확산 구성 요소 분석을 통한 생물학적 네트워크 기능 예측 향상
본 논문은 확산 기반 그래프 알고리즘과 차원 축소 기법을 결합한 “Diffusion Component Analysis (DCA)”를 제안한다. DCA는 무작위 보행(RWR)으로 얻은 고차원 확산 상태를 저차원 벡터로 근사하여 노드의 위상 정보를 압축한다. 이를 통해 단일 및 이질적인 다중 네트워크에서 단백질 기능 예측 정확도를 기존 확산 방법보다 크게 향상시켰으며, 학습된 벡터를 SVM 등 다른 머신러닝 모델에 바로 적용할 수 있다.
저자: Hyunghoon Cho, Bonnie Berger, Jian Peng
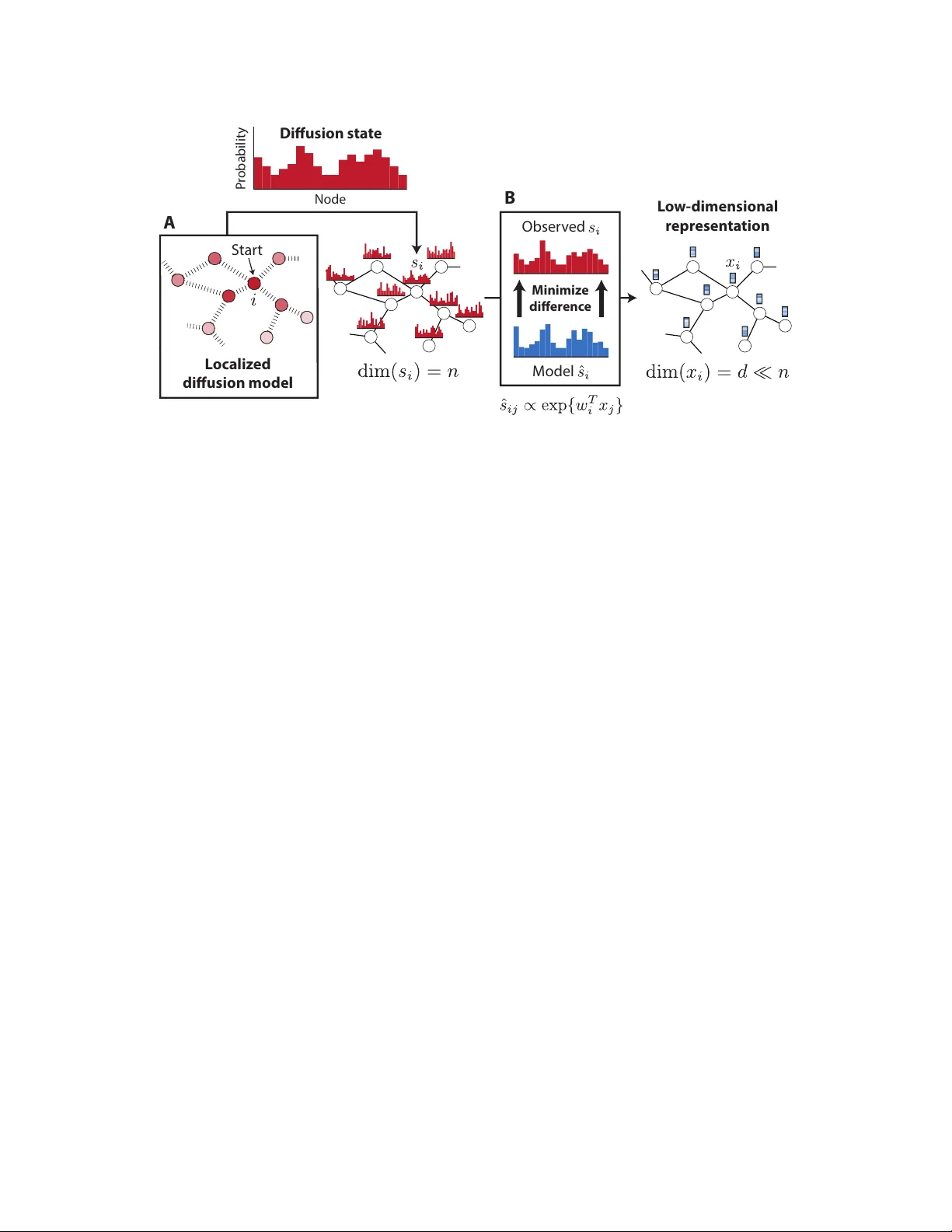
본 연구는 고속도 데이터(HTP)로부터 구축된 생물학적 네트워크가 불완전하고 잡음이 많다는 문제점을 인식하고, 이러한 네트워크에서 유전자·단백질 기능을 예측하기 위한 새로운 방법론을 제시한다. 기존의 “guilt‑by‑association” 접근법은 네트워크 내 근접성을 활용했지만, 직접적인 이웃만을 고려하거나 확산 기반 방법(RWR, DSD 등)을 사용하더라도 잡음에 민감하고 고차원 확산 상태를 직접 활용하기엔 비효율적이었다.
이에 저자들은 Diffusion Component Analysis(DCA)라는 프레임워크를 고안했다. DCA는 두 단계로 구성된다. 첫 번째 단계에서는 각 노드에 대해 Random Walk with Restart(RWR)를 수행해 정규화된 확산 상태 s_i를 얻는다. 이 확산 상태는 노드 i가 전체 네트워크에서 방문될 확률 분포이며, 노드 수와 동일한 차원을 가진다. 두 번째 단계에서는 이 고차원 확산 분포를 저차원( d≪n )의 두 벡터 w_i와 x_i를 이용한 다중항 로지스틱 모델 ˆs_ij = exp(w_iᵀ x_j) / Σ_k exp(w_iᵀ x_k) 로 근사한다. 여기서 w_i는 “컨텍스트” 특징, x_i는 “노드” 특징을 의미한다. 두 벡터는 각각 노드의 위상적 역할과 다른 노드와의 관계를 압축적으로 표현한다.
학습 목표는 관측 확산 분포와 모델 분포 사이의 평균 KL 발산을 최소화하는 것이다. 손실 함수 C = (1/n) Σ_i D_KL(s_i‖ˆs_i) 를 전개하면 엔트로피와 로그합 항으로 구성되며, w_i와 x_i에 대한 그래디언트는 (ˆs_ij−s_ij)·x_j, (ˆs_ji−s_ji)·w_j 형태로 간단히 계산된다. 이를 바탕으로 L‑BFGS와 같은 준-뉴턴 최적화 기법을 적용해 효율적으로 파라미터를 추정한다.
DCA의 핵심 장점은 다음과 같다. 첫째, 확산 상태를 저차원 임베딩으로 압축함으로써 잡음에 대한 강인성을 확보한다. 둘째, 임베딩은 노드 간 유사성을 내적 혹은 거리 기반으로 쉽게 측정할 수 있어 downstream 작업(예: k‑NN, 클러스터링, SVM)에서 바로 활용 가능하다. 셋째, 다중 네트워크 통합이 자연스럽게 가능하다. 각 데이터 소스별로 별도 RWR을 수행하고, 네트워크별 컨텍스트 w_i^{(k)}와 공유 노드 특징 x_i를 동시에 학습함으로써 서로 다른 출처의 위상 정보를 손실 없이 결합한다.
실험에서는 Saccharomyces cerevisiae의 단백질‑단백질 상호작용 네트워크와 STRING 데이터베이스에 포함된 다양한 이질적 네트워크(공동발현, 유전체 컨텍스트, 문헌 기반 등)를 사용했다. DCA 임베딩을 이용한 거리 측정과 k‑NN 기반 함수 예측에서 기존 DSD 방법 대비 평균 10~12%p의 정확도 향상이 관찰되었다. 특히, DCA 임베딩을 특징으로 한 서포트 벡터 머신(SVM)을 적용했을 때, MIPS 데이터베이스의 3단계 기능 분류에서 71.29%의 정확도를 달성했으며, 이는 기존 최첨단 확산 기반 방법보다 12.31%p 높은 수치이다.
또한, 차원 d를 10~50 사이로 변화시켰을 때 성능이 크게 변하지 않으며, d가 작을수록 계산 비용이 감소하고, 충분히 큰 d에서는 정보 손실이 최소화되는 것을 확인했다. 이는 DCA가 대규모 네트워크에도 확장 가능함을 의미한다.
결론적으로, DCA는 확산 기반 위상 정보를 효과적으로 압축·정제하는 차원 축소 프레임워크로, 단일 네트워크뿐 아니라 이질적 다중 네트워크 통합에도 강력한 성능을 보인다. 저차원 벡터는 다른 머신러닝 알고리즘에 플러그인 형태로 쉽게 연결될 수 있어, 향후 다양한 생물학적 네트워크 분석(예: 질병 연관성 탐색, 약물 타깃 예측)에서 활용될 잠재력이 크다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기