healthy and path ological tissues. Genome Bi olo gy , 9:R139, 2008. A. K lami and S. Kaski. Probabilistic approach t o detecting dep end encies b etw een data sets. Neur o c omputing , 72:39–46, 2008. A. K lami, S. Virtanen, an d S. Kaski. Ba y esian ex p onential family pro jections for coupled data sources. In P . Grunw ald and P . Spirtes, editors, Pr o c e e dings of the 26th Confer enc e on Unc ertainty in A rtificial Intel li genc e (UAI) , pages 286–293. AUAI Press, Corv alli s, Oregon, 2010. J. Kleinberg. An impossibility theorem for clustering. In S. Beck er, S. Thrun, and K. Ob erma yer, editors, Ad vanc es in Neur al Inf ormation Pr o c essing Systems 15 , pages 446–453 . MIT Press, Cambridge, MA, 2002. T. Kohonen. Self -Or ganizing M aps . Springer, Berlin, th ird edition, 2001. T. Kohonen . S elf-organized formation of topologically correct feature maps. Biolo gic al Cyb ernetics , 43:59– 69, 1982. D. Koller and N. F riedman. Pr ob abilistic Gr aphic al M o dels: Principles and T e chniques . MIT Press, USA , 2009. S. Kullback. Information The ory and Statistics . Wiley , New Y ork , 1959. K. Kurihara, M. W elling, and Y. W. T eh. Collapsed v ariational diric hlet process mixt ure mod els. In 20th International Joint Confer enc e on Art ificial Intel l igenc e (IJCAI 2007) , pages 2796–28 01. Morgan Kaufmann Publishers Inc, San F rancisco, CA, USA, 2007a. K. K u rihara, M. W elling, and N . Vlassis. Accelerated v ariational Dirichlet p rocess mix- tures. In B. Sch¨ olk opf, J. Platt, and T. Hoffman, editors, A dvanc es in Neur al Infor- mation Pr o c essing Systems 19 , pages 761–76 8. MIT Press, Cambridge, MA, 2007b. E. Laa jala, T. Aittoka llio, R. Lahesmaa, and L. L. Elo. Prob e-level estimation improv es the detection of differential sp licing in Affymetrix exon arra y studies. Genome bi olo gy , 10:R77, 2009. 81 BIBLIOGRAPHY K. Lage, N . T. Hansen, E. O. Karlb erg, A. C. Eklu n d, F. S . Ro que, P . K . Donaho e, Z. Sza- llasi, T. S. Jensen, and S. Brunak. A large-scale analysis of tissue-sp ecific pathology and gene expression of human disease genes and complexes. Pr o c e e dings of the National A c ademy of Scienc es, USA , 105:20870–2 0875, 2008. J. Lam b, E. D. Cra wford, D . Pec k, J. W. Mod ell, I. C. Blat, M. J. W rob el, J. Lerner, J.-P . Brunet, A. S ubramanian, K. N. Ross, M. Reich, H. Hieronym us, G. W ei, S. A. Armstrong, S. J. Hagga rty , P . A. Clemons, R . W ei, S. A. Ca rr, E. S. Lander, and T. R. Golub. The connectivity ma p: Using gene-expression signatures to connect small molecules, genes, and disease. Scienc e , 313:1929– 1935, 2006. E. Lander. The new genomics: G lobal views of biology . Scienc e , 274:536–53 9, 1996. D. A. Lauffenburger. Cell signaling pathw a ys as control mod u les: Complexity for sim- plicit y . Pr o c e e dings of the National A c ademy of Scienc es, U SA , 97:5031 –5033, 2000. M. Law, M. Figueiredo, and A. Jain. S im ultaneous feature selection and clustering using mixture models. IEEE T r ansactions on Pattern A nalysis and Machine Intel ligenc e , 26:1154 –1166, 2004. L. Lazzeroni and A. Owen. Plaid models for gene expression data. Stat istic a Sinic a , 12: 61–86, 2002. K.-A. Lˆ e Cao, I. Gonz´ alez, and S. D´ ejean. in tegrOmics: an R p ac k age t o unrav el rela- tionships b etw een tw o omics datasets. Bi oinformatics , 25:2855– 2856, 2009. H. Ledford. The cancer genome challenge. Natur e , 464:972–97 4, 2010. E. Lee, H.-Y. Chuang, J.-W. Kim, T. Ideker, and D . L ee. Inferring pathw a y activity to w ard precise d isease classification. PL oS Computational Biolo gy , 4:e1000217, 2008. D. Levine, D. Haynor, J. Castle, S . Stepaniants, M. Pel legrini, M. Mao, and J. Johnson. P athw ay and gene-set activ ation measurement from mRNA expression data: the tissue distribution of human pathw ays. Genome Biolo gy , 7:R93, 2006. T. R. Lezon, J. R. Banav ar, M. Cieplak, A. Maritan, and N. V. F edoroff. F rom the Cov er: Using the principle of entropy maximization to infer genetic interac tion netw ork s from gene expression patterns. Pr o c e e dings of the National A c ademy of Scienc es, USA , 103: 19033–1 9038, 2006. C. Li and W. H . W ong. Mo del-based analysis of oligon ucleotide arra ys: Expression index computation and outlier detection. Pr o c e e dings of the National A c ademy of Scienc es, USA , 98:31–36, 2001. X. Li, Z. He, and J. Zhou. Selection of optimal oligon ucleotide p robes for microarra ys using multiple cri teria, global alignment and parameter estimation. Nucleic A cids R ese ar ch , 33:6114–6123, 2005. Y. Li, P . Agarw al, and D . Ra jagopalan. A global pathw ay crosstalk netw ork. Bioinf or- matics , 24:1442–14 47, 2008. S. Liang, Y. Li, X . Be, S. How es, and W. Liu. Detectin g and profiling tissue-selective genes. Physiolo gic al Genomics , 26:158 –162, 2006. E. Lieb erman-Aid en , N. L. v an Berkum, L. Williams, M. Imak aev, T. Rago czy , A. T elling, I. Amit, B. R. La joie, P . J. Sab o, M. O. Dorsc hner, R. Sandstrom, B. Bernstein, M. A. Bender, M. Groudine, A. Gnirke, J. Stamato yannopoulos, L. A . Mirny , E. S . Lander, and J. Dekk. Comprehensiv e Mapping of Long-Range Interactio ns Reveals F olding Principles of the Human Genome. Scienc e , 326:289 –293, 2009. 82 BIBLIOGRAPHY X. Liu, M. Milo, N. D. Lawrence, and M. Rattray . A tractable p robabilistic mo del for Affymetrix prob e-level analysis across multiple chips. Bioinformatics , 21:3637 –3644, 2005. D. Lo ckhart, H . Dong, M. Byrne, M. F ollettie, M. Gallo, M. Chee, M. Mittmann, C. W ang, M. Kobaya shi, H. Horton, and E. B ro wn. Expression monitoring by hy- bridization to high- density oligon ucleotide arra ys. Natur e Biote chnolo gy , 14:1675–1680, 1996. J. E. Lucas, C. M. Carv alho, J. L.-Y. Chen, J.-T. Chi, and M. W est. Cross-study pro jections of genomic biomarkers : An ev aluation in cancer genomics. PL oS ONE , 4: e4523, 2009. M. Luk k, M. Kapu shesky , J. Nikk il¨ a, H . Parkinson, A. Goncalves, W. Hub er, E. Ukkonen, and A . Brazma. A global map of human gene expression. Natur e Biote chnolo gy , 28: 322–324 , 2010. S. C. Madeira and A. L. Olivei ra. Biclustering algorithms for b iologica l data analysis: a survey . IEEE/ACM T r ansactions on Computational Biol o gy and Bioinformatics , 1: 24–45, 2004. MAQC Consortium. T he microarray quality control ( MA QC) pro ject shows inter- and intrapla tform repro ducibility of gene expression measurements. Natur e Biote chnolo gy , 24:1151 –1161, 2006. E. Ma yr. What makes biolo gy unique?: c onsider ations on the autonomy of a scientific discipline . Cambridge Universit y Press, New Y ork, 2004. M. N . McCall, B. M. Bolstad, and R . A . Irizarry . F rozen robust multiar ra y anal ysis (fRMA). Bi ostatistics , 11:242– 53, 2010. J. D. McPherson. Next-generation gap. Natur e Metho ds , 6(S11):S2–5, 2009. B. H . Mecham, G. T. Klus, J. Strov el, M. A ugustus, D. Byrne, P . Bozso, D. Z. W et- more, T. J. M ariani, I. S. K ohane, and Z. Szal lasi. Sequence-matched p robes pro- duce increased cross-platform consistency and more rep rodu cible biological results in microarra y-based gene expression measurements. Nucleic A ci ds Re se ar ch , 32:e7 4, 2004a. B. H. Mecham, D. Z . W etmore, Z. Szallasi, Y. Sadovsky , I. Kohane, and T. J. Mariani. Increased measurement accuracy for sequence-verified microarra y prob es. Physiolo gic al Genomics , 18:308–315 , 2004b. R. Mei, E. Hubb ell, S. Bekiranov, M. Mittmann, F. C. Christians, M.-M. Shen, G. Lu, J. F ang, W.-M. Liu, T. Ryder, P . Kaplan, D. K u lp, and T. A . W ebster. Prob e selec- tion for h igh-density oligon ucleotide arra ys. Pr o c e e dings of the National A c ademy of Scienc es, USA , 100:11237–11 242, 2003. M. Milo, A. F azeli, M. Niranjan, and N. Lawrence. A probabilistic mod el for the extrac- tion of expression levels from oligonucleoti de arra ys. Bio chemic al So ci ety T r ansactions , 31:1510 –1512, 2003. D. Mon taner and J. Dopazo. Multidimensional gene set analysis of genomic d ata. PL oS One , 5:e10348, 2010. D. Monta ner, P . Minguez, F. A l-Shahrour, and J. Dopazo. Gene set internal coherence in the context of functional profiling. BMC Genomics , 10:197 , 2009. 83 BIBLIOGRAPHY Mouse Genome S equencing Consortium. Initial sequencing and comparativ e analysis of the mouse genome. Natur e , 420:520–56 2, 2002. P . M ¨ uller and F. A. Quintana. Nonparametric Bay esian Data Analysis. Statist ic al Scienc e , 19:95–1 10, 2004. C. Myers, D. Robson, A. Wible, M. Hibb s, C. Chiriac, C. Theesfeld, K. Dolinski, and O. T roy ansk ay a. Disco v ery of biological n etw orks from diverse functional genomic d ata. Genome Bi olo gy , 6:R114, 2005. S. Myllyk angas, S . Junn ila, A. Kokkola, R. Au tio, I. Scheinin, T. Kiviluoto, M.-L. Karjalainen-Lindsberg, J. Hollm ´ en , S. Knuutila, P . Puolakk ainen, and O . Monn i. In- tegrated gene copy number and expression microarra y analysis of gastric cancer high- ligh ts p otential target genes. International Journal of Canc er , 123:817– 825, 2008. S. Nacu, R . Critc hley-Thorne, P . Lee, and S. Holmes. Gene ex p ression netw ork analysis and applications to immunology. Bi oinformatics , 23:850–8 58, 2007. F. Naef and M. O. Magnasco. S olving the riddle of th e bright mismatc hes: Lab eling and effective bin ding in oligon ucleotide arrays. Physic al R eview E , 68:011906, 2003. D. Nam and S.-Y. Kim. G ene-set approach for expression p attern analysis. Briefings i n Bioinformatics , 9:189–197, 2008. J. Noirel, G. S anguinetti, and P . C. W right. Identifying differentially exp ressed subnet- w orks with MMG. Bioinformatics , 24:2792–2793, 2008. B. A. Nov ak and A . N. Jain. Pa thw ay recognition and augmentati on by computational analysis of microarra y ex pression data. Bioinformatics , 22:233–241, 2006. D. Nuyten and M. v an de Vijver. Using microarra y analysis as a prognostic and p redictive tool in oncology: focus on breast cancer and n ormal tissue toxicit y . Semi nars in R adiation Onc olo gy , 18:105 –114, 2008. P . N ymark, P . M. Lindholm, M. V . Korp ela, L. Lahti, S. R uosaari, S. Kaski, J. H ollm´ en, S. Anttila, V. L. Kinnula, and S. Knuutila. Gene expression profiles in asbestos-exp osed epithelial and mesothelial lung cell lines. BMC Genomics , 8:62, 2007. A. Ocana and A . Pandiella. Personali zed therapies in the cancer ”omics” era. Mole cular Canc er , 9:202 , 2010. Q. Pan, O. Shai, L. J. Lee, B. J. F rey , and B. J. Blenco w e. Deep surveying of alterna- tive splicing complexity in th e human transcriptome by high-throughp ut sequencing. Natur e Genetics , 40:1413 –1415, 2008. S. C. J. Park er, L. Hansen, H. O. Abaan, T. D. T ullius, and E. H. Margulies. Lo cal DNA top ography correlates with functional nonco ding regions of the human genome. Scienc e , 324:38 9–392, 2009. E. Parkhomenk o, D. T ritchler, and J. Beyene. Sparse canonical correlation analysis with application to genomic data integration. Statistic al Applic ations i n Genetics and Mole cular Bi olo gy , 8:1, 2009. H. Pa rkinson, M. Kapushesky , N . Kolesnik o v, G. R ustici, M. Sh o jatalab, N. Ab eygu- naw ardena, H. Berub e, M. D ylag, I. Emam, A. F arne, E. H ollow ay , M. Lukk, J. Mal- one, R. Mani, E. Pilic hev a, T. F. Rayner, F. R ezw an, A . Sh arma, E. Williams, X. Z . Bradley , T. Adamusiak, M. Brandizi, T. Burdett , R. Coulson, M. Kresty anino v a, P . Kurnosov, E. Maguire, S. G. Neogi, P . R occa-Serra, S.- A . Sansone, N. Sklyar, 84 BIBLIOGRAPHY M. Zhao, U. Sark ans, and A. Brazma. Arra y Express up date–from an archiv e of func- tional genomics exp eriments to th e atlas of gene expression. Nucleic A cids R ese ar ch , 37:D868–8 72, 2009. L. P arsons, E. H aque, an d H . Liu . S ubspace clustering for high dimensional d ata: A review. Sigkdd Explor ations , 6:90–105, 2004. H. Pearso n. G enetics: what is a gene? Natur e , 441:398–401, 2006. J. L. Phillips, S . W. H a yw ard, Y. W ang, J. V asselli, C. Pa vlovic h, H . P adilla-Nash, J. R . P ezullo, B. M. Ghad imi, G. D . Grossfeld, A. Rivera, W. M. Linehan, G. R . Cunha, and T. Ried. The Consequences of Chromosomal Aneu p loidy on Gene Expression Profiles in a Cell Line Mo del for Prostate Carcinogenesis. Canc er R ese ar ch , 61:8143 –8149, 2001. D. Pinkel and D. G. A lbertson. Arra y comparativ e genomic hybridization and its appli- cations in cancer. Natur e Genetics , 37:S11–17, 2005. A. Pola nski and M. Kimmel. Bi oinformatics . Springer, Germany , 2007. E. Prak and H. Kazazian Jr. Mobile elemen ts and t h e human genome. Natur e R eviews Genetics , 1:134–144, 2000. T. M. Przytyck a, M. S in gh , and D. K. Slonim. T ow ard the d ynamic in teractome: it’ s abou t time. Briefings i n Bi oinformatics , 11:15–29 , 2010. L.-X. Qin. A n integrativ e analysis of microRNA and mRN A Expression - a case study. Canc er I nformatics , 6:369–3 79, 2008. J. Quac ken bush. Co mputational analysis of microarra y data. Natur e R eviews Genetics , 2:418–4 27, 2001. J. R ac hlin, D. D. Cohen, C. Can tor, and S . Kasif. Biologi cal context netw orks: a mosaic view of th e interactome. Mole cular Systems Bi olo gy , 2:66, 2006. A. Ramasam y , A. Mondry , C. C. H olmes, and D. G. Altman. Key issues in cond ucting a meta-analysis of gene expression microarra y datasets. PL oS Me dicine , 5:e184, 2008. C. E. Rasmussen. The infin ite gaussian mixture model. In S. A. Solla, T. K. Leen, and K.-R. M ¨ uller, editors, A dvanc es i n Neur al Information Pr o c essing Systems 12 , pages 554–560 . MIT Press, Cambridge, MA, 2000. J. L. Reed, I. F amili, I. Thiele, and B. O. Palsson. T o w ards multidimensional genome annotation. Natur e R eviews Genetics , 7:130– 141, 2006. M. Reimers. Making informed c hoices ab out microarra y data analysis. PL oS Computa- tional Biolo gy , 6:e1000786, 2010. D. R eiss, N. Baliga, and R . Bonneau. Integrated biclustering of heterogeneous genome- wide d atasets for the inference of global regulatory netw orks. BMC Bioinformatics , 7: 280, 2006. S. Rogers, M. Girolami, C. Campb ell, and R . Breitling. The latent pro cess decomposition of cDNA microarra y data sets. I EEE/A CM T r ansactions on Computational Biolo gy and Bioinformatics , 2:143–156, 2005. S. Rogers, A . K lami, J. Sink konen, M. Girolami, and S. Kaski. Infinite factorization of multiple non- parametric views. Machine L e arning , 79:201, 2010. 85 BIBLIOGRAPHY R. R oth, P . Hevezi, J. Lee, D. Willhite, S. Lechner, A. F oster, a nd A. Zlotnik. Gene expression analyses revea l molecular relationships among 20 regions of the human CNS. Neur o genetics , 7:67–8 0, 2006. V. Roth and T. Lange. F eature selection in clustering problems. I n S. Thrun, L. Saul, and B. Sch¨ olkopf, editors, A dvanc es in Neur al Information Pr o c essing Systems 16 , pages 473–480 . MIT Press, Cambridge, MA, 2004. J. Russ and M. E. F ut sc hik. Comparison and consolidation of microarra y data sets of human tissue ex pression. BMC Genomics , 11:305, 2010. B. Sadiko v ic, M. Y oshimoto, K. Al-Romaih, G. Maire, M. Zielensk a, an d J. A. S quire. In vitro analysis of integrated global h igh-resolution DNA methylatio n profiling with genomic imbalance an d gene expression in osteosarcoma. PL oS One , 3:e283 4, 2008. Y. S aeys, I. Inza, and P . Larra ˜ naga. A review of feature selection techniques in bioinfor- matics. Bioi nformatics , 23:2507 –2517, 2007. K. Salari, R. Tibshirani, and J. R. Poll ac k. DR -Integrator: a new analytic t ool for integ rating DNA copy number and gene ex pression data. Bioi nf ormatics , 26:414–416, 2010. H. Sara, O. Kallioniemi, and M. Nees. A decade of cancer gene profiling: from molecular p ortraits to molecular function. Metho ds in M ole cular Biolo gy , 576:61– 87, 2010. R. S. Sa v age, Z. Ghahramani, J. E. Griffin, B. J. d e la Cruz, and D. L. Wild. Disco ve ring transcriptional modules b y Bay esian data in tegration. Bi oinformatics , 26:i158 –167, 2010. E. E. Schadt. Molecular netw orks as sensors and driv ers of common h uman d iseases. Natur e , 461:218– 223, 2009. M. Sch¨ afer, H. Sch w ender, S. Merk, C. Haferlach, K. Ickstadt, and M. Du gas. Integrated analysis of cop y num ber alterations and gene expression: a biv ariate assessment of equally directed abnormalities. Bioinformatics , 25:3228 –3235, 2009. I. Scheinin, S. Myllyk angas, I. Borze , T. Bohling, S . Knuutila, and J. Saharinen. CanGEM: mining gene copy num ber changes in cancer. Nucleic A ci ds Re se ar ch , 36:D830–8 35, 2008. U. Scherf, D. T. Ross, M. W altham, L. H. Smith, J. K. Lee, L. T anab e, K. W. Kohn , W. C. Reinhold, T. G. Myers, D. T. A ndrews, D. A . Scudiero, M. B. Eisen, E. A. Sausv ille, Y. Pommier, D. Botstein, P . O. Brown, and J. N . W einstein. A gene exp ression database for the molecular pharmacology of cancer. Natur e Genetics , 24:236 –44, 2000. B. Sch¨ olkopf and A. J. Smola. L e arning with kernels: supp ort ve ctor machines, r e gular- ization, optimization, and b eyond . MIT Press, USA, 2002. J. W. Schopf. F ossil evidence of A rchaean life. Philosophic al T r ansact ions of the R oyal So ciety of L ondon. Series B, 361:869–885, 2006. E. Schr¨ odinger. What is life? Mind and Matter . Cambridge Universit y Press, 1944. J. Sebat. Ma jor changes in our dna lead to major c hanges in o ur thinking. Natur e Genetics , 39:S3–5, 2007. E. Segal, A. Battle, and D. Koller. Decomp osing gene expression into cellular pro cesses. In R . B. Altman, A . K . Dunker, L. Hun ter, T. A. Jung, and T. E. Klein, editors, Pr o c e e dings of Pacific Symp osium on Bi o c omputing (PSB 2003) , p ages 89–100. W orld Scientific, S ingapore, 2003a. 86 BIBLIOGRAPHY E. Segal, M. S hapira, A. Regev, D. Pe’er, D. Botstei n, D. Koller, and N. F riedman. Mo d- ule netw orks: identif ying reg ulatory mo dules and th eir condition-sp ecific regulato rs from gene expression data. Natur e Genetics , 34:166–17 6, 2003b. E. Segal, B. T ask ar, A. Gasc h, N. F riedman, and D . Koller. Rich probabilistic models for gene expression. Bioinformatics , 17(S1):i243 –252, 2003c. E. S egal, H. W ang, and D. Koller. Disco vering molecular p athw ays from protein inter- action and gene expression data. Bioinformatics , 19(S1):i264–272 , 2003d. E. Segal, N . F riedman, D. Koller, and A. Regev. A mo dule map showing conditional activit y of exp ression mo dules in cancer. Nat ur e Genetics , 36:1090–1098 , 2004. D. F. Shanno. C onditioning of q uasi-Newton method s for function minimization. Math - ematics of Computation , 24:647–656 , 1970. C. E. Shann on . A mathematical theory of communication. Bel l System T e chnic al Journal , 27:379– 423, 623–656, 1948. A. J. Sharp, Z. Cheng, and E. E. Eichler. Structural v ariation of the human genome. Ann ual R eview of Genomics and Human Genetics , 7:407–442, 2006. R. S hen, A. Olshen, a nd M. Ladanyi. Integrativ e clustering of m ultiple genomic d ata types using a join t latent v ariable mo del with app lication t o breast and lung cancer subtype analysis. Bioinformatics , 25:29 06–2912 , 2009. M. Shiga, I . T ak iga w a, and H. Mamitsuk a. A nnotating gene function by co mbining expression data with a mod ular gene netw ork. Bioinformatics , 23:i468–478 , 2007. C. Sigg, B. Fischer, B. Ommer, V . Roth, and J. Buhmann . Nonnegative CCA for au- dio visual source separation. In Pr o c e e dings M LSP’ 07 IEEE International Workshop on Machine Le arning for Signal Pr o c essing , pages 253–258. IEEE Signal Pro cessing Society , Zurich, 2007. J. Sinkkonen, S . Kaski, and J. Nikkil¨ a. Discriminativ e clustering: Optimal contingency tables by lear ning metrics. In T. Elomaa, H . Mannila, and H. T oivo nen, editors, Pr o c e e dings of the ECML’02, 13th Eur op e an Confer enc e on Machine L e arning , pages 418–430 . Springer, Berlin, 2002. J. Sinkkonen, J. Nikk il¨ a, L. Lahti, an d S. Kaski. Asso ciative clustering by maximizing a Ba ye s factor. T echnical Rep ort A68, Helsinki Universit y of T echnology , Lab oratory of Computer and Information Science, Esp oo, Finland, 2003. J. Sinkkonen, S. Kaski, J. N ikkil¨ a, and L. Lahti. Asso ciative Clustering (AC): T echnical Details. T echnical Rep ort A84, Helsinki Univ ersit y of T ec hnology , Esp o o, Finland, 2005. E. Sliwersk a, F. Meng, T. Sp eed, E. Jones, W. Bunney , H. Akil, S. W atson, and M. Burmeister. SNPs on chips: the hidden genetic co de in expression arrays. Bi olo gic al Psychiatry , 61:13–16, 2007. J. S ommer. The d ela y in sharing researc h data is costing liv es. Natur e Me dicine , 16:744, 2010. C. Soneson, H. Lilljeb jorn, T. Fioretos, and M. F ontes. Integrativ e analysis of gene expression an d copy number alterations using canonical correlation analysis. B M C Bioinformatics , 11:191, 2010. 87 BIBLIOGRAPHY S. Sonnenburg, M. L. Braun, C. S . Ong, S. Bengio, L. Bottou, G. Holmes, Y. LeCun, K.- R. M ¨ uller, F. Pereira , C. E. Rasm ussen, G. R¨ atsch, B. S c h¨ olko pf, A. Smola, P . Vincen t, J. W eston, and R. Williamson. The need for open source softw are in machine learning. The Journal of Machine L e arning Re se ar ch , 8:2443–2466 , 2007. T. Sørlie, C. M. Perou, R. Tibshirani, T. A as, S. Geislerg, H. Johnsen, T. H astie, M. B. Eisen, M. van de Rijn, S. S. Jeffrey , T. Thorsen, H . Quist, J. C. Matese, P . O. Brown, D. Botstein, P . E. Lønning, and A .-L. Børresen-Daleb. Gene expression patterns of breast carcinomas distinguish tumor sub classes with clinical implications. Pr o c e e dings of the National A c ademy of Scienc es, USA , 98:10869– 10874, 2001. J. Stetefeld and M. A. Ruegg. Stru ctural and functional diversit y generated by alternative mRNA splicing. T r ends in Bi o chemic al Scienc es , 30:515–521 , 2005. V. Sto dden . The scie ntific metho d i n practice: Repro ducibility in th e computational sciences. MIT Slo an R ese ar ch Pap er , 4773–10, 2010. M. R. S tratton, P . J. Campb ell, and P . A. F utreal. The cancer genome. Natur e , 458: 719–724 , 2009. L. Str¨ omb¨ ac k and P . Lambrix. Representations of molecular pathw a ys: an ev aluation of SBML, PSI MI and BioP AX. Bioinformatics , 21:4401 –4407, 2005. J. M. Stuart, E. S egal, D. Koller, and S. K . Kim. A gene-co expression n et w ork for global disco ve ry of conserved genetic mo dules. Sc ienc e , 302:249 –255, 2003. A. I. Su, M. P . Cooke, K. A. Ching, Y. Hak ak, J. R. W alker , T. Wiltshire, A. P . Orth, R. G. V ega, L. M. Sapin oso, A. Moqrich, A. P atapoutian, G. M. Hampton, P . G. Sch ultz, and J. B. H ogenesc h. Large-scale analysis of the human and mou se transcrip- tomes. Pr o c e e dings of the National Ac ademy of Sci enc es, USA , 99:4465–447 0, 2002. A. I. Su, T. Wiltshire, S. Batalo v, H. Lapp, K. A. Ching, D. Block, J. Zh ang, R. So den, M. Hay ak aw a, G. Kreiman, M. P . Co oke, J. R. W alker, and J. B. Hogenesch. A gene atlas of th e mouse and h uman protein-enco ding transcriptomes. Pr o c e e dings of the National A c ademy of Scienc es, USA , 101:6062–606 7, 2004. J. Su, B.-J. Y oon , and E. R. Doughert y . Accurate and reliable cancer classification based on probabilistic inference of pathw a y activity . PL oS ONE , 4:e8161, 2009. A. S ubramanian, P . T ama yo , V. K. Mo otha, S. Mukherjee, B. L. Eb ert, M. A. Gillette, A. Pa ulo vich, S. L. Pomero y , T. R . Golub, E. S. Lander, and J. P . Mesiro v. Gene set enrichmen t analysis: A knowledge-based approac h for interpreting genome-wide expression p rofi les. Pr o c e e dings of the National A c ademy of Scienc es, USA , 102:155 45– 15550, 2005. S. Suth ram, J. T. Dudley , A. P . Chiang, R . Chen, T. J. Hastie, and A. J. Butte. Netw ork- based el ucidation of human d isease similarities rev eals common functional modules enric hed for plurip otent drug targets. PL oS Computational Biolo gy , 6:e100066 2, 2010. P . A. C. ’t Hoen , Y. Ariyurek, H. H. Thyges en, E. V reugdenhil, R. H. A . M. V ossen, R. X. de Menezes, J. M. Bo er, G.-J. B. v an Ommen, and J. T. den Du nnen. Deep sequencing-based expression analysis shows ma jor advances in robustness, resolution and inter-lab p ortability ov er five microarray platforms. Nucleic A cids R ese ar ch , 36: e141, 2008. P . T amay o, D. Slonim, J. Mesirov, Q. Zhu, S. K itareewan, E. Dmitrowsky , E. S. Lander, and T. R. Golub. I nterpreting p atterns of gene expression with self-organizing maps: Method s and application to hematop oietic differentia tion. Pr o c e e dings of the National A c ademy of Scienc es, USA , 96:2907–291 2, 1999. 88 BIBLIOGRAPHY A. T ana y , R . Sharan, and R . Shamir. Disco v ering statistically significan t biclusters in gene expression data. Bioinformatics , 18:S136–14 4, 2002. A. T anay , R. Sharan, M. Kupiec, and R . S hamir. Revealing mo dularity and organiza- tion i n the yeast mo lecular net w ork by integra ted analysis of highly heterogeneous genomewide data. Pr o c e e dings of the National Ac ademy of Scienc es , 101:2981 –2986, 2004. A. T anay , I. Steinfeld, M. Kupiec, and R. S h amir. Integrativ e analysis of genome-wide exp eriments in th e context of a large h igh-throughput data compen dium. Mole cular Systems Biolo gy , 1:000 2, 2005. A. L. T arca, S . Draghici, P . Kh atri, S. S. Hassan, P . Mittal, J.-S. K im, C. J. Kim, J. P . Kusanovic, and R . Romero. A no ve l signaling p athw ay impact analysis. Bioinformatics , 25:75–8 2, 2009. B. S. T aylo r, J. Barretina, N. D. So cci, P . DeCarolis, M. Ladanyi, M. Meyerson, S. S in ger, and C. Sander. F un ct ional copy-num b er alterations in cancer. PL oS ONE , 3:e3179 , 2008. The Cancer Genome Atlas Research Netw ork. Comprehensiv e genomic characteri zation defines human glioblastoma genes and core p athw ays. Natur e , 455:1061 –1068, 2008. The ENCODE Pro ject Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot pro ject. Natur e , 447:799–81 6, 2007. R. Tibshira ni, T. Hastie, B. Narasimhan, and G. Ch u. Diagnosis of m ultiple cancer typ es by shrunken centroids of gene expressio n. Pr o c e e di ngs of the National A c ademy of Scienc es, USA , 99:6567–6572 , 2002. M. M. Tice and D. R . Low e. Photosynthetic microbial mats in the 3,416-Myr-old o cean. Natur e , 431:549– 552, 2004. C. Tilstone. V ital statistics. Natur e , 424:610–61 2, 2003. N. Tishb y , F. C. Pereira , and W. Bialek. The information b ottlenec k meth o d. In 37th Ann ual Al l erton Confer enc e on Communic ation, Contr ol, and Computing , pages 368– 377. Universit y of Illinois, Urbana, Illinois, 1999. P . T¨ or¨ onen, M. K olehmainen, G. W ong, and E. Castr ´ en. Analysis of gene exp ression data using self-organizing maps. FEBS L ett ers , 451:14 2–146, 1999. O. G . T roy anska ya. Pu t ting microa rra ys in a context: Integrated analysis of div erse biological data. Briefings in Bioi nf ormatics , 6:34–4 3, 2005. Y. T u, G. Stolo v itzky , and U . Klein. Quanti tative n oise analysis f or gene expression microarra y exp eriments . Pr o c e e dings of the National Ac ademy of Scienc es, USA , 99: 14031–1 4036, 2002. J. T ukey . Explor atory data analysis . Ad dison-W esley , Reading, MA, 1977. I. Ulitsky and R. S hamir. I dentification of functional mo dules using netw ork top ology and high-throughp u t data. BMC Systems Bi olo gy , 1:8, 2007. I. Ulitsky and R. Shamir. Identif ying functional mod ules using expression profiles and confidence-scored p rotein interactions. B i oinformatics , 25:1158–1 164, 2009. L. J. v an ’t V eer and R. Bernards. Enabling p ersonalized cancer medicine th rough analysis of gene-expression p atterns. Natur e , 452:564 –570, 2008. 89 BIBLIOGRAPHY W. N. v an Wieringen and M. A. v an de Wiel. N onparametric testing for DNA cop y num ber induced d ifferenti al mRNA gene expression. Biometrics , 65:19–2 9, 2009. C. J. V ask e, S. C. Benz, J. Z. Sanborn, D. Earl, C. S zeto, J. Zhu, D. Haussler, and J. M. Stuart. Inference of p atient-sp ecific pathw a y activities from m ulti-dimensional cancer genomics data using P AR ADIGM. Bioinformatics , 26:i237–245, 2010. I. V astrik, P . D’Eustachio , E. Schmidt, G. Joshi-T ope, G. Gopinath, D. Croft, B. de Bono, M. Gillespie, B. Jassa l, S. Lewis, L. Matthews, G. W u, E. Birney , and L. Stein. Reac- tome: a know ledge b ase of biologic pathw ays and pro cesses. Genome Bi olo gy , 8:R39, 2007. J. C. V enter, M. D. Adams, E. W. Myers, P . W. Li, R . J. Mural, G. G. Sutton, H. O. Smith, M. Y and ell, C. A. Ev ans, R. A . Holt, J. D. Go cayne, P . Amanatides, R. M. Ballew, D. H. Huson, J. R. W ortman, Q. Zhang, C. D. Ko dira, X. H. Zh eng, L . Chen, M. Skupski, G. Subramanian, P . D. Thomas, J. Zhang, G. L. Gabor Miklos, C. Nelson, S. Brod er, A. G. Clark, J. Nadeau, V. A. McKusick, N . Zinder, A . J. Levine, R. J. Rob erts, M. Simon, C. Sla yman, M. Hu nk apiller, R. Bolanos, A. Delcher, I. Dew, D . F a- sulo, M. Flanigan, L. Florea, A. Halp ern, S. Hannenhalli, S. Kravitz, S. Levy , C. Mo- barry , K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley , K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramoulisw aran, R. Charlab, K. Chaturvedi, Z. Deng, V. D. F rancesco, P . D unn, K. Eilbeck, C. Ev angelista, A. E. Gabriel ian, W. Gan, W. Ge, F. Gong, Z. Gu, P . Guan, T. J. Heiman, M. E. Higgins, R.-R . Ji, Z. Ke, K. A. Ketch um, Z. Lai, Y. Lei, Z. Li, J. Li, Y. Liang, X. Lin, F. Lu, G. V . Merkulo v, N. Mil- shina, H. M. Moore, A. K. Naik, V. A. Nara yan, B. Neelam, D . Nu ssk ern, D . B. Ru sc h, S. Salzb erg, W. Sh ao, B. Shue, J. Sun, Z. Y. W ang, A. W an g, X. W an g, J. W ang, M.-H. W ei, R. Wides, C. X iao, C. Y an, A. Y ao, J. Y e, M. Zhan, W. Zhang, H . Zh an g, Q. Zhao, L. Zheng, F. Zh ong, W. Zhong, S. C. Zhu, S. Zhao, D. Gilb ert, S. Baumhueter, G. S pier, C. Carter, A. Crav c hik, T. W o o dage, F. Ali, H. An, A. Awe, D . Baldwin, H. Baden, M. Barnstead, I. Barrow , K . Beeson, D. Busam, A. Carver, A . Cen ter, M. L. Cheng, L. Curry , S. Danaher, L. Da v enp ort, R. Desilets, S. Dietz, K. Dod son, L. Doup , S. F er- riera, N. Garg, A. Gluecksmann, B. Hart, J. Haynes, C. Ha ynes, C. Heiner, S. Hladun , D. H ostin, J. H ouck, T. How land, C. Ibegwam, J. Johnson, F. Kalush, L. Kline, S. K o- duru, A. Lov e, F. Mann, D. Ma y , S. McCawl ey , T. McIntosh, I . McMullen, M. Moy , L. Moy , B. Murp hy , K. Nelson, C. Pfa nnkoch, E. Pratts, V. Puri, H. Qu reshi, M. Rear- don, R. Ro driguez, Y.-H . R ogers, D. R omblad, B. Ruh fel, R . Scott, C. Sitter, M. Small- w oo d, E. Stewart, R. St rong, E. Suh, R. Thomas, N . N. Tin t, S. Tse, C. V ech, G. W ang, J. W etter, S. Williams, M. Williams, S . Windsor, E. Winn-D een, K. W olfe, J. Zav eri, K. Zav eri, J. F. Abril, R . Guigo, M. J. Campb ell, K . V. Sjolander, B. K arlak, A . Ke- jariw al, H . Mi, B. Lazarev a, T. Hatton, A. Narechania, K . D iemer, A . Muruganuj an, N. Guo, S. Sato, V. Bafna, S. Istrail, R. Lip p ert, R. Sc hw artz, B. W alenz, S. Y o oseph, D. A llen, A. Basu, J. Baxendale, L. Blick, M. Caminha, J. Carnes-Stine, P . Caulk, Y.-H. Chiang, M. Coyne, C. Dahlke, A. D. Ma ys, M. Dom broski, M. D onnelly , D. Ely , S. Esparham, C. F osler, H . Gire, S. Glanowski, K . Glasser, A. Glodek, M. Gorokho v, K. Graham, B. Gropman, M. H arris, J. Heil, S. Henderson, J. H oov er, D. Jennings, C. Jordan, J. Jordan, J. Kasha, L. K agan, C. Kraft, A. Levitsky , M. Lewis, X. Liu, J. Lopez, D. Ma, W. Ma joros, J. McDaniel, S. Murphy , M. Newman, T. Nguyen, N. N guyen, M. No d ell, S. P an, J. Pec k, M. Peterson, W. Row e, R. Sanders, J. Scott, M. Simpson, T. Smith , A. Sprague, T. S tockw ell, R . T urner, E. V enter, M. W ang, M. W en, D. W u, M. W u, A. Xia, A. Zandieh, and X . Zhu. The Sequ ence of the Human Genome. Scienc e , 291:1304 –1351, 2001. H. Vino d. Canonical ridge and the econometrics of joint pro duction. Journal of Ec ono- metrics , 4:147–166 , 1976. 90 BIBLIOGRAPHY S. V olinia, M. Galasso, S. Costinean, L. T aglia v ini, G. Gam b eroni, A. D ru sco, J. Marc h- esini, N . Mascellani, M. E. Sana, R . Abu Jarour, C. Desp onts , M. T eitell, R. Baffa, R. Aq eilan, M. V . Iorio, C. T accioli, R. Garzon, G. Di Leva , M. F abb ri, M. Catozzi, M. Previati, S. Ambs, T. Pal umbo, M. Garo falo, A. V eronese, A. Bottoni, P . Gasparini, C. C. Harris, R. Visone, Y. P ek arsky , A. de la Chap elle, M. Blo omston, M. Dillhoff, L. Z. Rassenti, T. J. Kipps, K. Huebner, F. Pic hiorri, D. Lenze, S. Cairo, M. A. Buen- dia, P . Pineau, A. Dejean, N . Zanesi, S. Rossi, G. A. Calin, C. G. Liu, J. Pala tini, M. Negrini, A. V ecchione, A. Rosen b erg, and C. M. Cro ce. R eprogramming of miRN A netw orks in cancer and leukemia. Genome R ese ar ch , 20:589–599, 2010. S. W aaijenborg, P . C. V erselew el, d. W. Hamer, and A. H. Zwinderman. Quantif ying the A ssociation b etw een Gene Expressions and DNA-Markers by P enalized Canonical Correlation Analysis. Statistic al Applic ations in Genetics and Mole cular Biolo gy , 7:3, 2008. J. W atson and F. Crick. A structure for deoxyrib ose nucleic acid. Natur e , 171:737 –738, 1953. M. W est. Bay esian factor regression models in the ’large p, small n’ paradigm. Bayesian statistics , 7:723–732 , 2003. D. L. Wheeler, T. Barrett, D. A. Benson, S. H . Bry ant, K. Canese, D. M. Church, M. DiCuccio, R. Edgar, S. F ederhen , W. H elm berg, D. L. Kenton, O. Khov ayk o, D. J. Lipman, T. L. Madd en, D . R. Maglott, J. Ostell, J. U. Po ntius, K. D. Pruitt, G. D. Sch uler, L. M. Schriml, E. S equeira, S. T. Sh erry , K. Sirotkin, G. Starc henko, T. O. Suzek, R . T atu sov, T. A . T atusov a, L. W agner, , and E. Y aschenk o. Database resources of th e national cen ter for biotechnolo gy information. Nucleic A cids R ese ar ch , 33:D39– 45, 2005. D. J. Wilkinson. Ba y esian meth ods in bioinformatics an d computational systems b iology . Briefings in Bioinformatics , 8:109–116, 2007. D. M. Witten, R. Tibshirani, and T. Hastie. A p enalized matrix decomp osition, with applications to sparse p rincipal comp onents and canonical correlation analysis. Bio- statistics , 10:515–53 4, 2009. C. W u, R. Carta, an d L . Zhang. Sequence dep endence of cross-hybridization on short oligo microarra ys. Nuc leic A ci ds R ese ar ch , 33:e84, 2005. Z. W u and R. Irizarry . S tochastic models inspired by hybridization theory f or short oligon ucleotide arra ys. In P . E. Bourne and D. Gusfi eld, editors, Pr o c e e dings of the 8th Ann ual International Confer enc e on Computational Mole cular Biolo gy (RECOMB’04) , pages 98–106. ACM Press, New Y ork, 2004. V. W un derlic h. Early references to the mutational origin of cancer. International Journal of Epidemiolo gy , 36:24 6–247, 2007. Y. Y amanishi, J.-P . V ert, A. Naka ya, and M. Kanehisa. Extraction of correlated gene clusters from multiple genomic data by generalized kernel canonical correlation analy- sis. Bioi nf ormatics , 19:i32 3–330, 2003. Y. Y amanishi, M. Kotera, M. Kanehisa, and S. Goto. Drug-target interaction pred ic- tion from chemical , genomic and pharmacol ogical data in an i ntegra ted framew ork. Bioinformatics , 26:i246–25 4, 2010. F. Y ates. Contingency t ab les involving small num bers an d the χ 2 test. Journal of the R oyal Statistic al So ciety Supplement , 1:217–239, 1934. 91 BIBLIOGRAPHY C. L. Y auk, M. L. Berndt, A. Williams, and G. R. Douglas. Comprehensiv e comparison of six microarra y technologies. Nucleic Ac ids R ese ar ch , 32:e12 4, 2004. J. Zhang, R. P . Finney , R. J. Clifford, L. K. Derr, and K. H. Buetow. Detecting false expression signals in high-density oligon ucleotide arrays by an in silico approach. Ge- nomics , 85:297–30 8, 2005. L. Zhang, L. W ang, A. R a vindranathan, and M. Miles. A new algorithm for analysis of oligonucleo tide arrays: Application to exp ression profiling in mouse brain regions. Journal of Mole cular Bi olo gy , 317:225–235, 2002. W. Zh an g, Q. Morris, R. Chang, O. Shai, M. Bak o wski, N. Mitsak akis, N. Mohammad, M. R obinson, R. Zirngibl, E. Somogyi, N. Laurin, E. Eftekharp our, E. Sat, J. Grigull, Q. P an, W.-T. P eng, N. Krogan, J. Greenblatt, M. F ehlings, D. v an der Ko oy , J. Aubin, B. Bruneau, J. R ossan t, B. Blenco w e, B. F rey , and T. Hu ghes. The functional landscap e of mouse gene expression. Journal of Biolo gy , 3:21, 2004. X. Zhou and G. Gibson. Cross-sp ecies comparison of genome-wide expression patterns. Genome Bi olo gy , 5:232, 2004. D. Zhu, A . O. Hero, H. Cheng, R. Khanna, an d A. Swaroop. Net w ork constrained clustering for gene m icroarray data. Bi oinformatics , 21:4014– 4020, 2005. 92
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment