통합 mRNA와 DNA 복제수 분석을 통한 암 유전자 우선순위 선정 비교 리뷰
본 리뷰는 유전체 복제수(CN)와 전사발현(GE) 데이터를 동시에 활용해 암 관련 유전자를 우선순위화하는 12가지 알고리즘을 비교한다. 시뮬레이션 데이터와 실제 유방암·백혈병 데이터셋을 이용해 ROC‑AUC, 상위 200개 유전자에서의 진양성 비율, 실행 시간을 평가했으며, edira, Ortiz‑Estevez, pint/simcca, PREDA/SODEGI R이 전반적으로 우수한 성능을 보였다. 또한, R 패키지 intcomp을 통해 투명한 …
저자: Leo Lahti, Martin Sch"afer, Hans-Ulrich Klein
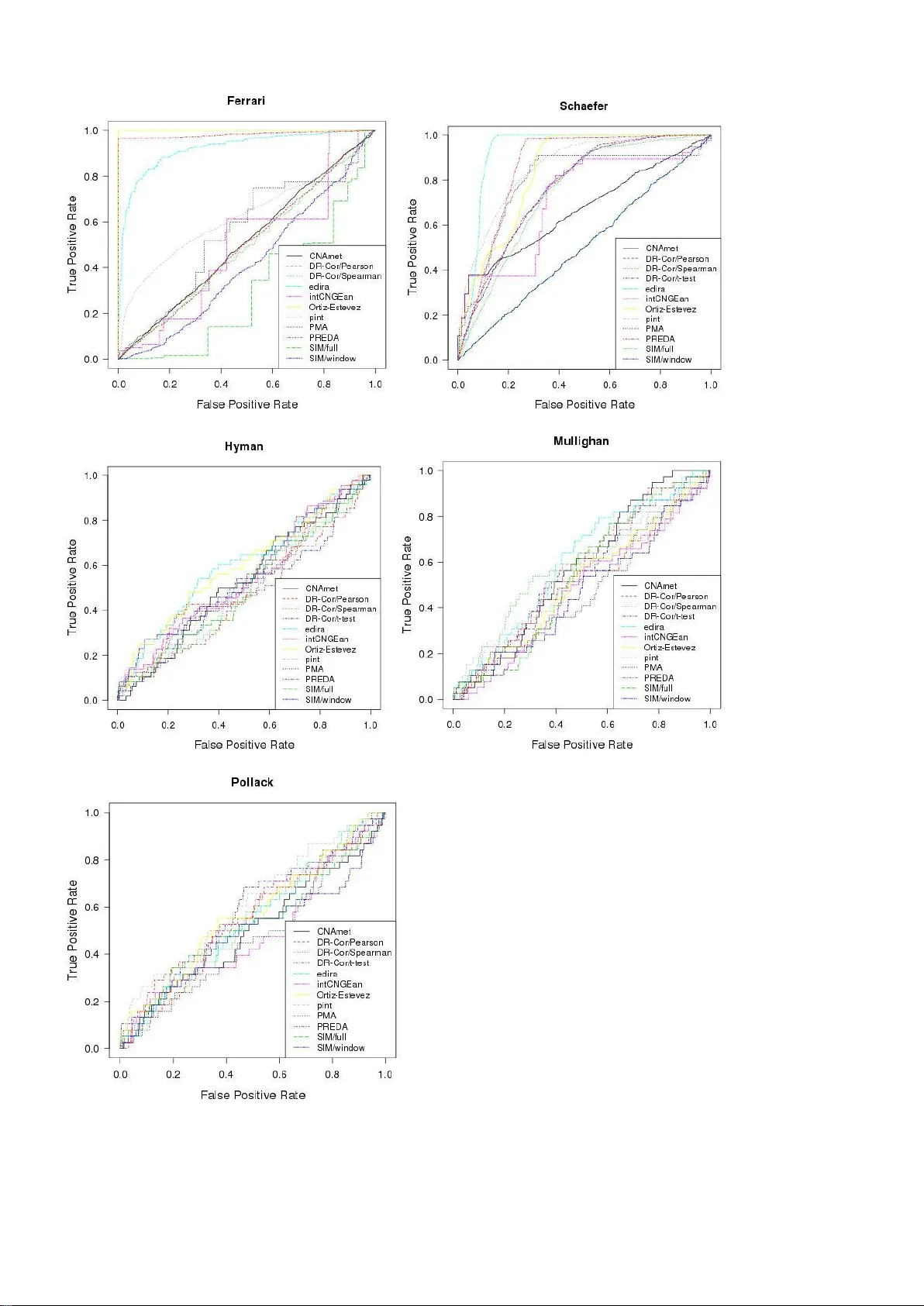
본 논문은 암 연구에서 복제수(CN)와 전사발현(GE) 데이터를 동시에 활용해 암 유전자를 우선순위화하는 다양한 통합 분석 방법을 체계적으로 비교한다. 먼저, PubMed와 Google Scholar, 그리고 Bioconductor 저장소를 통해 ‘gene expression’, ‘copy number’, ‘integration’ 등의 키워드로 검색한 결과, 현재 공개된 12개의 알고리즘을 선정하였다. 이들 알고리즘은 크게 두 단계 접근법, 회귀 기반, 상관 기반, 정준 상관(CCA) 및 그 변형, 잠재 변수 모델(예: pint/simcca, PREDA/SODEGI R) 등으로 구분된다. 각 방법은 CN 데이터를 어떻게 전처리하고, GE와 매핑하며, 통계적 유의성을 어떻게 평가하는지에 차이가 있다.
벤치마크는 두 개의 시뮬레이션 데이터셋과 세 개의 실제 암 데이터셋(두 개의 유방암, 하나의 급성 림프구성 백혈병)으로 구성되었다. ‘Schäfer’ 시뮬레이션은 정규 혼합분포를 이용해 16개의 블록에 복제수와 발현 변화를 부여하고, 잡음 수준을 다섯 단계로 조절했다. ‘Ferrari’ 시뮬레이션은 실제 신장암 데이터를 변형해 10개의 10 Mbp 블록에 복제수와 발현을 인위적으로 조정했다. 실제 데이터에서는 각각 전문가가 선별한 유방암 유전자 리스트와 Cancer Gene Census에 등재된 백혈병 유전자를 골드 스탠다드로 사용하였다.
성능 평가는 세 가지 지표로 이루어졌다. 첫째, 전체 유전자 리스트에 대한 ROC 곡선 아래 면적(AUC)이며, 이는 전체적인 진양성/위양성 비율을 반영한다. 둘째, 상위 200, 100, 50, 20개의 유전자에서 실제 암 유전자가 차지하는 비율(진양성 비율)으로, 실제 실험에서 검증 가능한 후보를 얼마나 잘 추출하는지를 평가한다. 셋째, 각 알고리즘의 실행 시간으로, 실용적인 측면을 고려한다.
결과적으로, 전체 AUC 평균 순위에서는 edira가 가장 높은 중위 순위를 차지했으며, Ortiz‑Estevez와 pint/simcca가 그 뒤를 이었다. 상위 200개 유전자에서의 진양성 비율에서는 pint/simcca가 가장 높은 순위를 기록했고, edira, Ortiz‑Estevez, PREDA/SODEGI R이 그 뒤를 따랐다. 실행 시간 측면에서는 edira와 PMA가 1분 이내로 가장 빠르게 수행되었으며, Ortiz‑Estevez는 최대 3분, 반면 permutation 기반 방법(CNAmet, DR‑Correlate 등)은 수십 분에서 수시간까지 걸렸다. 특히, 시뮬레이션 데이터는 해당 알고리즘의 설계 가정에 맞게 생성되었기 때문에 일부 방법이 과도하게 좋은 성능을 보였으며, 실제 데이터에서는 전반적인 진양성 비율이 낮아 골드 스탠다드 자체가 불완전함을 시사한다.
논문은 이러한 비교 결과를 바탕으로, 연구자가 자신의 데이터와 목적에 맞는 통합 방법을 선택할 때 고려해야 할 요소들을 제시한다. 또한, R‑패키지 intcomp을 공개함으로써 새로운 알고리즘, 데이터셋, 평가 지표를 손쉽게 추가·비교할 수 있는 투명하고 재현 가능한 벤치마크 환경을 제공한다. 이는 향후 암 유전자 탐색 및 기능적 해석 연구에 중요한 기반이 될 것으로 기대된다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기