크라이오ET 데이터 처리 전 과정을 자동화한 통합 워크플로우
본 논문은 전자크라이오단층촬영(CryoET) 데이터의 획득부터 서브톰그래프 평균화까지 전 과정을 자동화한 통합 파이프라인을 제시한다. 자동화된 틸트 시리즈 정렬, 퓨리에 기반 재구성, 입자별·틸트별 CTF 보정, 그리고 SGD 기반 초기 모델 생성과 입자·틸트별 정밀 정렬을 통해 인간 개입을 최소화하고, 정제된 단백질과 세포 내 복합체 모두에서 서브나노미터 해상도를 달성한다.
저자: Muyuan Chen, James M. Bell, Xiaodong Shi
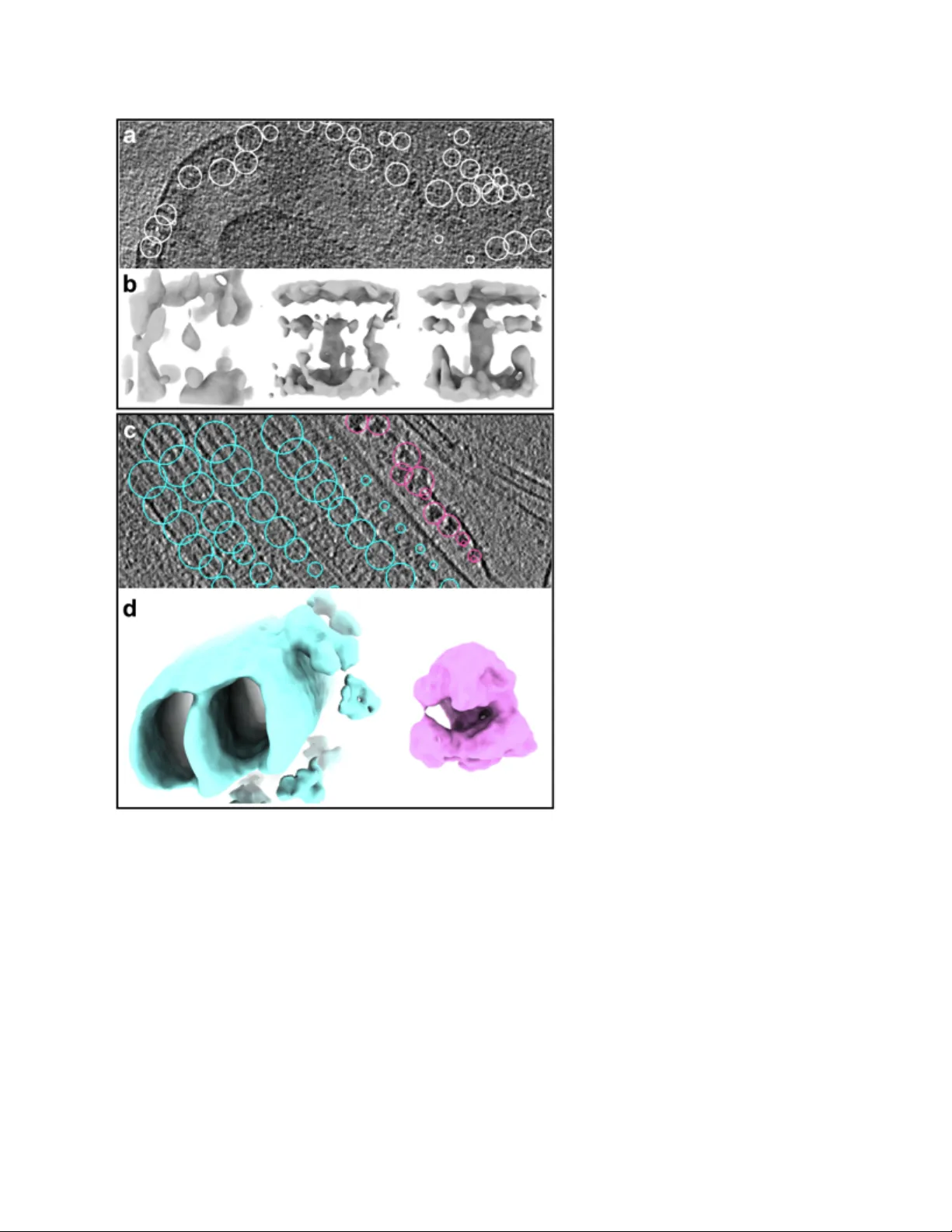
본 논문은 전자크라이오단층촬영(CryoET) 데이터 처리의 전 과정을 자동화하고 통합한 워크플로우를 EMAN2 환경에 구현하였다. 연구는 크게 네 부분으로 구성된다. 첫 번째는 자동 틸트 시리즈 정렬이다. 기존에는 fiducial 마커가 필요했으나, 저자들은 랜드마크 기반 반복 정렬 알고리즘을 개발하여 fiducial이 없거나 불완전한 경우에도 자동으로 정렬이 가능하도록 하였다. 이 알고리즘은 초기에는 512 × 512 픽셀로 다운샘플링된 이미지에서 가장 어두운 voxel을 랜드마크로 선택하고, 3D 좌표를 투영해 2D 패치를 추출한다. 이후 로컬 서브틸트 재구성을 통해 질량 중심을 보정하고, 전역 최적화 과정을 통해 모든 랜드마크가 일치하도록 변환 매개변수를 업데이트한다. 정렬 과정은 점진적으로 샘플링 해상도를 높이며, 이상치 랜드마크는 각 반복에서 제외한다.
두 번째 단계는 퓨리에 기반의 타일링 재구성이다. 전통적인 실공간 방법(SIRT 등)은 메모리와 연산량이 크게 소요되고, 가장자리 효과가 두드러진다. 저자들은 겹치는 정육면체 타일을 정의하고, 각 타일을 퓨리에 역변환한 뒤 가우시안 가중 평균을 적용해 전체 볼륨을 재구성한다. 이 방식은 메모리 사용을 최소화하면서도 고해상도 정보를 보존한다. 재구성된 톰그램은 시각화와 입자 선택을 위해 다운샘플링되지만, 서브톰그래프 평균화 단계에서는 원본 4 k × 4 k 틸트 이미지를 그대로 사용한다.
세 번째 단계는 입자·틸트별 CTF 보정이다. 기존의 틸트별 단순 CTF 보정은 얇은 시료에서는 충분하지만, 세포와 같은 두꺼운 시료에서는 입자 Z 위치에 따른 초점 차이가 크게 작용한다. 저자들은 전체 이미지의 전력 스펙트럼을 타일별로 나누어 각 타일의 중심 초점을 최적화하고, 이를 입자 3D 좌표와 틸트 기하학에 매핑해 각 틸트마다 정확한 초점값을 산출한다. 저 SNR 상황에서도 전체 이미지 정보를 활용해 안정적인 초점 추정이 가능하도록 설계되었다.
네 번째 단계는 초기 모델 생성과 서브톰그래프 정렬·평균이다. 초기 모델은 기존에 카탈로그 매칭에 의존하던 방식을 탈피해, 확률적 경사 하강법(SGD) 기반 비편향적 방법을 사용한다. 무작위로 선택된 입자 서브셋을 저해상도(≈100 Å) 필터링 후 평균화하고, 이를 반복적으로 정렬·평균·업데이트함으로써 30–50 Å 수준의 초기 모델을 빠르게 얻는다. 초기 모델이 확보되면 전통적인 서브톰그래프 정렬 알고리즘을 적용해 웨지 효과를 보정하고, 골드-스탠다드 FSC 검증을 통해 독립적인 홀드아웃을 수행한다.
마지막으로 입자·틸트별 정밀 정렬을 수행한다. 각 입자에 대해 모든 틸트 이미지의 5가지 회전·이동 파라미터를 독립적으로 최적화하고, 품질이 낮은 틸트는 가중치를 낮추거나 제외한다. 이렇게 얻어진 각 틸트별 정렬 파라미터를 이용해 새로운 가중 평균 3D 맵을 생성하고, 이를 반복적으로 업데이트한다. 결과적으로 기존 방법에 비해 해상도 제한 요인을 크게 감소시켜, 세포 내 14 Å 수준의 단백질 구조부터 정제된 복합체에서 3 Å 수준까지 다양한 시료에 적용 가능함을 입증한다.
성능 평가에서는 60장의 4 k × 4 k 틸트 시리즈(총 2 k × 2 k × 512 다운샘플 재구성)를 12코어 워크스테이션에서 전체 파이프라인을 실행하는 데 약 10⁴분(≈7 일)이 소요된다고 보고하였다. 이는 데이터 획득 시간과 동등하거나 그 이하이며, 실시간 자동화가 가능함을 시사한다. 실제 적용 사례로는 기존에 발표된 apoferritin 데이터와 E. coli 전체 세포에서 TolC‑AcrAB 복합체를 14 Å 해상도로 복원한 결과가 제시되었다.
요약하면, 이 통합 워크플로우는 자동 틸트 정렬, 퓨리에 타일링 재구성, 입자·틸트별 CTF 보정, SGD 기반 초기 모델링, 그리고 입자·틸트별 정밀 정렬을 하나의 파이프라인으로 결합함으로써 CryoET 데이터 처리의 효율성과 정확성을 크게 향상시킨다. 인간 개입을 최소화하고, 다양한 시료에 적용 가능한 범용성을 갖추어 CryoET 기반 구조생물학 연구의 생산성을 크게 증대시킬 것으로 기대된다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기